Advanced Glycation of Cellular Proteins as a Possible Basic Component of the “Master Biological Clock”
F. F. Severin1,2, B. A. Feniouk2,3, and V. P. Skulachev1,2,3*
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia; E-mail: skulach@genebee.msu.ru2Lomonosov Moscow State University, Institute of Mitoengineering, 119992 Moscow, Russia
3Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received May 23, 2013
During the last decade, evidence has been accumulating supporting the hypothesis that aging is genetically programmed and, therefore, precisely timed. This hypothesis poses a question: what is the mechanism of the biological clock that controls aging? Measuring the level of the advanced glycation end products (AGE) is one of the possible principles underlying the functioning of the biological clock. Protein glycation is an irreversible, non-enzymatic, and relatively slow process. Moreover, many types of cells have receptors that can measure AGE level. We propose the existence of a protein that has a lifespan comparable to that of the whole organism. Interaction of the advanced glycation end product generated from this protein with a specific AGE receptor might initiate apoptosis in a vitally important non-regenerating tissue that produces a primary juvenile hormone. This could result in the age-dependent decrease in the level of this hormone leading to aging of the organism.
KEY WORDS: biological clock, aging, protein glycation, melatonin, juvenile hormone, AGE, RAGEDOI: 10.1134/S0006297913090101
Abbreviations: AGE, Advanced Glycation End products; GRH, gonadotropin-releasing hormone; mROS, mitochondrial reactive oxygen species; RAGE, receptors of AGE.
Within the phenoptosis concept, aging is considered to be the last stage
of ontogenesis, the individual development of an organism [1-4]. However, if this is the
case, then both aging and ontogenesis as a whole should be managed by a
“master biological clock” as postulated by V. M. Dilman [5, 6] and later by A. Comfort [7]. Our knowledge of the molecular mechanism of the
measurement of time by living organisms has so far been limited to
circadian rhythm. This rhythm is determined by cyclic biochemical
reactions that take place in the epiphysis (in birds) or in the
suprachiasmatic nucleus of the hypothalamus (in mammals). The signals
formed by these organs cause fluctuations in concentration of certain
hormones in the blood, especially of melatonin. It seems obvious that
measurement of time on the scale of hours inherent to the circadian
mechanism is hardly suitable for time measurement on the scale of
decades.
Humans check time by comparing it with a reference device based on measuring the rate of radioactive decay. It is unlikely that such a mechanism could be used by living organisms in general. However, there are other spontaneous chemical processes that might be the basis of the mechanism measuring many years. L→D isomerization of amino acids in long-lived proteins is an example of this type of process. In whales, crystallins (lens proteins whose age is comparable to the maximum age of the animal, i.e. about two centuries) have been described [8]. Initially, all the amino acids in crystallin are L-isomers. Over many years, the L-amino acids spontaneously isomerize into D-isomers. This process is the fastest for aspartate (about 2% in 10 years). This means that in case of a 200-year-old whale, there are 40% aspartate residues that are present as D-isomers in crystallin. A counter of biological age might be formed from a protein similar to crystallin and some device that would measure D-aspartate content in the protein. Humans have a number of other proteins (besides crystallin) that are formed only once in a lifetime. Such proteins have been found in tooth enamel, white matter of the brain, aorta, arteries, skin, cartilage, bones, and tendons. Elastin is also a protein of this type [9].
Asparagine and glutamine deamidation in proteins can also be spontaneous. The rate of this process has been shown to depend on the protein conformation. It seems quite interesting that the rate of spontaneous deamidation varies from several hours to hundreds of years. N. I. and A. B. Robinson, who published a number of articles [10-12] and a special book [11a] devoted to the description of this phenomenon believe some “molecular clock” measuring the time of the protein’s life to be hidden in its amino acid sequence.
The process of accumulation of so-called AGE (Advanced Glycation End products) is another candidate for the role of the key component of the “master biological clock”. AGE embrace a group of diverse compounds that are formed in an organism as a result of complex chemical reactions, glycation of proteins and peptides being the first stage of this process (Fig. 1). These chemical reactions proceed slowly but inevitably in any aqueous solution containing proteins and reducing sugars. They result in the formation of characteristic colored products as well as covalent, enzymatically uncleavable cross-links in proteins.
