Directed Evolution and Characterization of Atrazine Chlorohydrolase Variants with Enhanced Activity
Y. Wang, X. Li, X. Chen*, and D. Chen*
Laboratory of Molecular Genetics, College of Life Sciences, Nankai University, 300071 Tianjin, China; fax: +86-22-2350-0133; E-mail: xiwenchen@nankai.edu.cn; chendefu@nankai.edu.cn* To whom correspondence should be addressed.
Received March 7, 2013; Revision received May 1, 2013
Atrazine chlorohydrolase (AtzA, EC 3.8.1.8) has attracted widespread interests as it catalyzes conversion of toxic atrazine to nontoxic hydroxyatrazine and can be used in the biodegradation of atrazine. To facilitate this application, a Haematococcus pluvialis-based method was applied to screen AtzA variants from a random mutagenesis library. Eight variants with enhanced enzyme activity were obtained. They showed 2.7- to 5.0-fold increase in specific activity compared with the wild type. Sequencing revealed that the two most active variants contained single substitution at Val12 and Leu395, respectively, while several improved variants contained substitutions at the four sites of Met315, His399, Asn429, and Val466 simultaneously, indicating that these residues contribute to the enzyme activity of AtzA. Kinetic analysis showed that five variants decreased the Km value 0.6- to 0.9-fold, whereas all the variants increased the catalytic efficiency (kcat/Km value) 2.5- to 4.1-fold compared to the wild type. The modeled three-dimensional structure showed that AtzA is comprised of a typical (β/α)8 domain of the amidohydrolase superfamily and a dual β-sheet domain. An iron ion and five ligand-binding residues are located in the β-barrel core of the (β/α)8 domain. Some substituted residues are involved in hydrogen bond formation in the (β/α)8-neighboring β-sheet.
KEY WORDS: atrazine chlorohydrolase (AtzA), DNA shuffling, error-prone PCR (EP-PCR), homology modeling, kinetic parameterDOI: 10.1134/S0006297913100040
Abbreviations: AtzA, atrazine chlorohydrolase; BBM, Bold’s basal medium; EP-PCR, error-prone PCR.
Atrazine (2-chloro-4-ethylamino-6-isopropylamino-1,3,5-triazine) is a
highly effective pre- and post-emergence triazine herbicide that was
widely used for the control of broadleaf weed species [1]. Because of its widespread usage, atrazine has been
found in both groundwater and surface soil and commonly exceeds the
health advisory level of 4.6 μM [2]. The
existence of atrazine in the environment has the potential to affect
aquatic organisms, terrestrial plants, and their ecosystems [3]. However, the degradation of atrazine is relatively
slow in the environment with an average half-life ranging from 4 to 57
weeks [4]. Therefore, removal of atrazine from a
contaminated environment becomes an urgent problem. Increasingly,
researches are now concentrated on the issue of biodegradation of this
toxic compound [5, 6].
A system of six enzymes from Pseudomonas sp. strain ADP that allow catabolism of the triazine herbicides as sources of nitrogen was elucidated in the early 1990s [7]. Atrazine chlorohydrolase (AtzA), which catalyzes the hydrolytic dechlorination of atrazine to hydroxyatrazine, is the first enzyme in the pathway leading to the mineralization and detoxification of atrazine in bacteria [8]. It belongs to a Fe(II)-dependent amidohydrolase superfamily [9] that has the typical β-barrel core within a (β/α)8 structural domain [10]. Functional AtzA has a subunit molecular weight of 60 kDa and was shown by gel filtration to consist of a holoenzyme with molecular mass of 245 kDa [8]. The gene encoding the enzyme was cloned from the bacteria and has been transformed into bacteria or plants with the purpose of bioremediation. An atrazine-mineralizing strain, Chelatobacter heintzii Cit1, was inoculated in soils and biodegraded atrazine successfully [11]. Overexpression of atzA in recombinant Escherichia coli could remediate atrazine-contaminated soil on field-scale level [12]. A modified bacterial atzA gene, p-atzA, was also transformed into alfalfa, Arabidopsis thaliana, and tobacco. All these transgenic plant species actively expressed p-atzA, and the isolated enzymes metabolize atrazine efficiently [13]. Recently, atzA genes from Pseudomonas sp. strain ADP and Arthrobacter strain AD1 were transferred into tobacco, respectively. The transgenic tobacco lines germinated and grew well in the presence of atrazine and could degrade atrazine effectively in soil [14].
Catalytic improvement of AtzA through protein engineering would contribute to efforts for atrazine biodegradation. Rational and irrational protein designs are two common strategies in protein engineering. However, the crystal structure of AtzA has not yet been determined and little information on the structure is available for the rational design of AtzA. Directed evolution can be used to improve the activity of enzymes without known structures [15]. This has been proved to be an extremely useful and efficient method to optimize biocatalysts for practical applications in a relatively short period of time [16]. Previously, combinatorial limited site saturation mutagenesis was performed on AtzA based on a putative structural model of the enzyme–substrate complex [17]. DNA shuffling was also performed on AtzA and TriA (melamine deaminase) to see if intermediates between the parental enzymes possess broader substrate specificities [18, 19].
