A Straightforward Experimental Approach to Expression, Purification, Refolding, and Enzymatic Analysis of Recombinant Dengue Virus NS2B(H)-NS3pro Protease
M. Junaid1,2, C. Angsuthanasombat2, J. E. S. Wikberg3, N. Ali4, and G. Katzenmeier2*
1Department of Pharmacy, University of Malakand, Chakdara, 18550 Pakistan; fax: +92-9467-6349; E-mail: juniphdr@gmail.com2Laboratory of Molecular and Cellular Microbiology, Institute of Molecular Biosciences, Mahidol University, Salaya, 73170 Thailand; fax: +662-441-9906; E-mail: katzenmeier.ger@mahidol.ac.th; chanan.ang@mahidol.ac.th
3Division of Pharmacology, Department of Pharmaceutical Biosciences, Uppsala University, 75124 Sweden; fax: +46-1855-9718; E-mail: jarl.wikberg@farmbio.uu.se
4Department of Pharmacology, Institute of Basic Medical Sciences, Khyber Medical University, Peshawar, Khyber Pakhtunkhwa, 25000 Pakistan; fax: +92-9192-17704; E-mail: niazpharmacist@yahoo.com
* To whom correspondence should be addressed.
Received February 14, 2013; Revision received April 23, 2013
Dengue virus threatens around 2.5 billion people worldwide; about 50 million become infected every year, and yet no vaccine or drug is available for prevention and/or treatment. The flaviviral NS2B-NS3pro complex is indispensable for flaviviral replication and is considered to be an important drug target. The aim of this study was to develop a simple and generally applicable experimental strategy to construct, purify, and assay a highly active recombinant NS2B(H)-NS3pro complex that would be useful for high-throughput screening of potential inhibitors. The sequence of NS2B(H)-NS3pro was generated by overlap extension PCR (SOE-PCR) and cloned into the pTrcHisA vector. Hexahistidine-tagged NS2B(H)-NS3pro complex was expressed in E. coli predominantly as insoluble protein and purified to >95% purity by single-step immobilized metal affinity chromatography. SDS-PAGE followed by immunoblotting of the purified enzyme demonstrated the presence of the NS2B(H)-NS3pro precursor and its autocleavage products, NS3pro and NS2B(H), as 37, 21, and 10 kDa bands, respectively. Kinetic parameters, Km, kcat, and kcat/Km for the fluorophore-linked protease model substrate Ac-nKRR-amc were obtained using inner-filter effect correction. The kinetic parameters Km, kcat, and kcat/Km for Ac-nKRR-amc substrate were 100 µM, 0.112 s–1, and 1120 M–1·s–1, respectively. A simplified procedure for the cloning, overexpression, and purification of the NS2B(H)-NS3pro complex was applied, and a highly active recombinant NS2B(H)-NS3pro complex was obtained that could be useful for the design of high-throughput assays aimed at flaviviral inhibitor discovery.
KEY WORDS: Dengue virus, NS2B(H)-NS3pro protease, purification, assay, substrateDOI: 10.1134/S0006297913080099
The threat to human health posed by infections with dengue virus has increased dramatically during the last decade. Currently 2.5 billion people, that is two fifth of the world population, are at risk to dengue, with an estimated 50 million infections occurring annually [1]. Dengue virus (DEN) is a member of the Flaviviridae family and a positive single-strand RNA virus that is transmitted to humans by Stegomyia aegypti (formerly Aedes) mosquitoes. Dengue causes a spectrum of diseases, which range from mild dengue fever to the more severe forms, dengue hemorrhagic fever and dengue shock syndrome [2, 3].
There are four serotypes of DEN (DEN1-4), which share approximately 65% identity of their genomes. Of these, DEN2 is associated with a higher prevalence of dengue hemorrhagic fever cases and is thus considered to be a more virulent strain. At present, there is neither a vaccine nor a specific antiviral drug available for the treatment of dengue diseases. However, a number of enzymatic functions encoded on the polyprotein, including the NS2B-NS3 serine protease, are considered as promising drug targets as their function is indispensable for viral replication and maturation [4-6].
