Effect of Mitochondria-Targeted Antioxidant SkQ1 on Programmed Cell Death Induced by Viral Proteins in Tobacco Plants
A. D. Solovieva1, O. Yu. Frolova2, A. G. Solovyev3,4, S. Yu. Morozov3, and A. A. Zamyatnin, Jr.3,5*
1Faculty of Biology, Lomonosov Moscow State University, Leninsky Gory 1/12, 119234 Moscow, Russia; fax: +7 (495) 939-43092Institute of Mitoengineering, Lomonosov Moscow State University, Vorobyevy Gory 1/73a, 119992 Moscow, Russia; fax: +7 (495) 939-5945
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Leninsky Gory 1/40, 119992 Moscow, Russia; fax: +7 (495) 939-0338
4Institute of Agricultural Biotechnology, Russian Academy of Agricultural Sciences, ul. Timiryazevskaya 42, 127550 Moscow, Russia; fax: +7 (499) 977-0947
5Institute of Molecular Medicine, Sechenov First Moscow State Medical University, ul. Trubetskaya, 8, 119991 Moscow, Russia; fax: +7 (495) 622-9632; E-mail: zamyat@belozersky.msu.ru
* To whom correspondence should be addressed.
Received April 25, 2013
Programmed cell death (PCD) is the main defense mechanism in plants to fight various pathogens including viruses. The best-studied example of virus-induced PCD in plants is Tobacco mosaic virus (TMV)-elicited hypersensitive response in tobacco plants containing the N resistance gene. It was previously reported that the animal mitochondrial protein Bcl-xL, which lacks a homolog in plants, effectively suppresses plant PCD induced by TMV p50 – the elicitor of hypersensitive response in Nicotiana tabacum carrying the N gene. Our studies show that the mitochondria-targeted antioxidant SkQ1 effectively suppresses p50-induced PCD in tobacco plants. On the other hand, SkQ1 did not affect Poa semilatent virus TGB3-induced endoplasmic reticulum stress, which is followed by PCD, in Nicotiana benthamiana epidermal cells. These data suggest that mitochondria-targeted antioxidant SkQ1 can be used to study molecular mechanisms of PCD suppression in plants.
KEY WORDS: mitochondria-targeted compounds, reactive oxygen species, hypersensitive response, ER stress, unfolded protein responseDOI: 10.1134/S000629791309006X
Abbreviations: C12TPP, dodecyl triphenylphosphonium; dpi, day post infiltration; ER, endoplasmic reticulum; PCD, programmed cell death; PSLV, Poa semilatent virus; PVX, potato virus X; ROS, reactive oxygen species; TMV, tobacco mosaic virus; UPR, unfolded protein response.
Programmed cell death (PCD) is one of the most effective defense
mechanisms in plants and other organisms devoid of mobile cells of the
immune system characteristic of mammals. By inducing PCD, plants limit
the spread of infection in infected cells, while new growth points are
formed from non-infected cells. The main characteristics of the
development of PCD include significant reprogramming of transcriptional
regulation, synthesis of protective compounds [1,
2], changes in the level of calcium ion content [3, 4], and strong increase in the
level of reactive oxygen species (ROS) [1, 5].
The respiratory chain of animal mitochondria is one of the major sources of ROS that cause cell death. In the case of plants, ROS are formed in mitochondrial electron transport chains, in particular, by NADPH-oxidase [6-8]. ROS formation in mitochondria depends on membrane potential; its reduction by 10-15% decreases the rate of ROS formation tenfold [9]. Chloroplasts, peroxisomes, and NADPH-oxidase of the plasma cell membrane also play a significant role in ROS generation in plants [10-12].
ROS formation is suppressed by antioxidants such as ascorbic acid, tocopherol, and glutathione, and by enzymes that regenerate oxidized forms of low molecular weight antioxidants. Antioxidants usually act cooperatively. For example, the interaction between ascorbic acid and glutathione has been thoroughly studied. Specific compartmentalization of antioxidant systems is also very important [13, 14]. Methods of fighting oxidative damage were proposed as early as the 1960s, when antioxidants were first used. However, due to membranes antioxidants are practically unable to penetrate into mitochondria and neutralize endogenous ROS produced in the electron transport chain.
