Targeted Delivery of Doxorubicin: Drug Delivery System Based on PAMAM Dendrimers
N. G. Yabbarov1*, G. A. Posypanova2, E. A. Vorontsov3, O. N. Popova2, and E. S. Severin3
1Bioengineering Center, Russian Academy of Sciences, pr. 60-letiya Oktyabrya 7/1, 117312 Moscow, Russia; fax: (499) 613-4818; E-mail: nikita_yabbarov@yahoo.com2National Research Center “Kurchatov Institute”, pl. Akademika Kurchatova 1, 123182 Moscow, Russia; fax: (499) 196-1704
3Russian Research Center of Molecular Diagnostics and Therapy, Simferopolsky Bulvar 8, 117149 Moscow, Russia; fax: (499) 613-2633
* To whom correspondence should be addressed.
Received November 27, 2012; Revision received March 13, 2013
Polyamidoamine (PAMAM) dendrimers of the second generation (G2) are branched polymers containing 16 surface amino groups that allow them to be used as universal carriers on creating systems for drug delivery. G2 labeled with fluorescein isothiocyanate (FITC) efficiently bound with the surface of tumor cells at 4°C and was absorbed by the cells at 37°C. The covalent binding to G2-FITC of a vector protein, a recombinant fragment of the human alpha-fetoprotein receptor-binding domain (rAFP3D), increased the binding and endocytosis efficiency more than threefold. Covalent conjugates of G2 with doxorubicin (Dox) obtained by acid-labile linking of cis-aconitic anhydride (CAA) without the vector protein (G2-Dox) and with the vector protein rAFP3D (rAFP3D-G2-Dox) were accumulated by the tumor cells with high efficiency. However, a selective effect was observed only in rAFP3D-G2-Dox, which also demonstrated high cytotoxic activity against the human ovarian adenocarcinoma SKOV3 cells and low cytotoxicity against human peripheral blood lymphocytes. Based on these results, rAFP3D-G2 conjugate is promising for selective delivery of antitumor drugs.
KEY WORDS: dendrimers, targeted drug delivery, endocytosis, alpha-fetoprotein, acid-labile linker, antitumorDOI: 10.1134/S000629791308004X
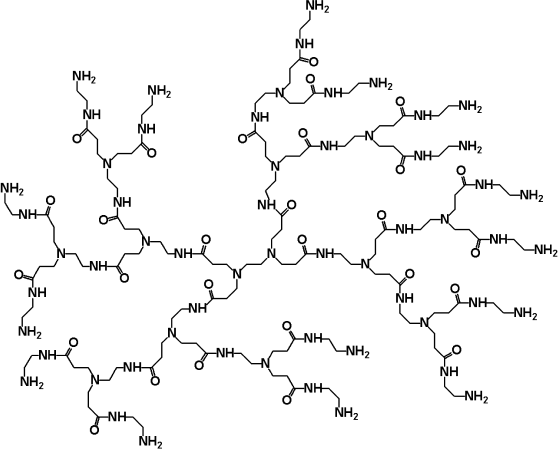
Dendrimers are branched spherically symmetric or asymmetric polymeric macromolecules characterized by a strictly controlled structure, definite molecular weight, monodispersity, and biocompatibility. Moreover, the biodegradation of dendrimers can be controlled. Dendrimers have a dendritically branched structure and consist of a core, which is the branching center, separate branches, and terminal chemical groups [1]. Over the past few years, dendrimers have been used with increasing frequency as carriers for delivery of biologically active compounds into cells [2] and also for the controlled release of drugs [3]. Earlier, the creation of conjugates and complexes of dendrimers with different substances due to encapsulation into the inner cavities or to interaction of hydrophobic, hydrophilic, and amphiphilic compounds with the surface groups of dendrimers were reported. Such hydrogen bonding, ionic, or hydrophobic interactions can concern fluorescent and radioactive labels, antitumor drugs, peptides, proteins, oligosaccharides, oligonucleotides, proteins, and nucleic acids [4]. These conjugations or complexing are associated with marked changes in pharmacokinetic parameters of preparations under testing; moreover, these parameters can be changed by different approaches: introduction of photo-, pH-, and ultrasound-sensitive links, changing the charge of surface chemical groups of the dendrimer molecules or of preparations to promote the absorption or release of the preparation, application of self-degrading dendritic molecules, etc. [5]. In the present work, we have used polyamidoamine dendrimers of the second generation (PAMAM G2) (Fig. 1). On the surface of G2 there are 16 amino groups that can be modified.
Fig. 1. Structure of PAMAM G2.
The covalent binding of pharmaceutical preparations with the surface groups of a dendrimer allows introduction of a significant number of molecules of a pharmaceutical preparation. The application of certain linking agents allows control the release of the preparation from the conjugate. Such links are hydrolyzed under the influence of specific conditions (acidic values of pH or the presence of specific proteases) that leads to rapid release of the pharmaceutical preparation with retention of its initial structure. As a pharmaceutical preparation, we have used the anthracycline antitumor antibiotic doxorubicin (Dox), which is widely used as a therapeutic agent. Doxorubicin causes cell death due to two main mechanisms: 1) intercalation into DNA through the incorporation of Dox between two neighbor nucleotides, which provides a reliable interaction with DNA and disturbs replication and transcription, and 2) binding and inhibition of topoisomerase II [6]. Due to presence in its structure of a quinone group, Dox is involved in redox processes generating free radicals responsible for DNA damages and lipid peroxidation. Thus, the efficiency of the antitumor action of Dox depends on its intracellular accumulation.
Although dendrimers are very promising as nanocarriers, their conjugates or complexes with pharmaceutical preparations lack a targeted action that limits their application. But the targeted influence is extremely important, in particular, in chemotherapy of oncological diseases. A joining to dendrimers of vector molecules capable of selective binding with specific receptors on the tumor cell surface and internalizing by these cells seems promising for increasing therapeutic efficiency and decreasing systemic toxicity of antitumor agents. Transferrin [7], folic acid [8], and also chimeric or humanized antibodies [9, 10] to surface antigens of tumor cells are often used as vectors.