The genomic RNA of dengue virus serotype 2 (DEN2) contains 10,723 nucleotides, which encodes a single polyprotein precursor of 3391 amino acid residues that upon cleavage yields three structural proteins, the capsid (C), membrane (M), and envelope (E) glycoproteins, and seven nonstructural proteins, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5, arranged in the order NH2-C-prM-E-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5-COOH [7].
The N-terminal 180-residue-long region of the nonstructural protein 3 (NS3) is a serine protease [8, 9]. Earlier studies have revealed that a 35-48-residue-long central hydrophilic region of NS2B, termed NS2B(H), is essential for proteolytic activity of the NS3 protease [10-12]. NS2B(H) is associated noncovalently with the NS3 protease (NS3pro), generating the NS2B(H)-NS3pro complex, which catalyzes cleavage at the NS2A/NS2B, NS2B/NS3, NS3/NS4A, and NS4B/NS5 junctions and in the capsid protein [13]. Thus, NS2B(H)-NS3pro represents an attractive target for anti-flaviviral drug development as reviewed extensively in the literature [5, 14].
Although several high-throughput screening studies have been conducted on the DEN protease, only a few small molecule inhibitors were found, which included charged [15-17] and uncharged [18-22] molecules. To the best of our knowledge, all of these efforts have thus far not resulted in any inhibitor of the DEN protease to enter the clinical development phases [23]. In previous studies, we created substrate peptide libraries and analyzed their interaction with dengue virus NS3 proteases using the proteochemometric approach, which resulted in detailed information on inhibitor binding in the prime and non-prime side region of the enzyme [24, 25].
In an earlier study where we employed multiple sequence alignments of the NS2B-NS3pro from DEN2 with the corresponding regions in other flaviviruses, namely Japanese Encephalitis Virus (JEV), West Nile Virus (WNV), and Yellow Fever Virus (YFV), we found that the DEN2 protease shows only 50.2% sequence identity (79.5% sequence similarity) with the JEV protease that is closest to WNV, with a 76.3% sequence identity (93.7% sequence similarity) [26]. Further, we identified four amino acid residues (positions 84 and 86 in NS2B and 132 and 155 in NS3) that were different in the putative binding pockets for all the four proteases. This makes the proteases of different flaviviruses quite different in terms of their binding mode, affinity, cleavage preferences, and cleavage efficiency [26].
Recently, residues in the DEN NS3 active site involved in key enzyme–substrate interactions were characterized by alanine scanning mutagenesis and assay with a synthetic peptide model substrate [27].
Four distinct substrate-binding pockets, S4-S1, have been identified in the flaviviral NS2B-NS3 proteases from X-ray crystal structures of the DEN3 and WNV protease–inhibitor complexes NS2B(H)-NS3pro–Bz-nKRR-H [28, 29]. These pockets accommodate the P4-P1 residues of protease substrates [30]. Ser135 forms a covalent bond with the aldehyde carbon, and the resulting hydroxyl of Bz-nKRR-H interacts with His51 of NS3. DEN2 has Ser at position 85, which may make a direct interaction with the P3 basic side chain. The preference for Arg at P2 likely also comes from interacting with Asp75 in the catalytic triad, whereas Lys at P3 does not make any ionic interactions.
To be able to develop inhibitors for the NS2B-NS3pro complex, it is required to have the enzyme available for activity assays. It is usually obtained with a two-step purification strategy involving immobilized metal-ion affinity chromatography (IMAC), followed by a time-, cost-, and yield-inefficient gel-filtration purification procedure [31-35]. In this study, we show how the enzyme can be easily expressed, purified by a simple and single-step purification process, refolded, and used for activity testing with a fluorogenic model peptide substrate optimized for screening of anti-flaviviral compounds. On top of this, while previous studies have commonly utilized a cleavage-resistant NS2B(H)-NS3pro fusion protein [22, 36], the method described herein offers the distinct advantage that enzymatic activity of the NS3 protease can be instantly assayed by monitoring proteolytic autocleavage on SDS-PAGE.