A new generation of antioxidants covalently bound to penetrating cations has been intensely studied in recent years [15]. One of these compounds, 10-(6′-plastoquinonyl)decyltriphenylphosphonium (SkQ1), consisting of plastoquinone and triphenylphosphonium connected via a C10 linker chain, is specifically accumulated in mitochondria, possesses high antioxidant activity, and is a regenerable antioxidant because its oxidized form is reduced by the electron transport chain in vivo [16-18]. Furthermore, SkQ1, due to its ability to interact with fatty acid anions, also acts as a mitochondria-targeted protonophore [19]. This property of SkQ1 results in mild uncoupling, leading to a decrease in membrane potential, which reduces ROS production [9, 19].
In studies on human fibroblasts and HeLa cells, extremely low concentrations of SkQ1 were shown to inhibit H2O2-induced apoptosis [15]. New drugs based on mitochondria-targeted antioxidants are being developed for the treatment of human diseases associated with oxidative stress [18, 19]. The first SkQ1-based medicine, Visomitin, used in treatment of “dry eye” syndrome, has been successfully tested and registered [20].
However, molecular mechanisms of the action of SkQ1 are not well studied in plant systems. Pea Pisum sativum L., variety Alpha was the main object of previously published studies. Leaf epidermis pellicles of pea seedlings were used for the experiments, and SkQ1 was found to suppress PCD caused by chitosan and cyanide in this experimental system [21, 22]. Moreover, SkQ1 was shown to slow the aging of rosette leaves in Arabidopsis plants [23].
In the present work we studied the effects of SkQ1 on the development of hypersensitive response (form of PCD) in tobacco plants induced by virus-specific proteins. SkQ1 was found to suppress PCD induced via the cascade of mitogen-activated protein kinases. At the same time, this compound does not suppress the development of endoplasmic reticulum (ER) stress that also leads to PCD.
MATERIALS AND METHODS
Recombinant clones and cloning. Genetic constructions pLH* [24], pRT-m-GFP5-ER [25], and pRT-18K [26] were described previously. Amplification of the p50, MEK2, and Bcl-xL genes as well as MEK2 mutagenesis for cloning in pLH* were performed using PCR and specific oligonucleotides (sequences available upon request). Cloning was performed using standard methods [27].
Transient expression using agroinfiltration or metal microparticle bombardment. For agroinfiltration, Agrobacterium tumefaciens cultures (strain C58C1) were grown and prepared according to the previously described method [28]. Nicotiana tabacum leaves were infiltrated by the cell suspensions.
Bombardment of Nicotiana benthamiana leaves with tungsten microparticles was performed in a PDS-1000 high-pressure helium system (Bio-Rad, USA) following the previously described method [29]. The samples were examined by microscopy using a TCS SP2 confocal laser scanning microscope (Leica, Germany). GFP fluorescence was excited by an argon laser (wavelength 488 nm) and detected in the range 500-530 nm.
Incubation of plant material in SkQ1 solutions. Cut off leaves of N. tabacum or N. benthamiana were first exposed to agroinfiltration or bombardment with metal microparticles, and then were incubated by placing the petioles into water or water solutions of SkQ1 or C12TPP. The solutions were changed daily.
RESULTS AND DISCUSSION
Effect of SkQ1 on mitochondria-mediated cell death. In tobacco plants carrying the resistance gene (N gene), a fragment of the gene of tobacco mosaic virus (TMV) replicase, which encodes a helicase domain, is an elicitor of hypersensitive response, a form of PCD in plants [30]. An experimental system including p50 protein (helicase domain of TMV replicase) and plants carrying the N gene is widely used for the analysis of molecular and cellular mechanisms involved in the induction of hypersensitive response. It was shown that p50 protein involves HSP90 cellular chaperone to activate the cascade of mitogen-activated kinases, including MEK2 protein kinase, which becomes activated due to phosphorylation and, in turn, phosphorylates two effector kinases, WIPK and SIPK, causing their activation [31, 32]. Precise functions of WIPK and SIPK protein kinases in the development of PCD remain unknown, even though it is clear that their activation leads to mitochondrial dysfunction, as the effect caused by the activation of the cascade including MEK2 and WIPK/SIPK can be inhibited by Bc1-xL protein [32, 33]. Bc1-xL is a membrane protein of human mitochondria that can block the outflow of cytochrome c from mitochondria that causes caspase activation. Besides that, this protein was previously shown to have antiapoptotic properties not only in animal, but also in plant cells [34-37].