We proposed that an approach based on use of the oncofetal protein alpha-fetoprotein (AFP) receptor as a target could be promising. The AFP receptor is a marker of malignant tumors [11, 12]. Application of natural or recombinant AFP as a vector for delivery of visualizing or cytotoxic preparations directly into the tumor would allow high diagnostic and therapeutic specificity and minimize side effects. Numerous experimental studies have confirmed that the natural human AFP, used as a vector for the targeted delivery of antitumor antibiotics into cancer cells, is a very efficient and selective system for delivery both in vitro and in vivo [13, 14].
AFP is a single-chained glycoprotein consisting of 609 amino acid residues with the molecular weight of 68-74 kDa according to different estimations. The primary structure of human AFP is subdivided into three domains. Each domain consists of about 200 amino acid residues. The site responsible for the receptor binding was detected in the third domain of the AFP molecule [15]. The application of rAFP3D as a vector molecule seems to be a solution for the problem of selective delivery of anticancer drugs into tumor cells [16].
MATERIALS AND METHODS
Reagents and equipment. PAMAM dendrimers of the second generation with terminal NH2-groups and ethylenediamine-based core were from Dendritech (USA); the BCA set, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), doxorubicin hydrochloride, cis-aconitic anhydride (CAA), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrasolium bromide (MTT), and fluorescein isothiocyanate (FITC) from Sigma-Aldrich (USA); RPMI 1640 culture medium from Gibco (USA); fetal bovine serum (FBS) from Hyclone (USA); Mowiol from Calbiochem (USA); culture flasks (25 cm2), 96- and 24-well plates from Corning-Costar (USA); dialysis sacks (CelluSeph, MWCO 1 kDa), Sephadex G-25, Superose 12, and Ni-IDA Sepharose were from GE Healthcare (USA); other reagents were from Fluka (Germany). The purification and analysis were performed using a Shimadzu LC-20 Prominence chromatography system (Japan) and a Luna C4 (2 × 100 mm) HPLC column (Phenomenex, USA), TSK gel G2000SWxl (7.8 × 300 mm) (Tosoh Bioscience, Japan), C18 (2.1 × 100 mm) (DrMaisch GmbH, Germany).
For rAFP3D expression, we used a commercial plasmid vector pET20 that contains a human AFP gene fragment encoding amino acid residues from 404 to 609 and comprising six additional histidine residues. The plasmid was kindly provided by A. N. Fedorov (Russian Research Center of Molecular Diagnostics and Therapy (RCMDT)). The human ovarian adenocarcinoma cell line was from the cell bank of RCMDT.
Preparation and analysis of rAFP3D. rAFP3D was expressed using a vector based on a commercial pET20 plasmid containing the human AFP gene fragment encoding amino acid residues 404-609 and containing six additional histidine residues.
The protein was expressed in the E. coli strain BL21(DE3)pUBS. The bacterial cells were transformed using the standard procedure [17]. The protein was located in inclusion bodies. The biomass was ultrasonicated, the inclusion bodies were precipitated by centrifugation, and rAFP3D was isolated as described earlier [18]. rAFP3D was purified on a column with Ni IDA Sepharose, and the purified rAFP3D was folded on the same column. The purity and homogeneity of rAFP3D was determined by reversed-phase HPLC using a Luna C4 column in a linear gradient of acetonitrile (5-60%, 40 min) in the presence of 0.1% trifluoroacetic acid at flow rate 0.2 ml/min; the protein was detected by absorption at 214 and 280 nm. The rAFP3D concentration was determined by using a BCA kit.
Preparation of FITC-labeled PAMAM dendrimers G2 (G2-FITC). All conjugates were synthesized using G2 with eight amino groups blocked with acetic anhydride [19]. For this, 0.1 g (0.03 mmol) of G2 was dissolved in 5 ml of anhydrous methanol, then 0.031 g (0.3 mmol) acetic anhydride and 0.062 g (0.6 mmol) triethylamine were added. The reaction mixture was incubated at 20°C for 12 h, and the methanol was evaporated under vacuum. The modified G2 was dissolved in water and dialyzed against water using 1-kDa MWCO dialysis membranes. The resulting product (G2) was lyophilized, and the number of terminal methyl groups was determined by 1H NMR (comparing the area of methyl group peak at 1.89 ppm and the total area of methylene protons (2.28-3.35 ppm)) and stored at 4°C.
1H NMR of the acetylated G2 derivative (300 MHz, D2O), δ ppm): 1.89 (24H, s, -C(O)CH3), 2.25-2.44 (56H, m, -C-CH2-C(O)-NH), 2.51-2.60 (28H, m, -N-CH2-CH2-NH), 2.66-2.81 (56H, m, -N-CH2-CH2-C(O)-), 2.95-3.06 (18H, -CH2NH2), 2.11-3.35 (70H, -C(O)NH-CH2).
The modified G2 was dissolved in 10 ml of phosphate-buffered saline (PBS) (1.7 mM KH2PO4, 5.2 mM Na2HPO4, 150 mM NaCl in H2O) to the concentration of 0.01 g/ml, pH was adjusted to 8, and a 10-fold molar excess of FITC in dimethyl sulfoxide (DMSO) was added. The reaction mixture was stirred at room temperature for 2 h and then for 12 h at 4°C. G2-FITC was purified from unreacted reagents by dialysis (twice for 4 h against the 1000-fold volume of PBS). The fluorescein content in the conjugate was determined by photocolorimetry at 498 nm using the known molar extinction coefficient ε498 = 6.8·104 liter·mol–1·cm–1 [20].