Fig. 1. Chemical formulas of the most common AGE [13, 14].
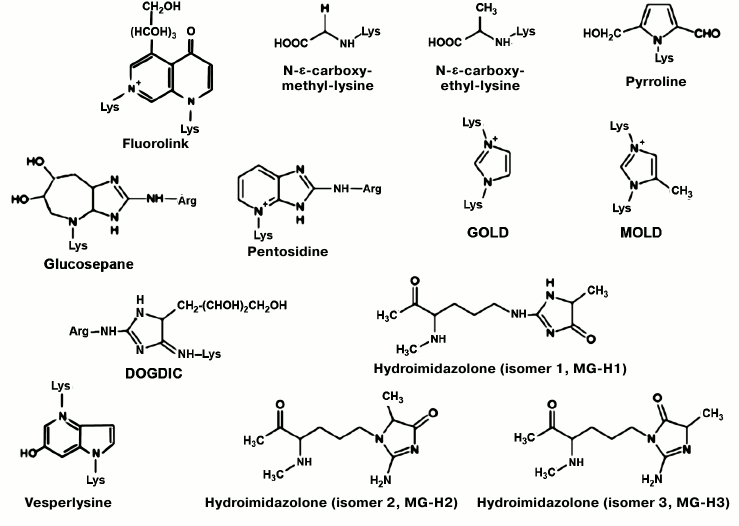
CHEMISTRY OF AGE PRODUCTION
In 1912, Louis Camille Maillard reported that reducing sugars react with amino acids in solution producing dark-colored products (melanoidins) [15]. Similar chemical reactions could be observed also in solutions of reducing sugars mixed with peptides and proteins. The “Maillard reaction”, being a redox process, is a complex network of successive and parallel reactions.
John E. Hodge [16] contributed greatly to the understanding of the chemistry of the Maillard reaction. He showed that the sugar aldehyde group reacts with amino groups producing N-glycosides at the initial stage of glycation (non-enzymatic glycosylation). Then N-glycosides undergo Amadori rearrangement, turning into 1-amino-1-deoxy-ketoses (Amadori compounds). These reactions are followed by further chemical transformations resulting in the formation of AGE [16].
Dicarbonyl products (glyoxal and methylglyoxal), formed as intermediate products in the course of the Maillard reaction, are of great importance. These highly active compounds, which are also formed in the cell as glycolysis byproducts, can react with proteins to form enzymatically uncleavable cross-links.
Comparison of various amino acid residues in peptides according to their ability to react with reducing sugars revealed that side chains of cysteine, lysine, and histidine, as well as the amino group of the N-terminal amino acid, have the highest relative activity. In the case of peptides reacting with protein AGE-products, the highest efficiency of cross-link formation was observed for arginine and tryptophan [17].
ROLE OF AGE PRODUCTS IN PATHOLOGY AND AGING
Just as increased concentration of reactive oxygen species (ROS) causes oxidative stress, increased concentration of sugars (glucose, fructose, deoxyglucose, and triose phosphates) and active dicarbonyl compounds (glyoxal and methylglyoxal) can cause “carbonyl stress” resulting in the increased rate of formation of AGE products, including cross links in proteins, which violate their structures and functions.
In the early 1980s, after AGE had been found to accumulate with age in certain tissues of living organisms, a theory of “non-enzymatic glycosylation as the cause of aging” was proposed [18].
Harmful effects of glycation were initially assumed to be associated with damage to long-lived proteins. Indeed, clear age-dependent increase in AGE concentration is observed in those body parts where proteins are not renewed or are renewed very slowly (e.g. in skin and cartilage collagen [19-21], in proteins of the lens [22, 23], and in some other tissues [24, 25]). In experiments on rats, aminoguanidine, an inhibitor of AGE formation, was shown to reduce manifestation of such signs of aging as decrease in elasticity of blood vessels and increase in heart size [26]. Furthermore, an important role of AGE was demonstrated in the development of atherosclerosis, cataract, diabetic nephropathy, retinopathy, neuropathy, and age-dependent vascular complications [13].