Two proteins, p50 and MEK2 protein kinase, were used to induce mitochondria-dependent cell death in plants. The p50 gene was amplified and cloned in the binary pLH* vector controlled by 35S promoter of cauliflower mosaic virus (Fig. 1a). The N. tabacum MEK2 gene was amplified and cloned for its further mutagenesis to produce a constitutively active mutant. Thus, two amino acid residues that are phosphorylated in the wild type protein were subjected to mutagenesis. Serine and threonine residues in positions 227 and 233, respectively, were substituted by residues of aspartic acid, and, thereby, their charge being analogous to the phosphate groups attached by the activating kinase. Such a mutant was previously shown to be constitutively active; it was also shown to cause hypersensitive response in tobacco leaves [31, 32]. The mutant MEK2DD gene was cloned in the pLH* vector controlled by 35S promoter (Fig. 1b). Agrobacterium tumefaciens cells were transformed with the constructions carrying the p50 and MEK2DD genes, and the resulting cultures were used for transient expression of these proteins in tobacco leaves using the agroinfiltration method.
Fig. 1. Expression cassettes used in this study. a) Organization of the Tobacco mosaic virus (TMV) genome and the expression cassette with the fragment of viral replicase, p50 protein. In a schematically presented TMV genome, the genes are shown as rectangles; molecular weights of the encoded proteins are shown in kilodaltons. The positions of conservative domains of the viral replicase are indicated as: MT, methyltransferase domain; HEL, helicase domain; POL, polymerase domain. The horizontal arrow indicates leaky termination codon of the gene of 126K protein leading to translation of the 183K protein. The expression cassette with p50 gene includes 35S promoter of cauliflower mosaic virus and transcription terminator marked “term”. b) Expression cassettes with MEK2DD and Bcl-xL genes. Introduced mutations are shown in the MEK2 gene.
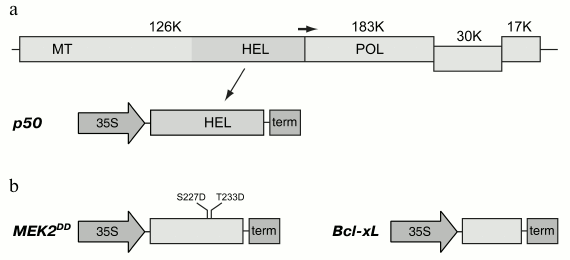
The culture of agrobacteria with cloned p50 gene was used for infiltration of the leaves of tobacco plants carrying the N gene (N. tabacum cv. Samsun NN), and the culture with the MEK2DD gene was used for infiltration of tobacco leaves of the same species, which, however, did not carry this resistance gene as its product is needed only at the first stage of the activation process leading to cell death (this stage includes recognition of p50 protein). Development of the hypersensitive response in the infiltrated areas of the leaves agroinfiltrated with p50 and MEK2DD could be observed on day 2 post-infiltration (Figs. 2a and 3a).
Fig. 2. Effect of SkQ1 on hypersensitive response in tobacco leaves (Nicotiana tabacum cv. Samsun NN). a) Hypersensitive response in the leaf infiltrated with agrobacterium culture carrying p50 gene. b) Inhibition of hypersensitive response: leaves agroinfiltrated with p50 were incubated in SkQ1 solutions. The left half of each leaf was infiltrated with culture carrying p50 gene, and the right half was infiltrated with the culture carrying pLH* vector without the expressed gene (negative control). The leaves were incubated either in control or in SkQ1 solutions of given concentrations. Arrows mark the area of the development of hypersensitive response. All the photos were taken on day 2 post-infiltration.

Fig. 3. Inhibition of hypersensitive response caused by incubation in SkQ1 solution and coexpression with Bcl-xL protein. a) Development of hypersensitive response in agroinfiltrated leaves of N. tabacum expressing MEK2DD. The leaves shown were incubated in water or 0.1 nM SkQ1 solution. b) Inhibition of hypersensitive response caused by p50 expression or Bcl-xL protein. The left half of the leaf was infiltrated with agrobacterium culture expressing p50, and the right half was co-infiltrated with the mixture of the two agrobacterial cultures, one of which expressed p50 and the other Bcl-xL. The photos were taken on day 2 post-infiltration.

