Fatal “Triad”: Lipotoxicity, Oxidative Stress, and Phenoptosis
A. V. Rzheshevsky
Restorative Medicine Center, ul. Mechnikova 8, 49000 Dnepropetrovsk, Ukraine; E-mail: alex-rjechewsky@mail.ru
Received June 6, 2013
Negative factors, such as the “magnificent” five that includes alcoholism, smoking, unhealthy food, lack of movement, and negative emotions, accompany a person almost from birth and trigger powerful internal biochemical reactions leading to disastrous consequences. Those new deleterious reactions force the organism to mobilize all of its internal reserves to neutralize, at least temporarily, the destructive effects of these negative factors. As a result of this continuous struggle for survival, body parts degenerate, starting from connective tissue protein molecules to entire newly formed organs (such as adipose tissue). Today we can state with certainty that the reason for the majority of widespread pathologies causing premature aging and death, such as atherosclerosis and arterial hypertension, is exactly those external negative factors that a person voluntary introduces into their life. However, the margin of safety that Nature enclosed in the human body is really amazing, allowing light-minded and self-destructive people to live up to 60 years and longer. It is quite possible that the lifespan will increase up to 100 years and more if a person stops destroying themself with negative emotions and bad habits, including unhealthy food and overeating. This article examines possible interconnection between unhealthy overeating and the theory of programmed aging and phenoptosis.
KEY WORDS: metabolic syndrome, insulin resistance, obesity, lipotoxicity, oxidative stress, phenoptosis, programmed agingDOI: 10.1134/S0006297913090046
Abbreviations: FFA, free fatty acids; IR, insulin resistance; MS, metabolic syndrome; ROS, reactive oxygen species.
In 1988 a Stanford University professor, the American endocrinologist
Gerald Reaven [1], turned his attention to the
connection between the disorders of carbohydrate and lipid metabolism
and cardiovascular diseases such as arterial hypertension and coronary
heart disease. He described the complex of pathological factors (which
he called “syndrome X”) often found in people suffering
from diseases of the cardiovascular and endocrine systems. He also
suggested reduced tissue sensitivity to insulin to be at the root of
all these problems. It is commonly believed that this was the starting
point for the large-scale study of the most complex pathological
complex known as “metabolic syndrome” (MS).
So, what exactly is metabolic syndrome? Here is how this concept is described by a publication of the World Health Organization (WHO): “When a person has central obesity (too much weight around the waist), abnormal blood fat levels (e.g. high triglyceride level or low HDL cholesterol), high blood pressure and high blood sugar at the same time, this is known as metabolic syndrome” [2]. WHO reports that people with MS have increased risk of the development of diabetes, heart disease, and stroke. It is quite clear that it was not by chance that all the above-listed factors – accumulation of fat around the waist, increased level of triglycerides and glucose in blood, high blood pressure – have been grouped together. They were so often found in the same people that finally doctors around the world paid attention to it, and subsequently medical organizations gained the right to bring them together and call them a syndrome, i.e. a group of factors that occur together more often than individually. Today, the prevalence of this syndrome is so high that it is referred to as “epidemic” in some international documents. This situation encourages the world medical community to pay more attention to this issue, and as a result MS has become by far one of the most studied and debated problems in modern medicine. Several years ago an international institute was organized dedicated solely to the study of this syndrome, and in April 2005 the I International Congress on MS was held in Germany.
What is the cause of such widespread MS and what leads to the development of this syndrome? Detailed analysis of the development of the entire pathological chain allows us to conclude that two interrelated adverse factors, unhealthy excessive diet and limited mobility, are the basis of all of the MS.
Already for many years we have been hearing the words of doctors and scientists about lack of exercise and improper diet being the cause of various bodily disorders. However, when not backed by hard evidence, such statements seem vague and do not have much effect on people. It was only in recent years that scientists have been able to clearly identify and describe this connection. It all starts from high-calorie food rich in simple carbohydrates (white bread, biscuits, fried potatoes, sugary drinks, etc.) and unhealthy saturated fats (present in pork, sausages, ham, pastries, butter, sour cream, etc.) In time, such a diet leads to fat being deposited in the body. Because of our topic, we will be particularly dealing with the abdominal (or central) obesity – folds of fat around the waist.
Today it is perhaps easier to meet on the street a “Bigfoot” or a living mammoth that a person over 45 years of age without fat deposits in the waist area. And if such a deposit did not turn into hypertrophied excess weight with a strongly protruded belly, then, as a rule, the majority of people consider such a phenomenon to be quite normal. In fact, often a person with some fat reserve can feel healthy and have a youthful appearance for many years. And yet, this abdominal (or central) obesity can serve as a clear signal that a person left the “road” of bodily healthy state and embarked on the path of various ailments and disorders, and their development is only a matter of time.
Why is it so obvious? The so-called insulin resistance is the cause of everything. Fatty waist folds reveal better than any analysis the problems with normal susceptibility of cells to insulin and the ability of glucose to penetrate into cells; it is because of these problems that carbohydrates, instead of compensating energy consumption and being deposited in muscles and liver, turn into fat and are deposited in adipose cells. However, waist folds prove to be only the visible part of an iceberg. Most commonly fat folds are accompanied by an internal, so-called visceral obesity, when fat deposits cover internal organs in the abdominal cavity. It is this phenomenon that was shown to be the most dangerous; it was also shown to be the main pathogenic factor of the most serious functional disorders.