MATERIALS AND METHODS
Construction of pTrcHisA/NS2B(H)-NS3pro of DEN2 by SOE-PCR. The plasmid encoding NS2B(H)-NS3pro containing NS2B(H) residues 48-95, followed by linker residues 121-130, and NS3pro residues 1-180, was constructed by a procedure described previously [10]. Briefly, the plasmid pTH/NS2B-NS3 precursor was obtained by gene synthesis, and it was used as a template for splicing by overlap extension (SOE)-PCR. The sequence of NS2B(H) was obtained by the primers 5′-TGCTCACTGGAGGATCCGCCGATTTGGAACTGGAG-3′ (nucleotides 4259 to 4293 in DEN2) and 5′-CTTCACTTCCCACAGGTACCACAGTGTTTGTTCTTCCTC-3′ (nucleotides 4399 to 4416).
For amplification of NS3pro, primers 5′-GAACAAACACTGTGGTACCTGTGGGAAGTGAAGAAAC-3′ (nucleotides 4492 to 4516) and 5′-CTTCTCTTTCAGGATCCCTAATCTTCG ATCTCTGGGTTG-3′ (nucleotides 5043 to 5081) were used (overlapping sequences are in bold letters, and the primer binding sites on DEN2 virus genome are indicated by numbers and underlined).
SOE-PCR was performed with a combination of both templates with the NS2B(H) forward and the NS3pro reverse primers, incorporating an overlapping region of 33 nucleotides. The PCR product was cut with BamHI and cloned into the expression vector pTrcHisA to yield the polyhistidine-tagged (His)6 fusion protein. The sequence of the construct was confirmed by automated DNA sequencing analysis with an ABI PrismTM Model 377 Sequencer (Perkin Elmer) using ABI PrismTM Dye Terminator Cycle Sequencing Ready Reaction Kit (Perkin Elmer).
Expression and purification of DEN2 NS2B(H)-NS3pro. The pTrcHisA construct containing the NS2B(H)-NS3pro sequence was transformed into E. coli (BL21) and cells were grown in 1 liter of Luria broth (LB) medium containing 100 µg/ml ampicillin, at 37°C with continuous shaking, until OD600 reached 0.6. Expression was induced by isopropyl β-D-1-thiogalactopyranoside at 0.2 mM, and the cells were incubated for 6 h at 37°C. The cells were harvested by centrifugation (6000g, 4°C, 10 min), and the pellet was resuspended in 30 ml cell lysis buffer (0.1 M Tris-HCl, pH 7.5, 0.3 M NaCl, 0.25 mg/ml lysozyme, 10 µg/ml DNase, and 5 mM MgCl2). The cells were kept at room temperature for 30 min and then lysed on ice by sonication using an XL Ultrasonic Processor (Misonix Inc., USA). The cell lysate was subjected to centrifugation (15,000g, 4°C, 30 min), and the soluble fraction was separated. The insoluble (inclusion bodies) fraction was washed two times with 20 ml cell lysis buffer containing 1% Triton X-100, and then two times with 20 ml cell lysis buffer (without Triton X-100) to remove Triton X-100. The pellet was suspended in 30 ml of denaturing buffer (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, 8 M urea) followed by a brief 15 s sonication using the ultrasonic processor, centrifugation at (15,000g, 4°C, 30 min), and then by filtration through 0.22 µm filters (Pall Corporation, USA).
The histidine-tagged NS2B(H)-NS3pro was then purified by immobilized metal-ion affinity chromatography (IMAC). Nickel-Sepharose HisTrap™ HP 5 ml columns (GE Healthcare, Sweden) were pre-equilibrated with 10 column volumes of denaturing buffer (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, 8 M urea), and the sample (30 ml from 1 liter of bacterial culture) was loaded at a flow rate of 1 ml/min using an FPLC pump (ÄKTA™FPLC™ system; GE Healthcare). The column was washed with 10 column volumes of degased washing buffer (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, 8 M urea, 20 mM imidazole). Protein was eluted with 10 column volumes of elution buffer (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, 8 M urea, 100 mM imidazole) at a flow rate of 1 ml/min. The elution was monitored by absorbance at 280 nm using a UV detector (ÄKTA™FPLC™ system; GE Healthcare), and fractions of 1 ml were collected. Aliquots of 20 μl of each fraction were analyzed by 15% SDS-PAGE and subsequent Western blotting using anti-hexahistidine antiserum (Invitrogen, USA) at 1 : 10,000 dilution.