Bcl-xL protein, whose gene was amplified and cloned in the binary pLH* vector, was used to confirm the fact that the response caused by the expression of these genes was mediated by mitochondria (Fig. 1b). Co-infiltration of tobacco leaves by agrobacterium cultures carrying p50 and Bc1-xL proteins led to the suppression of hypersensitive response induced by p50 (Fig. 3b). Likewise, inhibition of cell death could be observed on co-expression of MEK2DD and Bcl-xL (data not shown). These results are consistent with the literature [32, 33] and confirm the validity of the experimental approach used in this study.
To study the possible effect of SkQ1 on the development of hypersensitive response caused by p50, tobacco leaves agroinfiltrated with respective constructions as well as control construction (pLH* vector with no inserts) were placed into water or water solutions of 1 and 0.1 nM SkQ1. The same concentrations of dodecyl triphenylphosphonium (C12TPP) were used as a control. C12TPP is a lipophilic cation and C12 hydrophobic linker, but it has no antioxidant part. In the case of control experiments (leaves placed into water or C12TPP solution), development of hypersensitive response could be observed in the areas infiltrated with culture carrying p50 on the second day post infiltration, and on the third day post infiltration these parts of the leaves were completely dead (Fig. 2b). In the case of leaves placed into 1 and 0.1 nM SkQ1 solutions, no degradation of the leave tissue in the area infiltrated with p50 could be observed on the third day post infiltration (Fig. 2b). Similar data were obtained in experiments with MEK2DD (Fig. 3a). Thus, incubation of tobacco leaves in SkQ1 solution was shown to cause a significant retardation in the development of p50-induced hypersensitive response, this process being mediated by the N gene or developing directly via the cascade of mitogen-activated kinases.
Effect of SkQ1 on cell death induced by ER stress. Accumulation of significant amounts of unfolded or misfolded protein molecules in ER, excess of membrane proteins, as well as other factors, cause ER stress [38] resulting in the so-called “unfolded protein response” (UPR), which is supposed to compensate for functional damage to ER. However, this process causes cell death in case of prolonged exposure to the factors leading to ER stress [38, 39]. In both mammalian and plant cells, development of UPR is associated with an increase in the levels of a number of ER proteins participating in the folding of polypeptide chains, such as BiP protein, protein disulfide isomerase, calreticulin, and calmodulin [40, 41].
TGB3 protein (encoded by potato virus X (PVX)), a membrane protein necessary for the cell-to-cell spread of viral infection, has been recently shown to cause ER stress in the case of its high accumulation in plant cells [33, 42, 43]. TGB3 protein was found to activate bZIP60 transcription factor necessary for accumulation of ER proteins characteristic for UPR as well as SKP1 protein (a component of the E3 ubiquitin ligase complex) [33, 42, 43]. TGB3-induced development of UPR leads to cell death accompanied by an increase in the level of ROS and DNA fragmentation [33], but the molecular mechanism of cell death induction resulting from UPR remains unknown [44]. It is important to emphasize that this form of cell death is not associated with mitochondrial dysfunction, as it cannot be suppressed by Bcl-xL protein or other antiapoptotic proteins [33].
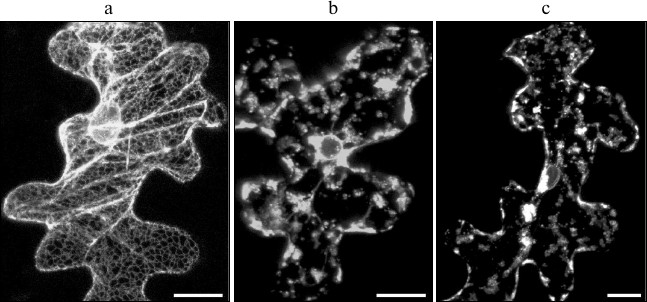
In this study, we have analyzed the possibility of SkQ1 effecting cell death caused by TGB3 protein. We have previously shown ER stress caused by the expression of Poa semilatent virus (PSLV) TGB3 protein to manifest morphologically in ER disintegration manifested in ER membranes losing their characteristic morphology and vesiculation [45]. The previously described marker m-GFP5-ER protein, a green fluorescent protein fused to the leader sequence directing it towards the ER lumen and to the C-terminal signal of retention in ER, was used to visualize ER morphology [25]. The marker m-GFP5-ER protein was coexpressed with TGB3 protein of PSLV in epidermal cells of N. benthamiana leaves after leaf bombardment with metal microparticles carrying expression vectors with cloned m-GFP5-ER and TGB3 genes. In the case of control experiments, when m-GFP5-ER was expressed in the absence of TGB3, GFP fluorescence was observed in the polygonal network of cortical ER and in nuclear membrane (Fig. 4a), which matches the previously described location of this marker protein [25]. However, coexpression of m-GFP5-ER and TGB3 caused significant changes in intracellular localization of the fluorescence. Instead of being seen in the structures characteristic for ER, fluorescence was observed in clusters of granular structures and in nuclear membrane (Fig. 4b). Incubation of leaves coexpressing m-GFP5-ER and TGB3 in 1 and 0.1 nM SkQ1 solutions did not cause the changes in ER marker localization when compared to leaves expressing m-GFP5-ER and TGB3 incubated in water (Fig. 4c). Thus, we can conclude that SkQ1 has no effect on TGB3-induced ER stress, which causes cell death independent of mitochondrial dysfunction.