Abdominal obesity plays such a purely negative role because, as it was discovered quite recently, adipose tissue is not a passive “warehouse” of accumulated fat, but an active endocrine organ capable of drastic interference with metabolic processes [3]. Recent research has shown fat deposits to produce many active compounds called “adipose derived hormones” or, shortly, adipokines [4]; these compounds include adiponectin, resistin, interleukin-1 and -6 (IL-1, IL-6), leptin, tumor necrosis factor-α ΤΝF-α), vaspin, visfatin, omentin, apelin, etc. Adipokines fulfill important regulatory, proinflammatory, immunomodulatory, and other functions.
It was in the middle of the last century that scientists first noticed that obesity and disorders in carbohydrate metabolism were accompanied by an increase in the levels of certain compounds in blood that was difficult to explain. Large-scale studies began only after the discovery in 1993 of TNF-α produced by adipocytes [5]. This research led to the understanding of the possible disastrous role of adipose tissue. The last doubts disappeared when obesity and insulin resistance were shown to be accompanied by a stable increase of the level of C-reactive protein, one of the most sensitive and widely recognized indicators of inflammation [6]. As of today, several dozens of adipokines have been discovered and studied. Let us briefly discuss some of them.
Leptin, one of the most thoroughly studied adipokines, was discovered in 1994 [7]. Regulation of metabolism of fatty acids by tissues due to activation of AMP-activated protein kinase is the main function of leptin. It also supports regulation of energy homeostasis and body weight control by reducing the synthesis and release of neuropeptide Y, which causes the feeling of hunger [8]. Leptin receptors are activated in the hypothalamus, which plays an important role in the integration of information on consumed food, the amount of energy stored as fat, glucose level in blood (these data support regulation of nutrition); data on energy accumulation and consumption are also gathered in the hypothalamus. Once leptin activates the receptors, orexigenic pathways, associated with neuropeptide Y (NPY) and agouti-related peptide (AGRP), are suppressed and the anorexigenic pathways associated with proopiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART) are induced [9].
In obesity, the entire physiological activity of leptin is distorted, and leptin resistance develops. Leptin resistance is assumed to be caused by disturbances in leptin transport and the ObRb signaling pathway, which includes overexpression of the suppressor of signaling pathway 3 (SOCS3), the inhibitor of the leptin signaling pathway [10]. Hypothalamic receptors stop responding to leptin, its level increases, which normally should result in the suppression of the feeling of hunger by blocking neuropeptide Y. However, it does not happen in obese people and the feeling of hunger does not decrease. Leptin level grows; it stimulates cellular immune response and causes proinflammatory reaction. Hyperleptinemia has been also found to stimulate the sympathoadrenal system, which, in turn, leads to increased blood pressure. Reduction of leptin sensitivity in the brain leads to accumulation of excessive amounts of triglycerides in adipose tissue as well as in muscles, liver, and pancreas, which disrupts the sensitivity of these tissues to insulin.
Resistin was discovered in 2001 by a group of scientists from the University of Pennsylvania (C. M. Steppan et al.); it was named “resistance hormone” [11]. These authors showed rosiglitazone, which increases tissue sensitivity to insulin, to significantly suppress resistin synthesis [11]. The following year, a group of Japanese scientists in experiments on obese mice showed the existence of an interconnection between increased resistin level and the suppression of insulin-mediated glucose uptake by target tissues [12]. The increase in resistin level was also shown to contribute to obesity: administration of exogenous resistin caused a steady increase in body weight in mice. Today, resistin level is assumed to play a significant role in obesity, insulin resistance, and also in the development of vascular disorders in humans.
Tumor necrosis factor-α (TNF-α) is perhaps the most pernicious enzyme of all presently known adipokines. This multifunctional proinflammatory cytokine synthesized by monocytes, macrophages, lymphocytes, hepatocytes, fibroblasts, and endothelial, epithelial, and other cells as well as by adipose tissue was discovered by E. Carswell et al. in 1975 in the serum of mice injected with bacterial products [13]. TNF-α has a self-stimulating effect and stimulates neighboring cells; it is also an endogenous pyrogen, i.e. a compound capable of causing pyrogenic effects – fever.
The natural physiological function of TNF-α is to form an inflammatory response to any pathogen penetrating the organism. It causes an increase in blood vessel diameter in the infected area and a local increase in blood flow, which prevents reproduction of harmful microorganisms. It concentrates functionally important proteins (immunoglobulins and others) in the infected area by increasing the blood vessels permeability and local fluid accumulation; it also enhances the flow of phagocytes to the inflammation area [14].
Fat deposits directly contribute to TNF-α changing from an organism defender to a destructive “aggressor”. Already in 1993 adipose cells were shown to produce TNF-α [5]. Increased TNF-α level plays an important, and perhaps even a leading role in reducing tissue sensitivity to insulin and developing of inflammatory processes. Numerous studies have been conducted that describe these processes in detail [15].
In liver cells, TNF-α violates carbohydrate and fat metabolism and exacerbates insulin resistance by suppressing the expression of genes responsible for glucose absorption and increasing the expression of genes regulating synthesis of fatty acids. It also contributes to similar processes in adipose tissue: suppression of the activity of genes involved in glucose metabolism and simultaneous increase in the activity of genes responsible for accumulation of triglycerides and adipogenesis. TNF-α disrupts normal functioning of the insulin receptor cascade and hampers glucose absorption by cells due to serine kinase activation in the substrate of insulin receptor-1 (IRS-1). High concentration of TNF-α also causes increased production of free radicals, such as hydrogen peroxide and others, which leads to oxidative stress [16].
Interleukin-6 (IL-6) is a cytokine, which, similarly to TNF-α, is involved in the activation of the immune system and repair mechanisms in case of inflammation and exposure to bacterial endotoxins, as well as during heavy physical exertion and various injuries. It is synthesized by activated macrophages and T-cells, endothelial cells, fibroblasts, and muscle and adipose tissues. It is involved in differentiation of B-lymphocytes and their transformation into immunoglobulin-secreting plasma cells; it also stimulates proliferation of cytotoxic T-lymphocytes, which directly destroy foreign objects [17].