The fractions containing NS2B(H)-NS3pro were pooled, the protein concentration was adjusted to 0.4 mg/ml, and the enzyme was refolded by dialysis at 4°C by using SPECTRA/POR dialysis membranes (6-8 kDa MWCO) (Spectrum Medical Industries, Inc., USA) against three batches of a 100-fold volume of buffer A (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl), one batch of a 200-fold volume of buffer B (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, 5% (v/v) glycerol), and one batch of a 200-fold volume of buffer C (0.1 M Tris-HCl, pH 9.5, 5% (v/v) glycerol, and 5% (v/v) ethylene glycol). Purified NS2B(H)-NS3pro was further concentrated to 1 mg/ml on centrifugal filter devices (Centricon 15 ml, 5 kDa MWCO; Millipore, USA) at 4°C. Protein concentrations were determined by using a Bradford protein micro-assay (Bio-Rad, USA) with bovine serum albumin (Sigma, USA) as calibration standard. Samples were stored in 50 mM Tris-HCl (pH 9.0) and 50% (v/v) glycerol, at –20°C.
Assay of DEN2 NS2B(H)-NS3pro enzymatic activity. Enzymatic activity of purified NS2B(H)-NS3pro was assayed using the synthetic peptide substrate acetyl-Nle-Lys-Arg-Arg-7-amino-4-methylcoumarin (Ac-nKRR-amc) (Peptides International, USA). Cleavage of amc from the peptide substrate was monitored using a Beckman Coulter DTX 880 Multimode Plate Reader (Beckman Coulter, USA) at excitation wavelength λ = 360 nm and emission wavelength λ = 485 nm. Assays were conducted on 96-well flat bottom black polystyrene microplates (Corning Life Sciences, USA) in a reaction volume of 100 µl containing 0.075 µM NS2B(H)-NS3pro, assay buffer (50 mM Tris-HCl, pH 9.5, 20% (v/v) glycerol), and substrate at concentrations ranging from 12.5 to 800 µM. Enzyme reaction mixtures were preincubated for 30 min at 37°C, and the reactions were started by addition of the substrate. Fluorescence release was monitored every 30 s over a period of 5 min, and relative fluorescence units (RFU) were converted to rates of product formation by calibration with free amc (Sigma). Inner filter effects were corrected as described in the literature [37]. The NS2B(H)-NS3pro kinetic parameters, Km, kcat, and catalytic efficiency kcat/Km, were calculated assuming Michaelis–Menten kinetics, v = Vmax·[S]/([S] + Km), with the aid of GraphPad Prism software. No significant hydrolysis of the peptide substrate was observed in the absence of the enzyme.
RESULTS AND DISCUSSION
Intensive efforts are devoted to the characterization of flaviviral enzymes, including the NS3 serine proteases, as potential targets for the rational discovery of antiviral drugs (for reviews see [5, 38] and references herein). In support of this, we here report a straightforward procedure for producing recombinant DEN2 NS2B(H)-NS3pro complex by overexpression in E. coli followed by a one-step purification procedure, and we report the kinetic parameters of the protease for a commercially available synthetic fluorogenic model peptide substrate, Ac-nKRR-amc.
The full-length NS2B-NS3 polyprotein region of DEN2 was obtained by time- and cost-efficient gene synthesis, and it was used as template for the generation of the NS2B(H)-NS3pro protease complex comprising residues 51-95 and residues 121-131 from the NS2B cofactor and N-terminal residues 1-180 of the NS3 protease domain. This construct has been shown to be catalytically active both with peptide substrates designed for in trans cleavage reactions and in proteolytic autocleavage leading to the formation of the NS2B(H)-NS3pro noncovalent complex [9].