Further research showed a significant proportion of the harmful effects of AGE accumulation to be associated with certain signals produced by RAGE (Receptor for Advanced Glycation End products) – a specific AGE receptor, as well as reactive oxygen species inducer. Thus, although the kinetics of glycation may be similar to kinetics of, for example, isomerization of amino acids in proteins (AGE level in elastin of the intervertebral discs increases with age in approximately the same proportion as the amount of D-aspartate [9]), the presence of AGE receptors makes them a far more likely candidate for the role of the “biological clock”.
AGE RECEPTORS
RAGE is to be the most thoroughly-studied AGE receptor. It is a multi-ligand membrane receptor of the immunoglobulin superfamily present in many cell types [27, 28]. In addition to RAGE, several other receptors binding AGE products have been discovered, including phagocytic receptors of macrophages (of the first and second types), oligosaccharyl transferase-48 (AGE-R1), phosphoprotein 80K-H (AGE-R2), and galectin-3 (AGE-R3) [29].
AGE receptors were originally assumed to bind and neutralize AGE. However, further studies showed AGE binding to RAGE receptor of macrophages and microglial cells triggers oxidative stress and causes activation of the p21ras/MAP-kinase signaling cascade, which in turn activates NF-κB factor [30]. As a result, the interaction of AGE with their receptors causes inflammatory response and oxidative stress. In this regard, the role of AGE as a biological clock is very likely to be realized via inflammatory response, ROS generation, and apoptosis induced by these factors.
For such a clock to control aging, the following components are needed: 1) a protein like crystallin or elastin whose lifetime is comparable to the human lifespan; 2) RAGE specific to the formation of AGE in this protein; 3) an apoptosis inducer triggered by the complex of RAGE and an AGE-containing protein. If these processes take place in tissue producing some primary juvenile hormone, and the ability this tissue for restoration is limited, it should result in the involution of the tissue with age and subsequent aging due to reduction of the level of this hormone in the organism.
This scheme assumes such long time periods as months and years are measured not by a single cell, but by a large group of cells, a special supracellular structure similar to the suprachiasmatic nucleus of the hypothalamus or the epiphysis producing melatonin, the hormone of circadian rhythm. It seems interesting that the level of melatonin, antioxidant and inducer of the entire group of enzymes of the cell antioxidant system, drastically decreases with age. Furthermore, melatonin has a geroprotective effect [31-34]. In other words, there is a possibility that circadian rhythm and general age are measured by the same organ. Reduction of the number of cells producing primary juvenile hormone might result in decrease in its level in blood and other tissues of the organism. We believe this could serve as a signal for increasing the concentration of ROS in mitochondria (mROS). This, in turn, stimulates apoptosis and causes reduction in the number of cells in these tissues, which results in deterioration of the physiological functions of the organism [4, 35, 36] (Fig. 2).
Fig. 2. Hypothetical mechanism of the “master biological clock” controlling ontogenesis in its last stage, the aging of the organism.
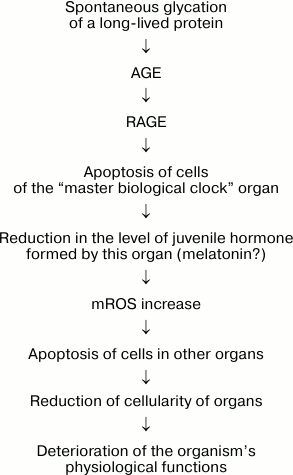
It should be emphasized that the primary juvenile hormone is likely to trigger a hormonal cascade composed of secondary, tertiary, and other juvenile hormones, which multiply the signal of the primary hormone and transmit this signal to other organs and tissues. Very recently, a mechanism controlling aging by one such secondary juvenile hormone, namely, gonadotropin-releasing hormone (GRH), has been discovered. In the group of the American cell biologist D. Cai [37], aging was shown to be accompanied by an increase in the number of microglial cells (playing the role of phagocytes in brain) of one of the sections of hypothalamus and by activation of transcription factor NF-κB in these cells. NF-κB causes microglia to produce tumor necrosis factor (TNF), which attacks neighboring neurons responsible for GRH synthesis. Neurons possess their own NF-κB activated by TNF. This activation in turn stimulates methylation of the GRH gene promotor, which blocks GRH synthesis by neurons. Without GRH, the hypophysis does not produce a tertiary juvenile hormone (gonadotropin) that required for the synthesis of sex hormones and a number of systems operating in a young organism and weakening with age. By blocking the above-described regulatory chain, the authors managed to prolong the life of mice and inhibit the development of such signs of aging as sarcopenia, osteoporosis, skin thinning, appearance of cross links in tendon tissue, and memory loss. Partial inhibition of the development of these symptoms could be achieved by subcutaneous administration of GRH to old mice. Within the scheme (Fig. 2), the events described by Cai et al. take place between the primary juvenile hormone and mROS in the cells of organs and tissues. It is noteworthy that neurons forming the secondary juvenile hormone GRH are localized in the hypothalamus, i.e. the same place as the suprachiasmatic nucleus with its “clock” of circadian rhythm.