IL-6 belongs to the category of early inflammation mediators, as its active synthesis starts immediately after exposure of cells to harmful bacteria and viruses. In the case of acute inflammatory processes, IL-6 level in blood plasma can be increased by several hundreds of times when compared to usual level in healthy people. Adipose tissue produces up to one third of IL-6 circulating in an organism, increasing its normal level almost 100 times and turning this useful cytokine into a harmful one. Such an increased IL-6 level significantly suppresses insulin activity in adipocytes and hepatocytes: similar to TNF-α, it suppresses normal function of the substrate of insulin receptor and glucose transporter GLUT-4 and consequently leads to insulin resistance. IL-6 inhibits the formation of one of the “useful” adipokines – adiponectin, which increases tissue sensitivity to insulin and improves glucose absorption by cells; it also simultaneously stimulates the synthesis of TNF-α, which proves to be a pathological factor in the progression of insulin resistance [18].
To understand how adipokines disrupt the normal function of insulin receptor, let us first describe this receptor and its functioning. An individual human cell is the beginning of human life and the last frontier of its security. It is not an easy task – to penetrate into the cell, and only physiologically vital compounds can easily do it. The cell membrane protects the cell from being penetrated by foreign objects. Receptors on the cellular surface play the role of “keys” opening the cell to needed compounds.
The insulin receptor consists of two extracellular α-subunits and two intracellular and transmembrane β-subunits; it belongs to the family of tyrosine kinase receptors because its intracellular part consists of an enzyme called tyrosine protein kinase. This part of the receptor contains a large number of tyrosine and serine residues. Once insulin is connected to the extracellular receptor part, tyrosine residues are activated by phosphorylation. Phosphorylation is probably the most common way to control the activity of proteins that regulate various intracellular processes. After interaction with insulin, autophosphorylated tyrosine residues transmit signal to other enzymes that constitute a signaling path providing glucose transport into the cell. Phosphate is transferred along the chain to the next signaling protein, which, in turn, transmits the bound phosphate further on, to the next protein, and so it moves into the cell until it reaches the final goal of the cascade – activation of phosphatidylinositol-3-kinase and moving of the special transporting protein GLUT-4 into the cell membrane, this process being followed by glucose transfer into the cell. In addition, interaction of insulin with an intracellular receptor triggers another signaling cascade parallel to the glucose-transporting one, which functions as the so-called mitogenic pathway (the name comes from the core kinase of this pathway, mitogen-activated protein (MAPK)) essential for proliferation, differentiation, and cell growth (see figure) [19]. It is worth noting that two parallel signaling cascades present in the same cell react differently to the same negative factors (such as discussed above proinflammatory adipokines), and in case of disturbances in signal transmission in the glucose transport chain, the signaling cascade of the mitogenic pathway is not affected, rather the opposite, it activates certain processes (including various inflammatory mechanisms) and can act as a negative factor in the pathogenesis of atherosclerosis [19].
Insulin signaling pathways. IRS-1, substrate of insulin receptor. Metabolic pathway: PI3K, phosphatidylinositol-3-kinase; Akt, protein kinase B; GLUT-4, glucose transporter. Mitogenic pathway: Ras, GTP-binding protein; Raf, serine/threonine protein kinase encoded by c-raf genes; MEK (MAPKK), kinase of mitogen-activated protein kinase; Erk (MAPK), kinase activated by extracellular signal; c-Myc and ELK-1, nuclear transcription factors. Antiapoptotic pathway: Bad, regulatory protein (inducer); BCL2, regulatory protein (inhibitor); GSK-3, glycogen synthase kinase
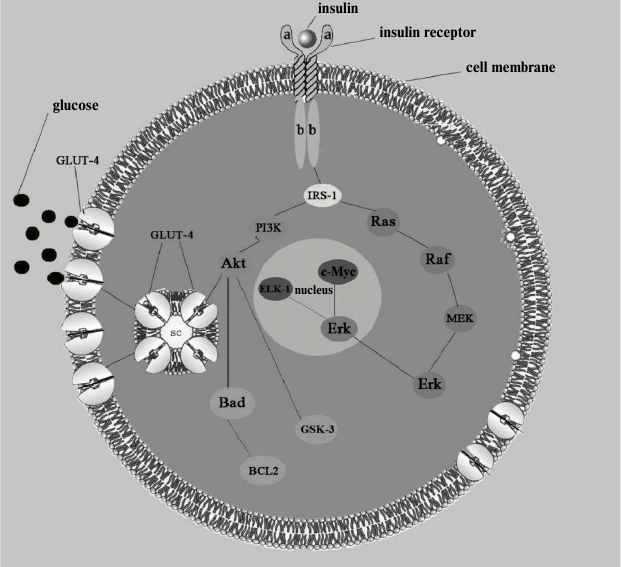
Now, after much research, we can quite confidently describe the exact mechanism of the disruption of insulin receptor function after which it can no longer provide assembly of the glucose transporter GLUT-4. Most researchers share the view that the receptor dysfunction usually develops after excessive phosphorylation of the substrate of insulin receptor, namely, of its inner serine residues [20]. It seems particularly noticeable that various negative factors (adipokines, fatty acid metabolites, etc.) affect this substrate in a targeted and focused manner, apparently because of it being the weakest link in the glucose transport signaling pathway. As a consequence, normal signal transmission from the receptor to the glucose transporter becomes impossible and delivery of glucose into the cell is hampered.