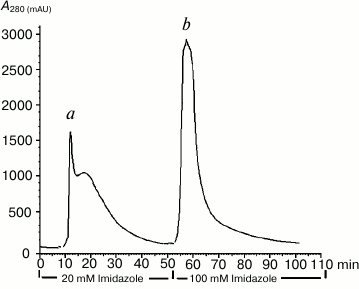
The recombinant NS2B(H)-NS3pro protein produced by E. coli was predominantly biosynthesized as an insoluble protein (inclusion bodies) that could be easily solubilized and purified in 8 M urea by a single step purification using metal-chelate affinity chromatography (IMAC) to >95% purity (Fig. 1).
Fig. 1. Purification of DEN2 NS2B(H)-NS3pro complex by Ni2+-affinity chromatography. The column was washed with 0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, and 20 mM imidazole (peak a). The NS2B(H)-NS3pro was eluted with elution buffer (0.1 M Tris-HCl, pH 8.0, 0.3 M NaCl, 100 mM imidazole) (peak b).
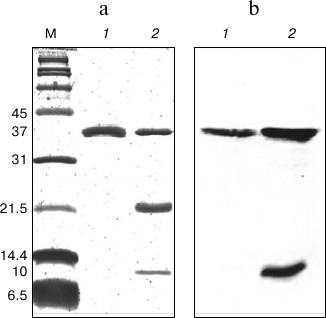
In SDS-PAGE and Western blot analyses (Fig. 2), the purified protein displayed a single band of 37 kDa representing the denatured intact NS2B(H)-NS3pro complex, which is enzymatically inactive and hence showed no autoproteolytic cleavage. Samples of the protein preparation after refolding displayed three bands of 37, 21, and 10 kDa corresponding to the NS2B(H)-NS3pro precursor, the NS3pro protease domain only, and the histidine-tagged NS2B(H) cofactor, respectively. In contrast to the corresponding protein from JEV, the NS2B(H)-NS3pro protein from DEN2 was largely insoluble (inclusion bodies) upon expression in E. coli, thus suggesting the existence of major conformational differences between the two proteins. In this study, the refolded enzyme was assayed by cleaving a commercially available model fluorophore-linked peptide substrate, Ac-nKRR-amc. The kinetic parameters, Km, kcat, and kcat/Km, were 100 ± 8.6 µM, 0.112 ± 0.003 s–1, and 1120 ± 66.78 M–1·s–1, respectively. It is well known that dengue protease has a strong preference for basic amino acid residues (Arg/Lys) at the P1 position, while the preferences for the other positions are Arg > Thr > Gln/Asn/Lys for P2, Lys > Arg > Asn for P3, and Nle > Leu > Lys for P4 positions [36]. In an earlier report, the internally quenched peptide Bz-RRRR-SAG-Y(NO2)-amide representing the DEN capsid protein cleavage site sequence was identified as an efficiently processed NS3 substrate [25].
Fig. 2. SDS-PAGE (a) and Western blot (b) analysis of DEN2 NS2B(H)-NS3(pro) after IMAC purification and refolding. a) 15% SDS-PAGE. Lanes: M, broad-range protein marker; 1) protein sample after IMAC purification; 2) protein sample after purification by IMAC and refolding by dialysis. b) Western blot profile using N-terminal anti-His antibody on 15% SDS-PAGE. Lanes: 1) protein sample after IMAC purification; 2) protein sample after IMAC purification and refolding by dialysis.
In our previous study on the JEV NS2B(H)-NS3pro protease [26], the kinetic parameters for the nKRR-amc substrate were determined for Km, kcat, and kcat/Km to, respectively, 32 μM, 0.01178 s–1, and 368.13 M–1·s–1. In the same study [26], the binding affinity for the substrate with a leucine at P3 (Boc-LRR-amc) was threefold higher when compared to that with a glycine at P3 (Boc-GRR-amc); however, the catalytic efficiency for Boc-GRR-amc was about 10-fold higher than for Boc-LRR-amc, thereby suggesting a significant contribution of this position to the catalytic mechanism, as seen in an earlier report for the DEN NS3 protease [36]. It has been shown that the NS3pro enzyme from DEN favors substrates with Arg-Arg at the P2-P1 positions, while the NS3 proteases from JEV and WNV prefer Lys-Arg at this position; data that thus suggest a greater similarity between the JEV and WNV protease than between DEN and JEV proteases [31].