Fig. 4. Analysis of effect of SkQ1 on ER stress induced by the expression of the membrane viral protein TGB3; this protein was temporarily expressed in epidermal cells of N. benthamiana leaf due to bombardment with metal microparticles. a) Subcellular localization of ER marker m-GFP5-ER. b) Localization of marker m-GFP5-ER protein when it is coexpressed with PSLV TGB3 protein. c) Localization of m-GFP5-ER coexpressed with TGB3 when the leaf was incubated in 0.1 nM water solution of SkQ1. Every image represents a superposition of a series of optical sections obtained by laser scanning confocal microscope. Scale bar, 20 μm.
Genomes of the majority of α-like plant viruses encode proteins whose expression can lead to the development or enhancement of hypersensitive response (one of the forms of PCD). These can be viral protein products different in their structure and functions: transport proteins [33], suppressors of posttranscriptional gene silencing [46-48], transcriptional factors [49], and others. Despite extensive research, functions of many of these proteins have still not been characterized [50-52]. In this work, we have used two experimental models in which expression of viral proteins activate PCD. The first experimental system included PCD induction through a cascade of mitogen-activated kinases, leading, as shown previously, to mitochondrial dysfunction [31, 32]. The second experimental system involved induction of UPR and ER stress leading to development of PCD [33, 45]. It should be noted that in the latter case the molecular mechanism of PCD induction remains not fully understood; in particular, the role of mitochondria in this signaling pathway has not been clarified [44].
In our experiments, mitochondria-targeted antioxidant SkQ1 effectively suppressed the development of PCD induced through the cascade of mitogen-activated kinases. Bcl-xL-mediated PCD suppression in this experimental system indicates that the suppression of PCD development takes place at the level of mitochondria. At the same time, SkQ1 had no effect on the development of ER stress caused by PSLV TGB3. These data are in good agreement with those obtained for the functional analog of PSLV TGB3 – PVX TGB3. In the experiments of Ye et al. [33], Bcl-xL could not suppress PCD induced by PVX TGB3. We conclude that despite the significant differences in their structure, both potex-like and hordei-like TGB3 induce ER stress leading to PCD independently of mitochondria.
Thus, the mitochondria-targeted antioxidant SkQ1 effectively suppresses PCD at the mitochondrial level not only in animals, but also in plants. However, SkQ1 proved to be ineffective for the suppression of PCD that develops without direct involvement of mitochondrial functions.
The authors would like to express their gratitude to V. P. Skulachev for the possibility to carry out this research and for his useful advice and criticism.
The authors are also grateful to the Mitotech LLC for providing SkQ1 and C12TPP.
REFERENCES
1.Shapiguzov, A., Vainonen, J. P., Wrzaczek, M., and
Kangasjarvi, J. (2012) Front. Plant Sci., 3, 292.
2.Janitza, P., Ullrich, K. K., and Quint, M. (2012)
Front. Plant Sci., 3, 271.
3.Eichmann, R., and Schafer, P. (2012) Front.
Plant Sci., 3, 200.
4.Howell, S. H. (2013) Annu. Rev. Plant Biol.,
64, 477-499.
5.Tripathy, B., and Oelmuller, R. (2012) Plant
Signal. Behav., 7, 1621-1633.
6.Robson, C. A., and Vanlerberghe, G. C. (2002)
Plant Physiol., 129, 1908-1920.