Appearance of such a defect in the insulin signaling pathway triggers self-initiated “vicious” cycles (increased glucose level, increased insulin secretion, production of proinflammatory cytokines, reduction of nitric oxide synthesis, activation of the sympathetic-adrenal system, etc.), which sooner or later will drive a person to their grave if decisive action is not taken [19].
However, several questions remain unresolved. Why do all the enzymes that play a pathological role in the development of insulin resistance (TNF-α, IL-6, diacylglycerol, etc.) phosphorylate serine amino acid residues leaving adjacent tyrosine residues unaffected? And why, in general, are these serine residues present in the receptor substrate, i.e. what is their physiological role? Perhaps they play the role of a regulator-inhibitor in transmission of phosphate from one protein to another, reducing, if necessary, phosphorylation and blocking glucose transfer into the cell.
It is also unclear what happens with the huge receptor reserve present in cells (the physiological norm of active receptors is known not to exceed 10% of the overall number). Felig et al. state in their book Endocrinology and Metabolism [21]: “Even though the number of insulin receptors was shown to vary from 50,000 (in adipocytes) to 250,000 (in hepatocytes), the maximum biological effects are observed when less than 10% of the receptors are active. The functional significance of such a “receptor reserve” is that in case of decrease in the number of receptors (e.g. due to obesity) accompanied by adequate increase in insulin concentration, the number of hormone–receptor complexes may reach the critical level required for triggering a biological response”. If such a receptor reserve serves as a safety net in case of any deviations, it remains unclear where it disappears when these deviations arise? Reduction in the ability of cells to take glucose observed in insulin resistance (IR) and hyperinsulinemia suggests the possibility of feedback between the hormone level and the number of receptors, when the rise in hormone level leads to a decrease in the number of receptors. It should be admitted that several aspects of the post-receptor mechanism of insulin signal still remain unclear.
The fatal role played by adipokines synthesized by adipose tissue in the development of insulin resistance has been proved by numerous studies, and today no one doubts it. Along with adipokines, fatty acids are the second major adverse factor strongly affecting IR [22].
It would be incorrect to say that all the fats taken by the organism are an absolute evil that causes only harm – the way it is done today by some people, especially girls and women. Just as in the case of adipokines, they have an important physiological function, namely providing skeletal muscles with energy during heavy physical exertion, enabling the conditions for organism formation at times of active growth, and maintaining the level of immune system activity and homeostasis of the entire organism. Execution of both male and female reproductive functions would be impossible without fats. And in addition, fatty acids are the main energy supplier for heart muscle – the myocardium receives more than a half of the total ATP from oxidation of fats [22].
Fats are stores in an organism in the form of triglycerides – compounds consisting of fatty acids and glycerol. Free fatty acids (FFA) are formed from triglycerides contained in adipose tissues by hydrolysis, and their level in blood is normally very low. Physiologically-conditioned increased FFA level is observed only during the period of active growth of an organism, i.e. up to 20-25 years. Excessive fat accumulation, similar to adipokines, turns fatty acids from being a favorable factor into a destructive one. Abdominal adipose tissue (i.e. fat deposits in the waist area and inside the abdominal cavity), due to the large number of blood vessels connected to general blood flow, is directly connected to FFA level in blood; that is why an increase in abdominal fat mass entails an increase of FFA level. Such an increase will surely have an adverse effect on glucose metabolism because the regulation of synthesis, transport, and consumption of glucose and fatty acids are closely interwoven [22]. And this is how it happens.
Adipose tissue cannot grow indefinitely. Its growth is limited by the number of adipose cells, which is determined during childhood and adolescence and hardly changes later in life. Long imbalance in energy distribution in the direction of excessive intake and accumulation of energy substrates (fats and glucose) will eventually lead to the situation when all the cells of adipose tissue will be overfilled with these substrates. From that moment on fat accumulates in tissues not intended for its storage and oxidation, primarily in skeletal muscles and liver [23]. Triglycerides in adipose (especially abdominal) tissue will start to break down rapidly, thus alleviating the burden of fat cells, and fatty acids will form a rapid continuous stream rushing into the bloodstream, poisoning the entire body.
This process will begin even before the adipose cells are completely filled. It will begin with the formation of an internal, visceral obesity, connected to the general bloodstream by multiple blood vessels. Fatty acids will start to enter liver and muscle tissue, where physiological oxidation of a large amount of fats is hardly possible, and as a result muscle and liver cells will be rapidly accumulating fats, this process leading to their fatty degeneration [24]. In the case of skeletal muscles, not just fat deposition develops as in adipose tissues, but cell mutation: due to excessive fat accumulation stem cells will differentiate not into new myocytes (muscle cells), but into defective adipose cells – preadipocytes [23]. The presence of these defective cells is typical of very old age – that is why an equality sign can be safely put between obesity and premature aging. And there will be several more such signs. Excessive fat oxidation in mitochondria of these cells, as well as in special cellular organelles, peroxisomes, will lead to the formation of a byproduct extremely harmful and dangerous for DNA, and the entire cell metabolites of fatty acids and widely known today free radicals, reactive oxygen species (ROS). This process will result in two new adverse factors, lipotoxicity and oxidative stress [25]. Let us consider the first factor.
The term “lipotoxicity” now refers to various negative effects of fatty acids on cellular structures [20]. As already mentioned, regulation of synthesis, transport, and consumption of fats and glucose are closely intertwined. And if fatty degeneration (obesity) is observed in tissues not intended for fat deposition and oxidation (in skeletal muscles, pancreas, and liver), then glucose metabolism in the cells of these organs will be disrupted. A competition will develop between energy substrates for the right to occupy a dominant position in the organism, which will end with complete victory of fats. Their oxidation rate constant, being higher than that of glucose, will help fats take the leading position in a cell. This directly affects the order and sequence of substrate oxidation in mitochondria: substrates with lower oxidation rate give way to those with higher rate [24].