Considering the scheme in Fig. 2, we can ask a question: which sugar is the source of AGE if these compounds are actually used for measuring age? Glucose seems to be a good candidate for this role as its concentration in blood is maintained mainly at the same level during the day (increases in its level following the consumption of food rich in glucose are relatively short-lived). However, among all the natural monosaccharides, glucose is characterized by equilibrium of aldehyde and cyclic isoforms, being maximally shifted in the direction of the cyclic isoform (only 0.2% of glucose is in the form of aldehyde capable of being an AGE precursor and, therefore, of participating in glycation of proteins). Thus, glucose is one of the least active sugars in relation to glycation. It seems very likely that glucose became the main “carbohydrate energy carrier” during evolution exactly due to this property [13]. On the other hand, the ability of the above-mentioned byproduct of glucose metabolism, methylglyoxal, to form AGE products exceeds the reactive ability of glucose 650-fold (in the reaction with β-alanine at 80oC) [38].
In the case of galactose, the amount of aldehyde form many times exceeds that of glucose [39]. Glycation of crystallins and serum albumin at the expense of galactose is much faster than that at the expense of glucose and fructose [40, 41]. Moreover, addition of galactose to the diet was shown to cause typical progeria (premature aging) [42-44], in which the mitochondrial path of apoptosis, when cytochrome c is released from mitochondria, plays an important role [45]. Development is progeria inhibited by salidroside, an inhibitor of RAGE-type receptors [46]. Metformin, inhibiting AGE formation from monosaccharides [47], is also known to be a geroprotector [48].
RAGE can be found in many cell types, in particular, in proinflammatory ones [27, 28]. According to in vitro data, not only AGE, but also a number of marker molecules and activators of inflammatory processes can be RAGE ligands: DNA and RNA, calcium-binding proteins of the S100 family, prions, and the non-histone chromosomal protein HMGB1, which together with RAGE play an important role in inflammatory (including septic and autoimmune) reactions and cancers [28, 49, 50]. In addition, some RAGE bind β-amyloid, a protein playing the key role in Alzheimer’s disease [27-29]. Also, RAGE knockout was shown to promote survival of animals with diabetes [51] or bacterial infection [28]. That would suggest diabetes (violation of sugar metabolism), inflammatory diseases, and Alzheimer’s disease to speed up the biological clock.
Finally, having accepted the hypothesis of the “master biological clock”, we can put forward the question of the scope of its application. It is natural to assume that such a clock might be used not only in aging, but also in the earlier stages of ontogenesis. This assumption is consistent with the data on the participation of RAGE in differentiation of neural tissue [52].
Thus, results of many experiments in the fields of gerontology, developmental biology, and medicine can be proposed using the hypothesis of an AGE-based “biological clock”. Therefore, there are good reasons to believe that this hypothesis will be tested in the near future.
Note added in proof: After this manuscript was accepted for publication, the paper of C. Menni et al. (C. Menni et al. (2013) Int. J. Epidemiol., July 8, doi: 10.1093/ije/dyt094) appeared. The authors showed that blood level of one of the protein glycation products, i.e. C-glycosyltryptophan, correlates with age and such age-related parameters as lung functional state and bone mineral density. The investigation was carried out on humans (6055 persons).
REFERENCES
1.Skulachev, V. P. (1997) Biochemistry
(Moscow), 62, 1191-1195.
2.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
3.Longo, V. D., Mitteldorf, J., and Skulachev, V. P.
(2005) Nat. Rev. Gen., 6, 866-872.