Altogether, in this study we expressed, purified, refolded, and tested the DEN2 NS2B(H)-NS3pro complex for its proteolytic activity using a model fluorogenic peptide substrate. The simple procedure reported here allows active DEN2 NS2B(H)-NS3pro to be easily produced in large quantities, e.g. for high-throughput screening and use in lead optimization of dengue protease inhibitors. Similar procedures may be applicable also to other flaviviral proteases of medical importance such as JEV and WNV proteases, for which also effective treatments are still lacking.
We thank Dr. Somphob Leetachewa for advice with FPLC separation, Somsri Sakdee for excellent technical work, and Anchalee Nirachanon for excellent secretarial assistance.
This work was supported by grants from the Thailand Research Fund (TRF) BRG4980008 (to GK) and the Swedish International Cooperation Development Agency (SIDA).
REFERENCES
1.World Health Organization (2009) Geneva: World
Health Organization (WHO) and the Special Program for Research and
Training in Tropical Diseases (TDR), pp. 147.
2.Monath, T. P. (1994) Proc. Natl. Acad. Sci.
USA, 91, 2395-2400.
3.Halstead, S. B. (1988) Science, 239,
476-481.
4.Melino, S., and Paci, M. (2007) FEBS J.,
274, 2986-3002.
5.Sampath, A., and Padmanabhan, R. (2009)
Antiviral Res., 81, 6-15.
6.Lescar, J., Luo, D., Xu, T., Sampath, A., Lim, S.
P., Canard, B., and Vasudevan, S. G. (2008) Antiviral Res.,
80, 94-101.
7.Zhang, L., Mohan, P. M., and Padmanabhan, R. (1992)
J. Virol., 66, 7549-7554.
8.Bazan, J. R., and Fletterick, R. (1989)
Virology, 171, 637-639.
9.Chambers, T. J., Nestorowicz, A., Amberg, S. M.,
and Rice, C. M. (1993) J. Virol., 67, 6797-6807.
10.Niyomrattanakit, P., Winoyanuwattikun, P.,
Chanprapaph, S., Angsuthanasombat, C., Panyim, S., and Katzenmeier, G.
(2004) J. Virol., 78, 13708-13716.
11.Falgout, B., Miller, H. R., and Lai, C. J. (1993)
J. Virol., 67, 2034-2042.
12.Chappell, K. J., Stoermer, M. J., Fairlie, D. P.,
and Young, P. R. (2008) J. Gen. Virol., 89,
1010-1014.
13.Falgout, B., Pethel, M., Zhang, Y. M., and Lai,
C. J. (1991) J. Virol., 65, 2467-2475.
14.Leyssen, P., De Clercq, E., and Neyts, J. (2003)
Clin. Microbiol., 13, 67-82.
15.Ekonomiuk, D., Su, X. C., Ozawa, K., Bodenreider,
C., Lim, S. P., Otting, G., Huang, D., and Caflisch, A. (2009)
J. Med. Chem., 52, 4860-4868.
16.Ekonomiuk, D., Su, X. C., Ozawa, K., Bodenreider,
C., Lim, S. P., Yin, Z., Keller, T. H., Beer, D., Patel,
V., Otting, G., Caflisch, A., and Huang, D. (2009) PLoS
Negl. Trop. Dis., 3, e356.
17.Ganesh, V. K., Muller, N., Judge, K., Luan, C.,
Padmanabhan, R., and Murthy, K. H. M. (2005) Bioorg. Med. Chem.,
13, 257-264.
18.Tomlinson, S. M., Malmstrom, R. D., Russo, A.,
Mueller, N., Pang, Y. P., and Watowich, S. J. (2009) Antiviral
Res., 82, 110-114.
19.Sidique, S., Shiryaev, S. A., Ratnikov, B. I.,
Herath, A., Su, Y., and Cosford, N. D. (2009) Bioorg. Med. Chem.
Lett., 19, 5773-5777.