Subsequent suppression of normal glucose absorption by cells will be caused by byproducts of fatty acid metabolism, such as ceramides and diacylglycerol, which activate protein kinase C resulting in excessive phosphorylation of serine residues of insulin receptor substrate and disturbed glucose transport into cells. In skeletal muscles excess fatty acids will cause cell degeneration and sarcopenia (loss of muscle mass), while in heart muscle tissue fatty acid metabolites acyl carnitine and acetyl-CoA will block ATP, the cell carrier produced by cells, and myocardial cells will experience energy “hunger”, which in time will result in such pathology as ischemic heart disease [22]. Ischemia will form another “vicious” cycle: oxidation of fatty acids will slow due to lack of oxygen, and their accumulated metabolites will further block ATP transport and aggravate ischemia.
Palmitic acid is the most dangerous for the organism as it directly contributes to the development of apoptosis – the mechanism triggering cell death. Negative effects of palmitate follow several directions simultaneously, so that a cell has very little chance of survival. First, this negative impact is caused by formation of an intermediate product of synthesis of sphingomyelins (phospholipids consisting of sphingosine, choline, and phosphoric and fatty acids) – ceramides, as well as compounds in cell membrane rafts: cholesterol, sphingomyelin, and palmitic acid, which cause membrane impermeability and inhibition of cell functioning [23]. Palmitate also enhances ROS generation [26]. And finally, palmitic acid has been recently found to bind calcium ions excessively accumulated in mitochondria and form mitochondrial lipid pores, which results in non-specific permeability of the inner mitochondrial membrane and release of the main catalyst of apoptosis cytochrome c, and also other apoptosis-inducing factors [27].
Palmitic acid is also the main adverse factor in the development of such severe and widespread pathology as atherosclerosis. It is palmitic low-density lipoproteins, due to their specific properties, that turn into “biological waste” in the bloodstream. An organism will try to utilize this waste with the help of macrophages, this process resulting in the appearance of soft (prone to rupture and blood clot formation) and hard (prone to necrosis) atherosclerotic plaques [28]. High levels of palmitic acid in blood will also block the beneficial effect of useful polyunsaturated fatty acids, such as the well-known ω-3 acids [28]. Besides the above-mentioned lipoproteins, large amounts of palmitic triglycerides will be formed in the liver as a result of esterification of palmitic acid. These triglycerides cannot be hydrolyzed because of the high melting temperature (48°C) and therefore will be accumulated in liver cells and provoke the development of fatty liver disease [24].
Increased levels of fatty acids and fat accumulation in non-adipose tissues result in the development of tissue insensitivity to insulin, i.e. insulin resistance (IR) [14]. At first it will be local, but if no measures are taken, IR will be rapidly and continuously growing, embracing new organs, and tissues, generating “vicious” cycles prone to self-stimulation [22]. For some time tissue IR will not be accompanied by an increase in glucose level in blood due to compensatory hyperinsulinemia, that is, increased insulin secretion. But it cannot last for very long, and eventually glucose level starts to rise, adding another destructive factor, glucose toxicity, to lipotoxicity.
Some people mistakenly believe glucose to be a harmless product whose excess can cause only accumulation of unnecessary body fat. However, this is not true. The negative effects of the consumption of simple carbohydrates and the increase in glucose level are numerous and diverse, and no specialists doubt them. Glucose toxicity developing simultaneously with lipotoxicity is manifested as follows. Normal glucose oxidation by mitochondria becomes impossible in tissues that have undergone fatty degeneration (these are first of all the main glucose consumers, skeletal muscles and liver). As a result, the organism will be forced to redirect this oxidation – it will now follow other pathways, in particular, the sorbitol pathway. Such abnormal glucose oxidation will not pass unnoticed to the organism. Increased sorbitol and fructose formation will be its byproduct, and it will cause excessive intracellular liquid accumulation, which may lead to osmotic shock, i.e. swelling and rupture of the cell membrane.
Sorbitol accumulation in neurons is particularly dangerous, for there it suppresses the synthesis of a vital cell component, myoinositol, leading to the disruption of nerve impulses. Alternative routes of glucose oxidation through byproducts will also contribute to an increase in protein kinase C activity and consequently to even greater dysfunction of insulin receptor and progression of insulin resistance. Thus one “vicious cycle” is complete.
It is also possible to trace the formation of another such cycle associated with glycosylation of proteins, which is observed on the increase in glucose level. Glycosylation is a natural physiological process of the attachment of carbohydrate residues to free amino groups of proteins. In hyperglycemia, so-called nonenzymatic glycosylation begins. The end products of this process are accumulated and bind to various proteins (hemoglobin, albumin, collagen, crystallin, and lipoproteins), disrupting their functions. For example, collagen glycosylation results in accumulation of byproducts in the extravascular matrix. As the organism considers these byproducts to be “alien”, an immune response develops: immune cells, i.e. macrophages, increase the secretion of proinflammatory cytokines (TNF-α, IL-6, etc.), the negative effect of which will, in turn, aggravate the already present insulin resistance. Hence another “vicious” cycle is closed [29].
It should be added that glycosylation and increased ROS production caused by excessive lipid and glucose oxidation directly contribute to damaging of collagen and another connective tissue protein, elastin. Many researchers consider this damage to be one of the main factors in human aging. This process increases the number of so-called “cross-links” within a molecule and between molecules; as a result, the protein ceases to be elastic. As collagen and elastin constitute about one-third of the organism’s proteins and belong to the family of long-lived proteins, their degenerative transformation will strongly affect the entire organism.