Synthesis of rAFP3D conjugate with PAMAM dendrimers G2 and FITC (rAFP3D-G2-FITC). Solution containing 0.5 mg (0.022 µmol) rAFP3D in 1 ml of PBS (pH 6.8) was supplemented with 2 mg (0.6 µmol) acetylated derivative of G2 containing eight amino groups, and the pH value was adjusted to 6.8. Then 0.042 mg (0.22 µmol) EDC was added (the stock was 10 mg/ml in PBS, pH 6.8) and the mixture was incubated at 4°C for 1 h. Then the pH of the reaction mixture was adjusted to 7.4, and the mixture was incubated with stirring at 4°C for 12 h. The conjugate was isolated on a column (10 × 300 mm) with Superose 12 with PBS (pH 7.4) as eluent. The molecular weight of the rAFP3D-G2 conjugate was determined by Laemmli electrophoresis in polyacrylamide gel and by HPLC on a TSK gel G2000SWxl column with PBS (pH 7.4) as eluent at rate flow 1 ml/min. The absorption was detected at 280 nm. The size of the rAFP3D-G2 conjugate (0.5 mg/ml solution in PBS, pH 7.4) was determined using a Zetasizer Nano ZS ZEN 3600 (Malvern) equipped with a He-Ne laser with wavelength 633 nm. The rAFP3D-G2 size was calculated using a Zetasizer program (ver. 6.32, Malvern).
To 2.6 mg (0.088 µmol) of the resulting rAFP3D-G2 conjugate, FITC stock solution in DMSO (0.17 mg (0.44 µmol), 2 mg/ml) was added. The reaction mixture was stirred for 2 h at room temperature and then for 12 h at 4°C. The conjugate was purified from unreacted reagents on a column (10 × 100 mm) with Sephadex G25. The fluorescein content in the conjugate was determined by photocolorimetry.
Synthesis of PAMAM dendrimer G2 conjugates with Dox (G2-Dox). The Dox-CAA derivative was prepared by the modified method of Shen and Ryser [17]. To do this, 10 mg (17.2 µmol) Dox-HCl was dissolved in 3 ml of 0.1 M NaHCO3 at 0°C. CAA (6.2 mg, 40 µmol) was dissolved in 300 µl of p-dioxane and slowly added to the Dox solution on ice while maintaining the reaction mixture pH at 8.5. The solution was incubated with intensive stirring for 20 min at 0°C, then for 20 min at 25°C. Then the reaction mixture was cooled on ice, supplemented with 1 M HCl until formation of a precipitate, centrifuged at 4°C for 10 min at 10,000g, then the supernatant was removed, and the precipitate was dissolved in PBS (pH 7.4) and stored at —70°C.
The amount of free Dox and also of its cis- and trans-isomers produced during the Dox-CAA synthesis was determined by HPLC using a C18 column and buffer A with the following composition: 3% (NH4)2CO3 in H2O–CH3OH–CH3CN (50 : 45 : 5 v/v) at rate flow 0.2 ml/min. The Dox derivatives were detected by absorption at 481 nm. The resulting Dox-CAA in PBS (pH 7.4) was supplemented with 10-fold molar excess of EDC, and the mixture was stirred at 4°C for 1 h.
G2 was dissolved in 1 ml of PBS to the concentration of 10 mg/ml, pH was adjusted to 7.4, and 0.01 g (1.5 mmol) Dox-CAA was added (the preparation and activation of Dox-CAA are described above). The reaction mixture was incubated with intensive stirring at 4°C for 12 h. The G2-Dox was purified from unreacted reagents by dialysis twice for 4 h against the 1000-fold volume of PBS. The purity of the G2-Dox was analyzed by HPLC using a C18 column in buffer A at flow rate 0.2 ml/min. The Dox derivatives were detected at 481 nm.
Synthesis of rAFP3D conjugate with PAMAM dendrimer G2 and Dox (rAFP3D-G2-Dox). The rAFP3D-G2 conjugate and Dox-CAA were prepared as described above. To 1 mg (0.069 µmol) rAFP3D-G2 in 2 ml PBS, 0.48 mg (0.69 µmol) Dox-CAA was added, and the mixture was incubated with intensive stirring at 4°C for 12 h. The conjugate was purified from unreacted reagents on a column (10 × 100 mm) with Sephadex G25.
The doxorubicin content in the rAFP3D-G2-Dox conjugate was determined by absorption at 481 nm using the known molar extinction coefficient ε481 = 10,410 liter·mol–1·cm–1 [21]. The rAFP3D concentration was determined with a BCA kit.
Study of doxorubicin release from G2-Dox conjugate. The rate of Dox release from the conjugate was determined in citrate-phosphate buffer (0.1 M citric acid, 0.2 M Na2HPO4) with pH values of 5.0, 5.5, 6.0, and 7.4 at 37°C. G2-Dox in PBS (pH 7.4) was added to 4.5 ml of the preheated citrate-phosphate buffer to the concentration of 0.1 mg/ml, and then the pH value of the solution was corrected. Solution aliquots were taken after 0, 1, 15, 60, 120, 240, 360, and 1440 min of incubation and frozen in liquid nitrogen. The amount of free Dox in the aliquots was determined by HPLC in buffer A using a C18 column at flow rate 0.2 ml/min. The detection was performed at 481 nm.
Cell cultures. Human ovarian adenocarcinoma SKOV3 cell line was maintained in plastic flasks in RPMI 1640 medium supplemented with 10% fetal bovine serum and gentamycin (50 µg/ml) in a CO2-incubator at 37°C in a humidified atmosphere containing 5% CO2. The cells were replated with Versene solution twice per week. Mononuclear leukocytes were isolated from peripheral blood of healthy donors by centrifugation in Ficoll-Paque gradient as described by Boyum [22].
Flow cytometry analysis of binding and endocytosis of rAFP3D-G2-FITC, rAFP3D-G2-Dox, and G2-Dox by tumor cells and lymphocytes. Before the experiment, the cells were preincubated for 2 h in serum-free RPMI 1640 medium. To analyze the binding, the conjugates at concentrations from 35 to 2500 nM were added on ice to the cell suspension and incubated at 4°C for 1 h. To study endocytosis, incubation was performed at 37°C. After the incubation, the cells were washed thrice with cold PBS and fixed in 2% p-formaldehyde. The fluorescence intensity of the cells was measured with an EPICS-XL flow cytometer equipped with an argon laser (exciting wavelength 488 nm, 525 nm band-pass filter for FITC and the 620 nm band-pass filter for Dox). For each sample (105 cells), the mean fluorescence intensity (MFI) was determined.