20.Johnston, P. A., Phillips, J., Shun, T. Y.,
Shinde, S., Lazo, J. S., Huryn, D. M., Myers, M. C., Ratnikov, B. I.,
Smith, J. W., Su, Y., Dahl, R., Cosford, N. D., Shiryaev, S. A., and
Strongin, A. Y. (2007) Assay Drug. Dev. Technol., 5,
737-750.
21.Mueller, N. H., Pattabiraman, N.,
Ansarah-Sobrinho, C., Viswanathan, P., Pierson, T. C., and Padmanabhan,
R. (2008) Antimicrob. Agents Chemother., 52,
3385-3393.
22.Leung, D., Schroder, K., White, H., Fang, N. X.,
Stoermer, M. J., Abbenante, G., Martin, J. L., Young, P. R., and
Fairlie, D. P. (2001) J. Biol. Chem., 276,
45762-45771.
23.Steuer, C., Gege, C., Fischl, W., Heinonen, K.
H., Bartenschlager, R., and Klein, C. D. (2011) Bioorg. Med.
Chem., 19, 4067-4074.
24.Prusis, P., Lapins, M., Yahorava, S., Petrovska,
R., Niyomrattanakit, P., Katzenmeier, G., and Wikberg, J. E. S.
(2008) Bioorg. Med. Chem., 16, 9369-9377.
25.Niyomrattanakit, P., Yahorava, S., Mutule, I.,
Mutulis, F., Petrovska, R., Prusis, P., Katzenmeier, G., and Wikberg,
J. E. S. (2006) Biochem. J., 397, 203-211.
26.Junaid, M., Chalayut, C., Torrejon, A. S.,
Angsuthanasombat, C., Shutava, I., Lapins, M., Wikberg, J. E., and
Katzenmeier, G. (2012) PloS One, 7, e36872.
27.Salaemae, W., Junaid, M., Angsuthanasombat, C.,
and Katzenmeier, G. (2010) J. Biomed. Sci., 17, 68.
28.Erbel, P., Schiering, N., Arcy, A. D., Renatus,
M., Kroemer, M., Lim, S. P., Yin, Z., Keller, T. H., Vasudevan, S. G.,
and Hommel, U. (2006) Nat. Struct. Mol. Biol., 13,
372-373.
29.Noble, C. G., Seh, C. C., Chao, A. T., and Shi,
P. Y. (2012) J. Virol., 86, 438-446.
30.Wichapong, A., Pianwanit, S., Sippl, W., and
Kokpol, S. (2010) J. Mol. Recognit., 23, 283-300.
31.Mueller, N. H., Yon, C., Ganesh, V. K., and
Padmanabhan, R. (2007) Int. J. Biochem. Cell Biol., 39,
606-614.
32.Luo, D., Xu, T., Hunke, C., Gruber, G.,
Vasudevan, S. G., and Lescar, J. (2008) J. Virol., 82,
173-183.
33.Rothan, H. A., Abdulrahman, A. Y., Sasikumer, P.
G., Othman, S., Rahman, N. A., and Yusof, R. (2012) J. Biomed.
Biotechnol., 2012, 1-6.
34.Phong, W. Y., Nicole, J., Moreland, N. J., Lim,
S. P., Wen, D., Prasad, N., Paradkar, P. N., and Vasudevan, S. G.
(2011) Biosci. Rep., 31, doi:10.1042/BSR20100142.
35.Yusof, R., Clum, S., Wetzel, M., Murthy, H. M.
K., and Padmanabhan, R. (2000) J. Biol. Chem., 275,
9963-9969.
36.Li, J., Lim, S. P., Beer, D., Patel, V., Wen, D.,
Tumanut, C., Tully, D. C., Williams, J. A., Jiricek, J., Priestle, J.
P., Harris, J. L., and Vasudevan, S. G. (2005) J. Biol. Chem.,
280, 28766-28774.
37.Liu, Y., Kati, W., Chen, C. M., Tripathi, R.,
Molla, A., and Kohlbrenner, W. (1999) Anal. Biochem.,
267, 331-335.
38.Ryan, M. D., Monaghan, S., and Flint, M. (1998)
J. Gen. Virol., 79, 947-959.