7.Murphy, M. P. (2009) Biochem. J.,
417, 1-17.
8.Venditti, P., Di Stefano, L., and Di Meo, S. (2013)
Mitochondrion, 13, 71-82.
9.Korshunov, S. S., Skulachev, V. P., and Starkov, A.
A. (1997) FEBS Lett., 416, 15-18.
10.Petrov, V. D., and Van Breusegem, F. (2012)
AoB Plants, 2012, pls014.
11.Balazadeh, S., Jaspert, N., Arif, M.,
Mueller-Roeber, B., and Maurino, V. G. (2012) Front. Plant Sci.,
3, 234.
12.Maruta, T., Noshi, M., Tanouchi, A., Tamoi, M.,
Yabuta, Y., Yoshimura, K., Ishikawa, T., and Shigeoka, S. (2012) J.
Biol. Chem., 287, 11717-11729.
13.Zechmann, B. (2011) Plant Signal. Behav.,
6, 360-363.
14.Koffler, B. E., Bloem, E., Zellnig, G., and
Zechmann, B. (2013) Micron, 45, 119-128.
15.Skulachev, V. P. (2007) Biochemistry
(Moscow), 72, 1385-1396.
16.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L.
E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Yu.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nipryakhina, O. K., Pashkovskaya, A.
A., Pletyushkina, O. Yu., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vysokikh, M.
Yu., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1273-1287.
17.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) Biochim. Biophys. Acta, 1787, 437-461.
18.Skulachev, M. V., Antonenko, Y. N., Anisimov, V.
N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov, M.
V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov, E.
Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F. F.,
Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M. Y.,
Zamyatnin, A. A., Jr., Zorov, D. B., and Skulachev, V. P. (2011)
Curr. Drug Targets, 12, 800-826.
19.Severin, F. F., Severina, I. I., Antonenko, Y.
N., Rokitskaya, T. I., Cherepanov, D. A., Mokhova, E. N., Vyssokikh, M.
Y., Pustovidko, A. V., Markova, O. V., Yaguzhinsky, L. S., Korshunova,
G. A., Sumbatyan, N. V., Skulachev, M. V., and Skulachev, V. P. (2010)
Proc. Natl. Acad. Sci. USA, 107, 663-668.
20.Yani, Ye. V., Katargina, L. A., Chesnokova, N.
B., Beznos, O. V., Savchenko, A. Yu., Vygodin, V. A., Gudkova, Ye. Yu.,
Zamyatnin, A. A., Jr., and Skulachev, M. V. (2012) Prakticheskaya
Meditsina, 59, 134-137.
21.Vasil’ev, L. A., Dzyubinskaya, E. V.,
Kiselevsky, D. B., Shestak, A. A., and Samuilov, V. D. (2011)
Biochemistry (Moscow), 76, 1120-1130.
22.Vasil’ev, L. A., Kiselevsky, D. B.,
Dzyubinskaya, E. V., Nesov, A. V., and Samuilov, V. D. (2012)
Biochemistry (Moscow), 77, 354-361.
23.Dzyubinskaya, E. V., Ionenko, I. F., Kiselevsky,
D. B., Samuilov, V. D., and Samuilov, F. D. (2013) Biochemistry
(Moscow), 78, 68-74.
24.Solovyev, A. G., Minina, E. A., Makarova, S. S.,
Erokhina, T. N., Makarov, V. V., Kaplan, I. B., Kopertekh, L.,
Schiemann, J., Richert-Poggeler, K. R., and Morozov, S. Y. (2013)
Biochimie, 95, 1360-7130.
25.Zamyatnin, A. A., Jr., Solovyev, A. G., Sablina,
A. A., Agranovsky, A. A., Katul, L., Vetten, H. J., Schiemann, J.,
Hinkkanen, A. E., Lehto, K., and Morozov, S. Y. (2002) J. Gen.
Virol., 83, 651-662.
26.Solovyev, A. G., Stroganova, T. A., Zamyatnin, A.
A., Jr., Fedorkin, O. N., Schiemann, J., and Morozov, S. Y. (2000)
Virology, 269, 113-127.
27.Sambrook, J., Fritsch, E. F., and Maniatis, T. A.
(1989) Molecular Cloning: A Laboratory Manual, 2nd Edn., Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
28.Zamyatnin, A. A., Jr., Solovyev, A. G., Bozhkov,
P. V., Valkonen, J. P., Morozov, S. Y., and Savenkov, E. I. (2006)
Plant J., 46, 145-154.