Laser confocal microscopy. The SKOV3 cells were plated onto cover glasses into 24-well plates (2.5·104 cells in 0.5 ml of culture medium per well) the day before the experiment. Before the experiment, the cells were preincubated for 2 h in serum-free RPMI 1640 medium. Then the medium was removed, and rAFP3D-G2-Dox, G2-Dox, or Dox (2 µM Dox) in 0.5 ml of the culture medium was added, and the samples were incubated for 4 and 24 h under standard conditions (in a CO2-incubator at 37°C in a humidified atmosphere containing 5% CO2). After the incubation, the cells were washed thrice with PBS, fixed in 4% p-formaldehyde, washed thrice with PBS supplemented with 50 mM glycine to decrease the autofluorescence, and embedded in Mowiol. The fluorescence intensity and distribution of rAFP3D-G2-Dox and G2-Dox in the cells was studied using a Carl Zeiss Cell Observer Z1 confocal microscope with a 100× objective. The photos were finally processed with the Photoshop program.
Study on cytotoxic activity of rAFP3D-G2-Dox and G2-Dox conjugates. To assess the cytotoxic activity, the cells were seeded into 96-well plates (5000 cells per well) the day before the experiment. The conjugates and free Dox were added to the cells at concentrations from 0.01-10 µM in triplets, and then incubated under standard conditions for 72 h. The cell vitality was determined using the MTT-test [23]. Four hours before the end of the incubation, 50 µl of MTT solution in the culture medium (1 mg/ml) was added into each well. Upon color development, the medium was removed, dropped formazan crystals were dissolved in 100 µl of DMSO, and the color intensity was determined by absorption at 540 nm with a microplate reader. Cell viability was determined as percent of untreated control. For designing the survival curves, calculation of IC50 values, and statistical treatment of the results the OriginPro (OriginLab Corporation) program was used.
RESULTS
Characteristics of rAFP3D. The recombinant protein rAFP3D corresponding to the fragment containing amino acid residues 404-609 of human AFP was isolated from the inclusion bodies by metal-chelate chromatography. The rAFP3D was renatured and finally purified by immobilization on Ni-IDA Sepharose. As determined by HPLC (Fig. 2a) and by electrophoresis (Fig. 2b), the molecular weight of rAFP3D is 22.5 kDa.
Fig. 2. The molecular weight determination of rAFP3D-G2. a) Gel-permeation chromatography: the peak eluting at 7.6 min corresponds to rAFP3D-G2 and that at 7.99 min to rAFP3D. As standards of molecular weights, we used the following substances: 1) cytochrome c (12.4 kDa); 2) lysozyme (14 kDa); 3) rAFP3D (22.5 kDa); 4) peroxidase (40.2 kDa); 5) ovalbumin (40.3 kDa); 6) BSA (67 kDa); 7) IgG (150 kDa). b) Electrophoresis in polyacrylamide gel: 1) molecular weight standards; 2) G2; 3) AFP3D; 4) AFP3D-G2; 5) AFP3D-G2-FITC; 6) AFP3D-G2-Dox.
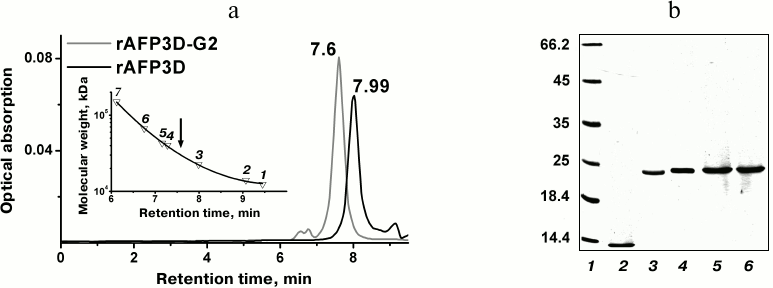
Synthesis and characteristics of G2-FITC and rAFP3D-G2-FITC conjugates. An acetylated derivative of G2 with eight amino groups was used in all stages of the work. COOH-groups of rAFP3D were activated with EDC. rAFP3D was conjugated with G2 according to the scheme presented in Fig. 3 (1). According to data of dynamic light scattering, the rAFP3D-G2 conjugate size was ~4 nm. By data of gel filtration (Fig. 2a), the molecular weight of the rAFP3D-G2 conjugate was ~30 kDa. According to electrophoresis, the rAFP3D-G2 sample did not contain rAFP3D aggregations, and the molecular weight of the conjugate was close to the weight of unmodified rAFP3D (Fig. 2b). Thus, in the rAFP3D-G2 conjugate one molecule of rAFP3D is bound with two G2 molecules.
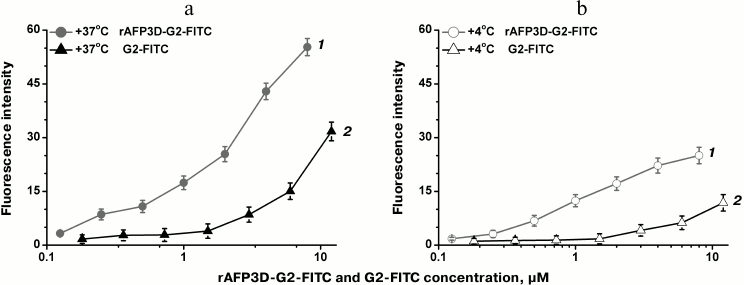
To study binding and endocytosis by tumor cells of rAFP3D and G2, their FITC-derivatives were synthesized; the ratio of G2 and FITC was similar in both conjugates (to exclude the influence of different numbers of free amino groups on the absorption by the cells (Fig. 3 (2)). The G2/FITC ratio in the G2-FITC conjugate was 1 : 3, and the rAFP3D/FITC ratio in the rAFP3D-G2-FITC conjugate was 1 : 6. The binding and endocytosis efficiency was assessed by flow cytometry. On analyzing the cells treated with G2-FITC and AFP3D-G2-FITC, values of the mean fluorescence intensity of the cells were normalized to FITC. Figure 4 presents the binding and endocytosis of rAFP3D-G2-FITC and G2-FITC by the SKOV3 line cells of human ovarian adenocarcinoma. The binding and endocytosis of G2-FITC by the tumor cells was significantly lower than the same parameters for rAFP3D-G2-FITC.