It would be wrong to think that only unhealthy people, whose glucose level in blood is chronically and constantly raised, are affected by glucose toxicity. Even a single sharp increase in glucose level observed after consumption of simple carbohydrates (sweet drinks, biscuits, sweets, chips, beer) causes a strong proinflammatory reaction and triggers oxidative stress in the organism of a completely healthy person.
This fact was confirmed several years ago by a group of American researchers from the Department of Endocrinology, Diabetes, and Metabolism of the University of Buffalo [30] who described this process. They first gave completely healthy people a drink composed of 75 g of glucose dissolved in water, and then they took blood samples for analysis after 1, 2, and 3 h. The results of the analysis showed a sharp increase in different proinflammatory factors (AP-1, Egr-1, and others), as well as the well-known nuclear transcription factor NF-κB, one of the most potent stimuli of inflammation, could be observed for three hours after glucose intake. In addition, they discovered that oxidative stress increases dramatically after glucose intake: production of extremely dangerous superoxide radicals by leukocytes was shown to be 140% higher than norm, while concentration of one of the most effective and beneficial antioxidants, α-tocopherol (vitamin E) was reduced [31]. Some American researchers showed that consumption of excessively fat food causes similar effects [32]. So now everyone who wants to eat sweets or fast food can visualize the processes that this food will induce in their body.
Many years of research on intracellular processes led the Russian Academician, Professor of Moscow State University V. P. Skulachev, to conclude that aging and death can be programmed by the human genome [33-37]. His theory can be summarized as follows: appearance of the first deviations from the natural norm activates the process of self-destruction in the human body. This mechanism developed in the course of evolution so that an individual with various damages that accumulated with age would not transmit his possibly damaged genes to descendants, and therefore the genetic program that was developing for millions of years would not be violated.
Thus, nature discards those individuals that become potentially dangerous for the future population, and it is not an individual life but the well-being and normal development of the community as a whole that is most important to a species. According to the Russian Academician, this cruel law is applicable to all species, and it means that any complex biological system (from unicellular organisms to humans) will be subject to self-destruction if it becomes unnecessary or dangerous for the existence of a system of higher hierarchy – the community of individuals of the species, the overall population [34].
Muir and Howard [38] showed in their study of a Japanese aquarium fish the fatal role that even a single individual with a “damaged” DNA can play for the entire biological species. The gene of human growth hormone was introduced into the fish, and the transgenic males proved to be more attractive for females because of their size. However, the reproductive function of these fish was found to be damaged, and calculations showed that even one such transgenic fish with a high degree of probability can cause degeneration and death of the entire population after a certain number of generations. According to V. P. Skulachev, it is ROS that play the role of “samurai sword”, which helps the organism to “commit biochemical suicide”. As ROS are constantly produced in mitochondria, the first stage of this process was given the name mitoptosis [34].
Mitochondria that for any reason begin to produce large quantities of ROS that cannot be neutralized by an antioxidant system sacrifice themselves for the security of the entire cell. The interaction of ROS with one of the proteins of the inner mitochondrial membrane results in the formation of a nonspecific channel – a pore due to which the mitochondrial membrane becomes permeable, and, therefore, the mitochondrion dies. According to Skulachev, “It is noteworthy that the death of a mitochondrion producing excessive amounts of ROS requires no proteins other than those that are already present in the mitochondrion. It seems that a mitochondrion that could not detoxify its own ROS commits suicide so as to rid the cell of possible troubles. In other words, prolonged opening of the permeable membrane pore is nothing but a suicide of a mitochondrion that has become dangerous for the cell. In this way the purity of the mitochondrial population in a cell can be maintained” [34]. According to Skulachev, this mechanism has a specific function, to prevent cellular DNA from being damaged by dangerous ROS.
If liquidation of several mitochondria does not help, and ROS are still produced in threatening amounts, the second stage of protection of the organism begins, the self-destruction of the whole cell, apoptosis: “If more and more mitochondria turn into super-producers of ROS and “open Kingston valves”, their concentrations increase, and the cell containing many defective mitochondria undergoes apoptosis. As a result, cells with mitochondria producing too many ROS are eliminated from a tissue” [34]. If destructive processes continue, the number of cells that have committed suicide becomes critical and leads to the death of an organ, and then – of an entire organism, i.e. phenoptosis. These two terms – “mitoptosis” (self-destruction of mitochondria) and “phenoptosis” (self-destruction of an organism) have been put into wide use by Academician Skulachev, and now they can be found in international scientific studies on biochemistry.
If we look at all the pathologies resulting from unhealthy and excessive diet from the perspective of the above-described hypothesis, we can see that they quite logically fit into the theory of programmed self-destruction formulated by Skulachev. This is how this sad scenario can be realized. If a person, while being not so physically active, consumes too much food rich in fats and carbohydrates, this situation directly contradicts the main natural function of fats and carbohydrates, providing the organism’s energy requirements. Fats consumed with food are supposed to fill particularly high and prolonged energy costs, such as those that face professional athletes, or full-time manual workers, or members of polar expeditions after hours of difficult trekking. These mechanisms were designed by evolution millions of years ago, when the first mammals left seas for the land and because of life in new conditions developed muscles of a new type and a new, insulin-dependent mechanism of delivering energy substrates to these muscles. This is how Prof. V. N. Titov described these evolutionary changes in the first land animals: “Formation of a new function in the course of phylogenesis, the function of prolonged and intense muscle activity (motion) was the cause of marked changes not only in physiology, but also morphology in multicellular organisms. This led to: a) formation of skeletal striated myocytes from smooth muscle cells; b) formation of specialized adipocytes from LCT (loose connective tissue); these adipocytes could start accumulating fatty acids in the form of triglycerides in a single large lipid droplet in the cytosol. Later this led to differentiation of functionally different β-cells from α-cells of islets of Langerhans; these β-cells started synthesis and deposition of insulin” [28]. If a person does not understand such a simple and obvious point that the primary function of food is to supply the body with necessary compounds, not to be a source of pleasure, then sooner or later such person will become unnecessary and even dangerous for the entire human population. And the population will get rid of him by programmed phenoptosis, which will be realized through a variety of cardiovascular, endocrine, and other pathologies.