4.Skulachev, V. P. (2012) Biochemistry
(Moscow), 77, 689-706.
5.Dilman, V. M. (1978) Mech. Ageing. Dev.,
8, 153-173.
6.Dilman, V. M. (1982) Master Biological Clock
[Russian translation], Znanie, Moscow.
7.Comfort, A. (1979) The Biology of
Senescence, 3rd Edn., Elsevier, New York.
8.George, J. C., Bada, J., Zeh, J., Scott, L., Brown,
S. E., O’Hara, T., and Suydam, R. (1999) Can. J. Zool.,
77, 571-580.
9.Sivan, S. S., Van El, B., Merkher, Y., Schmelzer,
C. E., Zuurmond, A. M., Heinz, A., Wachtel, E., Varga, P. P., Lazary,
A., Brayda-Bruno, M., and Maroudas, A. (2012) Biochim. Biophys.
Acta, 1820, 1671-1677.
10.Robinson, A. B., and Robinson, N. E. (1991)
Proc. Natl. Acad. Sci. USA, 88, 8880-8884.
11.Robinson, N. E., and Robinson, A. B. (2004)
Mech. Ageing. Dev., 125, 259-267.
11a.Robinson, N. E., and Robinson, A. B. (2004) Molecular Clocks:
Deamination of Asparaginyl and Glutaminyl Residues in Peptides and
Proteins, Althouse Press, Cave Junction, Oregon (http://book.deamidation.org/MolecularClocks.pdf).
12.Robinson, N. E., and Robinson, A. B. (2001)
Proc. Natl. Acad. Sci. USA, 98, 4367-4372.
13.Krautwald, M., and Munch, G. (2010) Exp.
Gerontol., 45, 744-751.
14.Biemel, K. M., Friedl, D. A., and Lederer, M. O.
(2002) J. Biol. Chem., 277, 24907-24915.
15.Maillard, L. (1912) C. R. Hebd. Seances Acad.
Sci., 154, 66-68.
16.Hodge, J. E. (1953) J. Agr. Food Chem.,
1, 928-943.
17.Munch, G., Schicktanz, D., Behme, A., Gerlach,
M., Riederer, P., Palm, D., and Schinzel, R. (1999) Nat.
Biotechnol., 17, 1006-1010.
18.Monnier, V. M., Stevens, V. J., and Cerami, A.
(1981) Prog. Food Nutr. Sci., 5, 315-327.
19.Dyer, D. G., Dunn, J. A., Thorpe, S. R., Bailie,
K. E., Lyons, T. J., McCance, D. R., and Baynes, J. W. (1993) J.
Clin. Invest., 91, 2463-2469.
20.Sell, D. R., Kleinman, N. R., and Monnier, V. M.
(2000) FASEB J., 14, 145-156.
21.DeGroot, J., Verzijl, N., Wenting-Van Wijk, M.
J., Bank, R. A., Lafeber, F. P., Bijlsma, J. W., and TeKoppele, J. M.
(2001) Arthritis Rheum., 44, 2562-2571.
22.Araki, N., Ueno, N., Chakrabarti, B., Morino, Y.,
and Horiuchi, S. (1992) J. Biol. Chem., 267,
10211-10214.
23.Frye, E. B., Degenhardt, T. P., Thorpe, S. R.,
and Baynes, J. W. (1998) J. Biol. Chem., 273,
18714-18719.
24.Schleicher, E. D., Wagner, E., and Nerlich, A. G.
(1997) J. Clin. Invest., 99, 457-468.
25.Simm, A., Wagner, J., Gursinsky, T., Nass, N.,
Friedrich, I., Schinzel, R., Czeslik, E., Silber, R. E., and Scheubel,
R. J. (2007) Exp. Gerontol., 42, 668-675.
26.Corman, B., Duriez, M., Poitevin, P., Heudes, D.,
Bruneval, P., Tedgui, A., and Levy, B. I. (1998) Proc. Natl. Acad.
Sci. USA, 95, 1301-1306.
27.Basta, G. (2008) Atherosclerosis,
196, 9-21.
28.Ramsgaard, L., Englert, J. M., Manni, M. L.,
Milutinovic, P. S., Gefter, J., Tobolewski, J., Crum, L., Coudriet, G.