Fig. 3. Schemes of synthesis of the conjugates: rAFP3D-G2 (1), rAFP3D-G2-FITC (1a), rAFP3D-G2-Dox (1b); G2-FITC and G2-Dox (2); Dox-CAA (3).
Fig. 4. Binding and endocytosis by SKOV3 cells of rAFP3D-G2-FITC (1) and G2-FITC (2). a) Endocytosis at 37°C; b) binding at 4°C.
Synthesis and characteristics of G2-Dox and rAFP3D-G2-Dox conjugates. To prepare the conjugate, Dox was modified with acid-labile cis-aconitic anhydride CAA (Fig. 3 (3)). From HPLC data, the resulting Dox-CAA contained about 3% of free Dox. The total yield of Dox-CAA was about 90%. Upon activation of the COOH-groups, Dox-CAA was conjugated with the acetylated derivative of G2 containing eight amino groups and with rAFP3D-G2.
1H-NMR of Dox-CAA (300 MHz, DMSOd6), δ (ppm): 7.90 (2H, m, Dox), 7.62 (1H, s, Dox), 5.28 (1H, d, Dox), 4.94 (1H, m, Dox), 4.88 (2H, m, Dox), 4.60 (2H, d, -C(O)CH2-, Dox), 4.20 (1H, q, Dox), 4.00-3.95 (4H, m, H at the 3C of the carbohydrate residue, cis/trans-Dox-CAA, 3H at -O-CH3, Dox), 3.60 (1H, d, Dox), 3.01-2.82 (2H, m, Dox), 2.15 (2H, m, Dox), 1.89 (1H, t, Dox), 1.70 (1H, dd, Dox), 1.17 (3H, d, Dox). On integration, the cis/trans-derivatives of the cis-aconitic acid residue at Dox did not result approximately integer values because of the presence in the mixture of cis- and trans-Dox-CAA, δ (ppm): 5.69 (s, -CH, cis-CAA), 3.03 (m, -CH2, cis-CAA), 8.86 (d, NH, cis-CAA); 6.43 (s, -CH, trans-CAA), 3.19 (m, -CH2, trans-CAA), 7.58 (d, NH, trans-CAA).
Conjugates with three Dox molecules per G2 molecule displayed good stability in water. Introduction of more than three Dox molecules per G2 increased the hydrophobicity of the conjugates and decreased their solubility. Because the activated derivative of Dox-CAA can react with amino groups of both G2 and rAFP3D, to determine the amount of Dox capable of binding directly to rAFP3D, Dox-CAA was conjugated with rAFP3D under the same conditions.
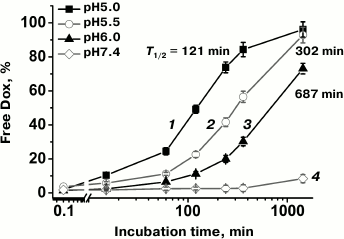
Release of Dox from G2-Dox conjugate. The release of Dox from the G2-Dox conjugate was studied by HPLC upon incubation of G2-Dox samples in citrate-phosphate buffer with pH values of 5.0, 5.5, 6.0, and 7.4 at 37°C. Results of the experiment are shown in Fig. 5. At neutral pH values, G2-Dox was stable: after 24 h of incubation only 8% of free Dox was recorded in the solution. At pH 6.0, the halftime of free Dox release (the time of release from the conjugate of 50% free Dox) was 687 min, and in 24 h 75% of Dox was in the free state. At pH 5.5, the half-release time decreased to 302 min, and in 24 h 90% of Dox was in the free state. At pH 5.0, the half-release time was 121 min, and in 24 h 93% of Dox was in the free state.
Fig. 5. Release of Dox from the G2-Dox conjugate at different pH values: 1) 5.0; 2) 5.5; 3) 6.0; 4) 7.4. T1/2 is the half-time for Dox release.
Endocytosis and intracellular distribution of rAFP3D-G2-Dox and G2-Dox. The endocytosis of rAFP3D-G2-Dox and G2-Dox was studied using flow-cytometry and confocal microscopy. Mononuclear leukocytes of human peripheral blood lacking AFP receptors on their surface and containing 95% unstimulated lymphocytes [24] were used as control cells.
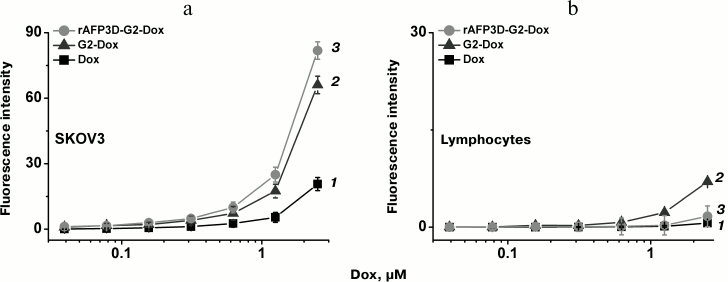
Upon 1 h of incubation of the SKOV3 line cells at 37°C, the accumulation by the cells of rAFP3D-G2-Dox determined by the Dox fluorescence was comparable to the accumulation of G2-Dox and was significantly higher (more than fivefold) than the accumulation of free Dox (Fig. 6a). The accumulation of the rAFP3D-G2-Dox and G2-Dox conjugates and of free Dox in the lymphocytes was lower than in the tumor cells (Fig. 6b). The accumulation of G2-Dox in lymphocytes was about fivefold higher than the accumulation of rAFP3D-G2-Dox and free Dox.
Fig. 6. Uptake of rAFP3D-G2-Dox, G2-Dox, and free Dox by ovarian adenocarcinoma SKOV3 cells (a) and in human peripheral blood lymphocytes (b) evaluated by fluorescence intensity of the cells. 1) rAFP3D-G2-Dox; 2) G2-Dox; 3) Dox.
The intracellular distribution of rAFP3D-G2-Dox, G2-Dox, and Dox in the SKOV3 cells was studied using confocal microscopy after incubation for 4 and 24 h.