There is an indirect confirmation of this hypothesis: improper nutrition leads to the development of the same processes that can be observed in normal aging: sarcopenia (decrease of muscle mass); suppression of autophagy; degenerative transformation of connective tissue proteins and increased ROS generation; shortening of DNA ends (telomeres); formation of defective cells; and leptin resistance [20, 29, 39]. Another observation also confirms the connection between excessive nutrition and phenoptosis: formation of numerous self-initiated “vicious” cycles, the appearance of which is similar to triggering a chain reaction, which is continued and enhanced till the death of an organism. Launching of this chain reaction can be traced starting from that critical point when adipose tissue reaches such size and properties that it starts secreting into blood more destructive factors (adipokines and fatty acids) than an organism is capable of neutralizing without severe consequences. Once such a “Rubicon” is crossed, pathological processes start to prevail over physiological ones; they grow with time like a snowball, gaining power and giving rise to endless new destructive biochemical reactions.
Development of obesity and subsequent death of an organ such as the liver can serve as a good illustration of this entire process: 1) initial excess of fatty acids leads to triglyceride accumulation of hepatocytes; 2) non-metabolizable triglycerides, being accumulated as a “dead weight”, form fatty liver disease (hepatic steatosis); 3) this process triggers an avalanche of numerous negative factors: increased activity of cytochrome P450 2E1, increased generation of reactive oxygen species, lipid peroxidation, activation of proinflammatory cytokines and nuclear transcription factor NF-κB, mitochondrial DNA damage, and increased expression of PPARγ receptors, release of malondialdehyde and granzyme B, induction of Fas-ligand and subsequent cell death; 4) dead hepatocytes, when accumulated in the intercellular space, initiate the formation of inflammatory response and the transition of steatosis to steatohepatitis; 5) fibrosis and subsequent cirrhosis are the final stages of this process [40-42].
What attracts our attention and can confirm both Academician Skulachev’s theory and the connection between this theory and unhealthy diet? Perhaps the fact that there is one phenomenon that plays the most important role in all these processes in liver: it is mitochondrial dysfunction (damage of the mitochondrial genome and mitochondrial mutations), which always ends with mitoptosis, mitochondrial death; in liver, due to the very high number of mitochondria in hepatocytes, this process reaches such proportions that it becomes critical to the fate of this organ [40].
All these successive biochemical reactions are so strikingly similar to the triggering of self-destruction that it is impossible to imagine any other comparison. It remains to add that violation and damping of such an important function as reproduction, which develops prematurely due hormonal disturbances (for example, polycystic ovary syndrome) connected to improper nutrition, also fits well into the theory.
Whether there is any sense in such a cruel evolutionary method, everyone can judge for himself when observing today many people, who treat their health extremely thoughtlessly, and pass on to their descendants already “damaged” genes. As a result, miserable children are born – children genetically predisposed to alcoholism, obesity, and various mental, endocrine, and other deviations. By analogy with apoptosis and phenoptosis, we can suggest the following stages of the development of this process: “ethnoptosis”, when the number of “unhealthy” people in a particular population reaches critical values and the entire nation disappears; and “genoptosis”, when the same scenario is realized on the scale of the entire planet. But let us hope that this scenario will not come true. Rather, on the contrary, the most beneficial situation for the population option will develop when the life of people prone to self-destruction will become shorter and shorter in subsequent generations until they will finally start dying before having offspring, and, as a result, at some point they will disappear together with their “sick” genes.
Proceeding from the aforesaid, it is evident that changing the lifestyle will be the “first line of defense” and the best prevention of disorders developing due to improper nutrition and lack of physical activity. This lifestyle change should be based on an increase in physical activity and dietary adjustments that should include two important components. The first one embraces excluding (or minimizing) from the diet harmful products containing refined grains and refined sugar. Every year numerous data of research on the harmful effects of refined sugar appear, and it seems fairly safe to assume that rather sooner than later sugar and sugar-containing products will be completely forbidden for sale and production. It also seems logical to reduce meat and high-fat dietary products (such as sour cream, cheese, and butter) rich in palmitic acid, which, as we recall, is the major negative factor in the development of atherosclerosis and cell death [28]. At the same time, it would be appropriate to include in the diet fish and linseed and olive oil. Fish and oils contain useful polyunsaturated acids, which, being antagonists of palmitic acid, will help to prevent vascular problems [29]. The American Heart Association and the National Heart, Lung, and Blood Institute give similar recommendations on healthy diet in their joint report: “Diet therapy includes low consumption of saturated fats, trans fats, cholesterol, and simple carbohydrates (sugars), increased consumption of fruits, vegetables, and whole-grain products; excessive (extreme) consumption of carbohydrates or fats should be avoided” [43]. There is also a second component of a healthy diet – a significant reduction in the total amount of food consumed at the end of the reproductive period (45-50 years of age). This measure is supposed to reduce oxidative processes in mitochondria and as a consequence to reduce ROS generation. Following these simple recommendations will most likely help people to live to very old age and perhaps live until the time of the invention of an effective “youth elixir”, with which it will be possible to overcome or significantly postpone such a nasty thing as premature aging.