29.Morozov, S. Yu., Fedorkin, O. N., Juttner, G.,
Schiemann, J., Baulcombe, D. C., and Atabekov, J. G. (1997) J. Gen.
Virol., 78, 2077-2083.
30.Erickson, F. L., Dinesh-Kumar, S. P., Holzberg,
S., Ustach, C. V., Dutton, M., Handley, V., Corr, C., and Baker, B. J.
(1999) Philos. Trans. R. Soc. Lond. B Biol. Sci., 354,
653-658.
31.Jin, H., Liu, Y., Yang, K. Y., Kim, C. Y., Baker,
B., and Zhang, S. (2003) Plant J., 33, 719-731.
32.Takabatake, R., Ando, Y., Seo, S., Katou, S.,
Tsuda, S., Ohashi, Y., and Mitsuhara, I. (2007) Plant Cell
Physiol., 48, 498-510.
33.Ye, C. M., Chen, S., Payton, M., Dickman, M.
B., and Verchot, J. (2013) Mol. Plant. Pathol., 14,
241-255.
34.Lacomme, C., and Santa Cruz, S. (1999) Proc.
Natl. Acad. Sci. USA, 96, 7956-7961.
35.Kawai-Yamada, M., Jin, L., Yoshinaga, K., Hirata,
A., and Uchimiya, H. (2001) Proc. Natl. Acad. Sci. USA,
98, 12295-12300.
36.Mitsuhara, I., Malik, K. A., Miura, M., and
Ohashi, Y. (1999) Curr. Biol., 15, 775-778.
37.Qiao, J., Mitsuhara, I., Yazaki, Y., Sakano, K.,
Gotoh, Y., Miura, M., and Ohashi, Y. (2002) Plant Cell Physiol.,
43, 992-1005.
38.Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005)
J. Clin. Invest., 115, 2656-2664.
39.Zhang, K., and Kaufman, R. J. (2006)
Neurology, 66, S102-S109.
40.Oh, D. H., Kwon, C. S., Sano, H., Chung, W. I.,
and Koizumi, N. (2003) Biochem. Biophys. Res. Commun.,
301, 225-230.
41.Urade, R. (2009) Biofactors, 35,
326-331.
42.Ye, C., Dickman, M. B., Whitham, S. A., Payton,
M., and Verchot, J. (2011) Plant Physiol., 156,
741-755.
43.Ye, C. M., Kelly, V., Payton, M., Dickman, M. B.,
and Verchot, J. (2012) Mol. Plant, 5, 1151-1153.
44.Tabas, I., and Ron, D. (2011) Nat. Cell
Biol., 13, 184-190.
45.Solovyev, A. G., Schiemann, J., and Morozov, S.
Y. (2012) Sci. World J., 2012, 416076.
46.Brigneti, G., Voinnet, O., Li, W. X., Ji, L. H.,
Ding, S. W., and Baulcombe, D. C. (1998) EMBO J., 17,
6739-6746.
47.Scholthof, H. B., Scholthof, K. B., and Jackson,
A. O. (1995) Plant Cell, 7, 1157-1172.
48.Yelina, N. E., Savenkov, E. I., Solovyev, A. G.,
Morozov, S. Y., and Valkonen, J. P. (2002) J. Virol., 76,
12981-12991.
49.Lukhovitskaya, N. I., Solovieva, A. D., Boddeti,
S. K., Thaduri, S., Solovyev, A. G., and Savenkov, E. I. (2013)
Plant Cell, 25, 960-973.
50.Canto, T., MacFarlane, S. A., and Palukaitis, P.
(2004) J. Gen. Virol., 85, 3123-3133.
51.Lukhovitskaya, N. I., Yelina, N. E., Zamyatnin,
A. A., Jr., Schepetilnikov, M. V., Solovyev, A. G., Sandgren, M.,
Morozov, S. Y., Valkonen, J. P., and Savenkov, E. I. (2005) J. Gen.
Virol., 86, 2879-2889.
52.Gushchin, V. A., Lukhovitskaya, N. I., Andreev,
D. E., Wright, K. M., Taliansky, M. E., Solovyev, A. G., Morozov, S.
Y., and MacFarlane, S. A. (2013) J. Gen. Virol., 94,
230-240.