The fluorescence intensity of the cells incubated with G2-Dox for 4 and 24 h was lower than the fluorescence of the cells treated with rAFP3D-G2-Dox and free Dox during the same time intervals (Fig. 7). The increase in incubation time to 24 h significantly increased the fluorescence of the cells treated with Dox and rAFP3D-G2-Dox and less pronouncedly increased the fluorescence of the cells treated with G2-Dox. The G2-Dox fluorescence was mainly located in the cytoplasm after incubation for 4 h (Fig. 7e) and for 24 h (Fig. 7k). In the cells treated with Dox and rAFP3D-G2-Dox, the Dox fluorescence was distributed between the nucleus and cytoplasm, but was mainly located in the nucleus (Fig. 7, c, g, i, m).
Fig. 7. Intracellular distribution of Dox, G2-Dox, and rAFP3D-G2-Dox in SKOV3 cells after incubation for 4 h (c-h) and for 24 h (i-n) in the presence of Dox (c, d, i, j), the G2-Dox conjugate (e, f, k, l), and the rAFP3D-G2-Dox conjugate (g, h, m, n). a, b) Control cells. a, c, e, g, i, k, m) Fluorescence confocal microscopy. b, d, f, h, j, l, n) Differential integral contrast. The bar corresponds to 10 µm. The section is made at the middle height of the cells.

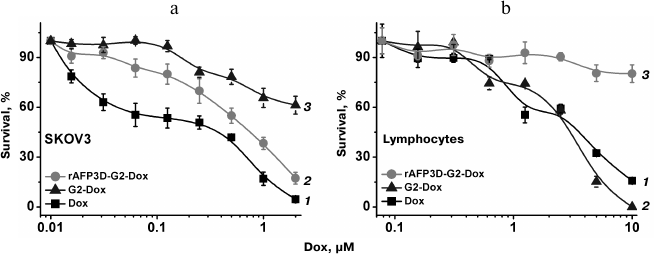
Cytotoxic activity of G2-Dox and rAFP3D-G2-Dox. The cytotoxic activity of the conjugates was evaluated using the MTT-test after incubation for 72 h of SKOV3 cells and peripheral blood mononuclear leukocytes with G2-Dox, rAFP3D-G2-Dox, and free Dox. The rAFP3D-G2-Dox conjugate manifested high cytotoxic activity to the tumor SKOV3 cells (Fig. 8a, curve 2). The IC50 value for this conjugate was 0.615 µM. The G2-Dox cytotoxicity to the SKOV3 cells was noticeably lower (Fig. 8a, curve 1), the IC50 for G2-Dox being 4.5 µM. For lymphocytes the G2-Dox toxicity was close to that of free Dox (Fig. 8b, curves 1 and 2), the IC50 values being 2.7 and 3 µM, respectively. The toxicity of rAFP3D-G2-Dox for the control cells was significantly lower (Fig. 8b, curve 3), the IC50 value for rAFP3D-G2-Dox being 64 µM.
Fig. 8. Survival of SKOV3 cells of human ovary adenocarcinoma (a) and human peripheral blood lymphocytes (b) after incubation for 72 h in the presence of Dox and its conjugates with G2 and rAFP3D-G2. 1) Dox; 2) G2-Dox; 3) rAFP3D-G2-Dox.
DISCUSSION
Over the past few years, PAMAM dendrimers have been used with increasing frequency as carriers of various drugs [5]. PAMAM dendrimers of the second generation, unlike older generations’ dendrimers, have low toxicity; therefore, we used just them for our studies. We used partially acetylated G2 as a polymeric carrier for Dox conjugated with the surface amino groups of the dendrimer due to CAA. The resulting amide bond is stable at neutral pH values, but at pH < 7.4 Dox is released from the conjugate as an initial, unmodified drug, which was shown by studies on the release of Dox. With decrease in the pH value, the rate of Dox release increased. After 24 h of incubation at pH values of 5.0, 5.5, and 6.0, 75-93% of the Dox was in a free state, whereas at pH 7.4 only 8% of Dox was released from the conjugate. Thus, the acid-labile link between the antibiotic and carrier ensured the stability of the conjugate in the solution and the release of doxorubicin within cellular compartments with acidic pH values: lysosomes and late endosomes.
The G2-Dox conjugate was efficiently internalized by the tumor cells. The entrance of the conjugate into the cells during 1 h of incubation was fivefold higher than the absorption of free Dox. However, the cytotoxicity of the G2-Dox conjugate to the SKOV3 line cells of human ovarian adenocarcinoma was low: the IC50 value for G2-Dox was nine times lower than the IC50 for Dox. The study by confocal microscopy on the internalization of free Dox and G2-Dox in the same cells for longer time intervals, for 4 and 24 h, revealed a significant difference in the penetration and in the localization of these preparations: the accumulation in the cells of free Dox was significantly higher. And the fluorescence of free Dox was observed mainly in the nuclei, i.e. in the location of its target – DNA. G2-Dox was distributed uniformly through the whole cytoplasm as point inclusions; even upon incubation for 24 h the nuclei contained minimal amounts of Dox.
The entrance mechanism of the PAMAM dendrimers into the cells significantly depends on the charge of the surface groups. Cationic dendrimers interact most efficiently with the plasma membrane of the cells [19]. To prepare conjugates with FITC and Dox, we used the G2 derivative with eight acetamide groups. This modification increased the hydrophobicity of G2, which theoretically would be able to improve the penetration of G2-FITC or G2-Dox into the cells. However, this modification is associated with a significant decrease in the G2 charge, which significantly weakens the G2 tropism to the plasma membrane. Because Dox also is rather hydrophobic, it was supposed that G2-Dox would be able to enter the cells not only through endocytosis, but also by diffusion, and that is partially conformed by our data on the intracellular distribution of fluorescence in the cells treated with G2-Dox (Fig. 7, e, k).