M., Piganelli, J., Zamora, R., Vodovotz, Y., Enghild, J. J., and Oury,
T. D. (2011) PLoS One, 6, e20132.
29.Singh, R., Barden, A., Mori, T., and Beilin, L.
(2001) Diabetologia, 44, 129-146.
30.Lander, H. M., Tauras, J. M., Ogiste, J. S.,
Hori, O., Moss, R. A., and Schmidt, A. M. (1997) J. Biol.
Chem., 272, 17810-17814.
31.Anisimov, V. N. (2003) Molecular and
Physiological Mechanisms of Aging [in Russian], 1st Edn., Nauka,
St. Petersburg.
32.Anisimov, V. N., Popovich, I. G., Zabezhinski, M.
A., Anisimov, S. V., Vesnushkin, G. M., and Vinogradova, I. A. (2006)
Biochim. Biophys. Acta, 1757, 573-589.
33.Karasek, M. (2007) J. Physiol. Pharmacol.,
58, 105-113.
34.Pierpaoli, W., and Bulian, D. (2005) Ann. N.
Y. Acad. Sci., 1057, 133-144.
35.Skulachev, V. P., Severin, F. F., and Feniouk, B.
A. (2013) Life without Aging [in Russian], EKSMO, Moscow (in
print).
36.Severin, F. F., and Skulachev, V. P. (2009)
Uspekhi Gerontol., 22, 37-48.
37.Zhang, G., Li, J., Purkayastha, S., Tang, Y.,
Zhang, H., Yin, Y., Li, B., Liu, G., and Cai, D. (2013) Nature,
497, 211-216.
38.Namiki, M. (1988) Adv. Food Res.,
32, 115-184.
39.Dworkin, J. P., and Miller, S. L. (2000)
Carbohydrate Res., 329, 359-365.
40.Swamy, M. S., Tsai, C., Abraham, A., and Abraham,
E. C. (1993) Exp. Eye Res., 56, 177-185.
41.Ledesma-Osuna, A. I., Ramos-Clamont, G., and
Vazquez-Moreno, L. (2008) Acta Biochim. Polon.,
55, 491-497.
42.Song, X., Bao, M. M., Li, D. D., and Li, Y. M.
(1999) Mech. Ageing Dev., 108, 239-251.
43.Liang, Y. X., Wang, Z., Li, D. D., Jiang, J. M.,
and Shao, R. G. (2003) Biomed. Environ. Sci., 16,
323-332.
44.Deng, H. B., Cheng, C. L., Cui, D. P., Li, D. D.,
Cui, L., and Cai, N. S. (2006) Biomed. Environ. Sci., 19,
432-438.
45.Lu, J., Wu, D. M., Zheng, Y. L., Hu, B., and
Zhang, Z. F. (2010) Brain Pathol., 20, 598-612.
46.Mao, G. X., Deng, H. B., Yuan, L. G., Li, D. D.,
Li, Y. Y., and Wang, Z. (2010) Biomed. Environ. Sci., 23,
161-166.
47.Bujak-Gizycka, B., Suski, M., Olszanecki, R.,
Madej, J., and Korbut, R. (2009) Folia Medica Cracoviensia,
50, 21-33 (Polish).
48.Anisimov, V. N. (2008) Molecular and
Physiological Mechanisms of Aging [in Russian], 2nd Edn., Nauka,
St. Petersburg.
49.Stern, D., Yan, S. D., Yan, S. F., and Schmidt,
A. M. (2002) Adv. Drug. Deliv. Rev., 54, 1615-1625.
50.Sims, G. P., Rowe, D. C., Rietdijk, S. T.,
Herbst, R., and Coyle, A. J. (2010) Annu. Rev. Immunol.,
28, 367-388.
51.Nielsen, J. M., Kristiansen, S. B., Norregaard,
R., Andersen, C. L., Denner, L., Nielsen, T. T., Flyvbjerg, A., and
Botker, H. E. (2009) Eur. J. Heart Fail, 11, 638-647.
52.Kim, J., Wan, C. K., O’Carroll, S. J.,
Shaikh, S. B., and Nicholson, L. F. B. (2012) J. Neurosci. Res.,
90, 1136-1147.