REFERENCES
1.Reaven, G. M. (1988) Diabetes, 37,
1595-1607.
2.Mendis, S., and Webber, D. (eds.) (2005)
Avoiding Heart Attacks and Strokes: Don’t Be a Victim
– Protect Yourself, WHO, Geneva.
3.Shvartz, V. Ya. (2009) Probl. Endokrinol.,
55, 38-44.
4.Solntseva, A. V. (2009) Med. Novosti,
3, 7-11.
5.Hotamisligil, G., Shargill, N., and Spiegelman, B.
(1993) Science, 259, 87-91.
6.Takebayashi, K., Suetsugu, M., and Matsutomo, R.
(2006) South Med. J., 99, 23-27.
7.Zhang, Y., Proenca, R., and Maffei, M. (1994)
Nature, 372, 425-432.
8.Gaillard, S., and Gaillard, R. (2007) Obesity
& Metabolism, 3, 191-205.
9.Wynne, K., Stanley, S., and McGowan, B. (2005)
J. Endocrinol., 184, 291-318.
10.Koerner, A., Kratzsch, J., and Kiess, W. (2005)
Best Pract. Res. Clin. Endocrinol. Metab., 19,
525-546.
11.Steppan, C. M., Bailey, S. T., Bhat, S., Brown,
E. J., Banerjee, R. R., Wright, C. M., Patel, H. R., Ahima, R. S., and
Lazar, M. A. (2001) Nature, 409, 307-312.
12.Shojima, N., Sakoda, H., Ogihara, T., and
Fujishiro, M. (2002) Diabetes, 51, 1737-1744.
13.Carswell, E. A., Old, L. J., Kassel, R. L.,
Green, S., Fiore, N., and Williamson, B. (1975) Proc. Natl. Acad.
Sci. USA, 72, 3666-3670.
14.Goossens, G. H. (2008) Physiol. Behav.,
94, 206-218.
15.Charo, I. F., and Ransohoff, R. M. (2006) N.
Engl. J. Med., 354, 610-615.
16.Denke, M. A. (2007) N. Engl. J. Med.,
357, 2526-2532.
17.Pedersen, B. K. (2007) Biochem. Soc.
Trans., 35, 1295-1297.
18.Trayhurn, P., and Wood, I. S. (2005) Biochem.
Soc. Trans., 33, 1078-1081.
19.Kaydashev, I. P. (2011) Mezhdunarod.
Endokrinol. Zh., 3, 35-40.
20.Goossens, G. H. (2008) Physiol. Behav.,
94, 206-218.
21.Felig, F., Baxter, J. D., Brodus, A. E., and
Fromen, L. A. (1985) Endocrinology and Metabolism [Russian
translation], Meditsina, Moscow.
22.Velkov, V. V. (2008) Laboratoriya,
1, 16-19.
23.Tereshina, E. V. (2007) Uspekhi Gerontol.,
20, 59-65.
24.Titov, V. N. (2012) Kardiol. Vestnik,
2, 24-32.
25.Ivashkin, V. T., and Mayevskaya, M. V. (2010)
Ros. Zh. Gastroenterol. Gepatol. Koloproktol., 1,
4-13.
26.Listenberger, L. L., Ory, D. S., and Schaffer, J.
E. (2001) J. Biol. Chem., 276, 14890-14895.
27.Belosludtsev, K. N., Belosludtseva, N. V., and
Mironova, G. D. (2005) Biochemistry (Moscow), 70,
815-821.
28.Titov, V. N. (2012) Ateroskleroz
Dislipidemii, 3, 49-57.
29.Tereshina, Ye. V. (2006) Gerontol.
Geriatr., 5, 38-48.
30.Aljada, A., Ghanim, H., Mohanty, P., Kapur, N.,
and Dandona, P. (2002) J. Clin. Endocrinol. Metab., 87,
1419-1422.
31.Drapkina, O. M., and Chaparkina, S. O. (2007)
Ros. Med. Vesti, 3, 67-76.
32.Mohanty, P., Hamouda, W., Garg, R., Aljada, A.,
Ghanim, H., and Dandona, P. (2000) J. Clin. Endocrinol. Metab.,
85, 2970-2973.
33.Skulachev, V. P. (1998) Soros Obrazovat.
Zh., 8, 2-7.
34.Skulachev, V. P. (2001) Soros Obrazovat.
Zh., 6, 4-10.
35.Skulachev, V. P. (1999) Soros Obrazovat.
Zh., 9, 1-7.
36.Skulachev, V. P. (1996) Soros Obrazovat.
Zh., 3, 4-16.
37.Skulachev, V. P. (1997) Biochemistry
(Moscow), 62, 1191-1195.
38.Muir, W. M., and Howard, R. D. (2001) Amer.
Nat., 158, 1-16.
39.Bergman, R. N. (2005) N. Engl. J. Med.,
353, 2201-2209.
40.Babak, O. Ya., and Kolesnikova, Ye. V. (2011)
Sovrem. Gastroenterol., 3, 56-63.
41.Buyeverova, Ye. L. (2002) Ros. Zh.
Gastroenterol. Gepatol. Koloproktol., 4, 225-229.
42.Kravchenko, N. A. (2012) Eksp. Klin. Med.,
2, 25-31.
43.Grundy, S. M., Hansen, B., Smith, S. C., Cleeman,
J. I., and Kahn, R. A. (2004) Circulation, 109,
551-556.