PAMAM dendrimers containing cationic groups can be internalized by cells due to absorptional endocytosis [25], clathrin- and caveolin-macropinocytosis [19], caveolin-dependent endocytosis [26], caveolin-independent endocytosis [27], and also by clathrin-dependent endocytosis [28]. According to the literature data, the penetration mechanism of cationic PAMAM dendrimers depends on the dendrimer generation and the cell type. Cationic dendrimers of earlier generations are internalized by the cells of some human carcinomas and of mouse melanoma through caveolin-dependent endocytosis [26, 29, 30]. Now we cannot definitely indicate the real pathway of G2-Dox internalization by the adenocarcinoma SKOV3 line cells: this is a subject requiring a special study. However, only in the case of the clathrin-dependent endocytosis is G2-Dox delivered into cellular compartments with acidic pH values, i.e. into the late endosomes and lysosomes. As judged by the intracellular distribution of G2-Dox (Fig. 7, e, k) and data on the cytotoxicity to the SKOV3 line cells, the G2-Dox conjugate seems not to enter these compartments. At neutral pH values the G2-Dox conjugate was very stable: after incubation at 37°C for 24 h only 8% of the Dox was released from the conjugate. Just this seems to cause the low cytotoxicity of the conjugate for the SKOV3 line cells. Moreover, because of the hydrophobicity of Dox, the maximal number of antibiotic molecules joined to G2 on the conjugation did not exceed three, i.e. the efficiency of G2 as a carrier was also relatively low. To increase the delivery efficiency, we used as a vector protein the recombinant receptor-binding fragment of human alpha-fetoprotein rAFP3D, which had to: 1) provide the delivery into endosomes and lysosomes of the construction containing the antitumor agent, and 2) ensure the selectivity of this delivery.
Earlier it was shown that the use of vector molecules of natural AFP [13, 31] and of its receptor-binding fragment located in the third domain [16] significantly increases the efficiency of pharmaceutical preparation delivery into tumor cells. This is due to expression of the AFP receptors in cells of malignant tumors, whereas they are absent in normal tissues.
We found that the binding and endocytosis efficiency of the fluorescein-labeled conjugate rAFP3D-G2-FITC by the SKOV3 tumor cells was significantly higher than the binding and endocytosis efficiency of G2-FITC lacking the vector protein (Fig. 4). However, the absorption by the same cells of the rAFP3D-G2-Dox conjugate after 1 h of incubation was only slightly higher than the absorption of G2-Dox (Fig. 6a). The accumulation in the tumor cells of rAFP3D-G2-Dox was more than five times higher than the accumulation of free Dox in the same cells. On increasing the incubation time, the accumulation by the cells of the rAFP3D-G2-Dox conjugate steadily increased (Fig. 7, g, h, m, n). And the fluorescence was located both in the cytoplasm and nuclei, but after 24 h of incubation it was mainly in the nuclei. This suggested a gradual release of Dox from the rAFP3D-G2-Dox conjugate due to acidic pH values in late endosomes and to diffusion of the free antibiotic into the cell nuclei. We think that the probable joining of Dox molecules to rAFP3D by the protein amino groups does not significantly influence the ability of rAFP3D to bind the AFP receptors on the tumor cell surface, similarly to the lack of influence of two FITC molecules joining to rAFP3D [32].
The absorption of the rAFP3D-G2-Dox conjugate by the control cells, peripheral blood lymphocytes lacking AFP receptor on their surface, was low: it was 50 times lower than the absorption by the SKOV3 cells (Fig. 6b) and more than three times lower than the absorption by the lymphocytes of the G2-Dox conjugate lacking the vector protein. It seems that the accumulation of rAFP3D-G2-Dox in the peripheral blood mononuclear leukocytes is mainly contributed by monocytes: although they represent only 5% of mononuclear leukocytes, they intensively absorb AFP [33] and rAFP3D (unpublished data).
The rAFP3D-G2-Dox conjugate was highly cytotoxic for the SKOV3 tumor cells (Fig. 8a) and was poorly toxic for the lymphocytes (Fig. 8b), as differentiated from the G2-Dox conjugate. The cytotoxicity of G2-Dox for the lymphocytes was comparable to the toxicity of free Dox and close to that of G2-Dox for the SKOV3 adenocarcinoma: the IC50 value of G2-Dox was 4 µM for the SKOV3 cells and 3 µM for the lymphocytes.
The introduction into the conjugate of the vector protein rAFP3D significantly decreased the toxicity of the conjugate for the control cells (Fig. 8b). Thus, the introduction of rAFP3D into the construction markedly increased the selectivity of action of the G2-Dox.
Based on comparing the data on the Dox release from the conjugate and on the intracellular distribution of rAFP3D-G2-Dox, we suppose that the conjugate should display cytotoxicity more slowly than free Dox. Thus, during 4 h at pH 5.5 (the pH value in the late endosomes) more than 40% of Dox was released from the rAFP3D-G2-Dox conjugate (Fig. 5). Dox released from the conjugate leaves endosomes/lysosomes and migrates into the nucleus (Fig. 7, g, h, m, n). Only after 24 h of incubation at pH 5.0-5.5 Dox is virtually completely released from the conjugate. Free Dox penetrates into the cell by diffusion and is rapidly accumulated in the nucleus (Fig. 7, c, d), where it manifests its cytotoxicity. And the cytotoxicity of free Dox for the tumor cells in vitro was actually two times higher than the cytotoxicity of the rAFP3D-G2-Dox conjugate.
Our findings show selectivity of action on tumor cells of the rAFP3D-G2-Dox conjugate containing rAFP3d as a vector molecule, and this has been confirmed by studies on the binding, endocytosis, intracellular distribution, and cytotoxicity of the conjugate for the tumor cells and peripheral blood lymphocytes from healthy donors. Thus, we conclude that rAFP3D-G2 is promising as an efficient system for targeted delivery of Dox. Possibly, the efficiency of this system can be increased by application of more hydrophilic pharmaceutical preparations.
REFERENCES
1.Tomalia, D. A., Baker, H., Dewald, J., Hall, M.,
Kallos, G., Martin, S., Roeck, J., Ryder, J., and Smith, P. (1985)
Polymer J., 17, 117-132.
2.Aulenta, F., Hayes, W., and Rannard, S. (2003)
Eur. Polymer J., 39, 1741-1771.
3.Svenson, S., and Tomalia, D. A. (2005) Adv. Drug
Deliv. Rev., 57, 2106-2129.
4.Boas, U., and Heegaard, P. M. H. (2004) Chem.
Soc. Rev., 33, 43-63.
5.Sampathkumar, S.-G., and Yarema, K. J. (2007) in
Nanotechnologies for the Life Sciences, Wiley-VCH Verlag GmbH &
Co. KGaA.
6.Sinha, B. K., and Politi, P. M. (1990) Cancer
Chemother. Biol. Response Modif., 11, 45-57.
7.Agarwal, A., Saraf, S., Asthana, A., Gupta, U.,
Gajbhiye, V., and Jain, N. K. (2008) Int. J. Pharmac.,
350, 3-13.
8.Ward, B. B., Dunham, T., Majoros, I. J., and Baker,
J. R., Jr. (2011) J. Oral Maxillofac. Surg., 69,
2452-2459.
9.Chen, K., and Mitchell, D. (2012) in Glioma
(Yamanaka, R., ed.) Springer, New York, Vol. 746, pp. 121-141.
10.Miyano, T., Wijagkanalan, W., Kawakami, S.,
Yamashita, F., and Hashida, M. (2010) Mol. Pharmac., 7,
1318-1327.
11.Moro, R., Tcherkassova, J., and Song, E. (2005)
IVD Technology [Online] Available at:
http://www.ivdtechnology.com/article/new-broad-spectrum-cancer-marker.
12.Nitsvetov, M. B., Moskaleva, E. Y., Posypanova,
G. A., Makarova, O. V., Stepanov, V. A., Rogov, K. A., Koromyslova, I.
A., Karaulov, A. V., Severin, S. E., and Severin, E. S. (2005)
Immunologiya, 26, 122-125.
13.Moskaleva, E. Y., Posypanova, G. A., Koromyslova,
I. A., Shmyrev, I. I., Krivonos, A. V., Myagkikh, I. V., Feldman, N.
B., Finakova, G. V., Katukov, V. Y., Luzhkov, Y. M., Nakachian, R.,
Andreani, J., Severin, E. S., and Severin, S. E. (1996) Tumor
Target., 2, 299-306.
14.Severin, S. E., Posypanova, G. A., Katukov, V.
Y., Shmyrev, I. I., Luzhkov, Y. M., Gerasimova, G. K., Zhukova, O. S.,
Vorozhtsov, G. N., Kaliya, O. L., Lukyanets, E. A., and Severin, E. S.
(1997) Biochem. Mol. Biol. Int., 43, 1081-1089.
15.Posypanova, G. A., Gorokhovets, N. V., Makarov,
V. A., Savvateeva, L. V., Kireeva, N. N., Severin, S. E., and Severin,
E. S. (2008) J. Drug Target., 16, 321-328.
16.Godovannyi, A. V., Savvateeva, M. V.,
Sotnichenko, A. I., Yabbarov, N. S., Klimova, O. V., Gnuchev, H. V.,
and Severin, S. E. (2011) Mol. Med., 1, 44-48.
17.Sambrook, J., and Russell, D. G. (2001)
Molecular Cloning: A Laboratory Manual, 3rd Edn., Cold Spring
Harbor Laboratory Press, New York.
18.Sharapova, O. A., Yurkova, M. S.,
Laurinavichyute, D. K., Andronova, S. M., Fedorov, A. N., Severin, S.
E., and Severin, E. S. (2011) J. Chromatogr. A, 1218,
5115-5119.
19.Perumal, O. P., Inapagolla, R., Kannan, S., and
Kannan, R. M. (2008) Biomaterials, 29, 3469-3476.
20.Butcher, E. C., and Weissman, I. L. (1980) J.
Immunol. Meth., 37, 97-108.
21.Tian, Y., Bromberg, L., Lin, S. N., Alan Hatton,
T., and Tam, K. C. (2007) J. Contr. Rel., 121,
137-145.
22.Boyum, A. (1968) Scand. J. Clin. Lab.
Invest., 97 (Suppl.), 77-89.
23.Denizot, F., and Lang, R. (1986) J. Immunol.
Meth., 89, 271-277.
24.Torres, J. M., Laborda, J., Naval, J., Darracq,
N., Calvo, M., Mishal, Z., and Uriel, J. (1989) Mol. Immunol.,
26, 851-857.
25.Kitchens, K. M., Kolhatkar, R. B., Swaan, P. W.,
and Ghandehari, H. (2008) Mol. Pharm., 5, 364-369.
26.Saovapakhiran, A., D’Emanuele, A., Attwood,
D., and Penny, J. (2009) Bioconj. Chem., 20, 693-701.
27.Qi, R., Mullen, D. G., Baker, J. R., and Banaszak
Holl, M. M. (2009) Mol. Pharm., 7, 267-279.
28.Kitchens, K. M., Foraker, A. B., Kolhatkar, R.
B., Swaan, P. W., and Ghandehari, H. (2007) Pharm. Res.,
24, 2138-2145.
29.Seib, F. P., Jones, A. T., and Duncan, R. (2007)
J. Contr. Rel., 117, 291-300.
30.Albertazzi, L., Serresi, M., Albanese, A., and
Beltram, F. (2010) Mol. Pharm., 7, 680-688.
31.Severin, S. E., Posypanova, G. A., Sotnichenko,
A. I., Moskaleva, E. Y., Feldman, N. B., Grigoriev, M. I., Severin, E.
S., and Petrov, R. V. (1999) Dokl. Akad. Nauk SSSR, 366,
561-564.
32.Sharapova, O. A., Posdnyakova, N. V.,
Laurinavichyute, D. K., Yurkova, M. S., Posypanova, G. A., Andronova,
S. M., Fedorov, A. N., Severin, S. E., and Severin, E. S. (2010)
Bioorg. Khim., 36, 760-768.
33.Esteban, C., Trojan, J., Macho, A., Mishal, Z.,
Lafarge-Frayssinet, C., and Uriel, J. (1993) Leukemia, 7,
1807-1816.