In this study, error-prone PCR (EP-PCR) and DNA shuffling were combined to construct a mutagenesis library of atzA. The mutagenesis library was then transformed into Haematococcus pluvialis for screening AtzA variants. Eight variants with enhanced enzyme activity were obtained. Characterization of these improved variants and the modeled structure contribute to understanding of the structure–function relationship of AtzA.
MATERIALS AND METHODS
Strains, plasmids, culture media, and growth condition. Plasmid pMD4 containing atzA from Pseudomonas sp. ADP (accession number U55933) was kindly provided by Prof. Michael J. Sadowsky of the University of Minnesota, USA. Haematococcus pluvialis strain H1 was purchased from the Salt Research Institute of China. Escherichia coli strains DH5α-FT and BL21(DE3) and plasmids pCAMBIA-1301 and pET41a(+) were kept in our lab. Escherichia coli strains were cultured on Luria–Bertani (LB) medium at 37°C with antibiotics as needed. Haematococcus pluvialis and its transformants were cultured on Bold’s basal medium (BBM) [20] containing different concentrations of atrazine under a 16/8 h day/night cycle at 25°C with light intensity of 65 μmol photons·m–2·s–1. The cultures were periodically shaken at least once a day during the growth.
Tolerance test of H. pluvialis to atrazine. When the culture of H. pluvialis grew to logarithmic phase (1·106 cells/ml), it was collected by centrifugation at 2604g for 5 min. Fifty microliters of the alga was transferred into a 24-well tissue culture plate. Each well contained 2 ml liquid BBM medium with 0, 0.05, 0.1, 0.2, 0.5, or 1.0 mg/liter atrazine. After a 3-week incubation, the growth state was observed.
Construction of mutagenesis library and screening for improved variants. EP-PCR was conducted according to previous research [21] with minor modification and was carried out in 25 μl reaction mixture containing 1× PCR buffer, 7.5 mM MgCl2, 0.2 mM of each dATP and dGTP, 1.0 mM of each dTTP and dCTP, 0.3 μM of primer pairs of atzA-F (5′-CCGAGCTCATGCAAACGCTCAGCATCC-3′, SacI restriction site underlined) and atzA-R (5′-AATCTAGACTAGAGGCTGCGCCAAGC-3′, XbaI restriction site underlined), 0.01 ng/μl pMD4 DNA, 0.01% Triton X-100, 1.0 μM NaNO2 or 37.5 μM CoCl2 as mutagen, and 0.05 U/μl rTaq DNA polymerase (Takara Biotechnology, China): 94°C for 7 min, 50 cycles of 94°C for 40 s, 58°C for 32 s, 72°C for 110 s and a final extension at 72°C for 7 min. EP-PCR products induced by these two mutagens were mixed equimolarly for DNA shuffling according to Stemmer [22]. Approximately 3 μg of mixed EP-PCR products were digested with DNase I for 20 min at 15°C, and fragments of between 50- and 200-bp were then excised from 1% agarose gel. The fragment at concentration of 40 ng/μl was reassembled in a PCR reaction using rTaq DNA polymerase without primers. The reassembly PCR reaction was conducted at 94°C for 5 min followed by 25 cycles of 94°C for 40 s, 58°C for 32 s, 72°C for 110 s, and a final extension at 72°C for 7 min. Fragments of approximately 1.4 kb were excised from 0.7% agarose gel to become the template of the next round PCR. Finally, PCR with primers was conducted using 40 ng/μl reassembled fragments as template. The procedure was 94°C for 7 min, 30 cycles of 94°C for 40 s, 58°C for 32 s, 72°C for 110 s, and a final extension at 72°C for 7 min. Final mutated products were purified, ligated with pCAMBIA-13, and then transformed into DH5α-FT for library construction. Approximately 1.5·105 colonies were collected. Plasmids were then extracted and transformed into H. pluvialis by electroporation according to a published protocol [23] with minor modification. Briefly, 1 μl plasmid containing 0.4 μg p1301-atzA was mixed with competent cells of H. pluvialis. The mixture was placed into a disposable electroporation cuvette with 0.1 cm gap (Bio-Rad, USA). Then three consecutive pulses with field strength of 6 kV/cm were delivered at intervals of 10 s. The transformed algal cells were first cultivated in liquid BBM medium for 24 h for rehabilitation and then cultivated on solid BBM medium with 0.5 mg/liter atrazine for screening. After single colonies of the alga appeared on plates, they were separated into 10-ml flasks which contained 2 ml liquid BBM medium with 0.5 mg/liter atrazine. When growing into logarithmic phase (1·106 cells/ml), 100 μl of the alga were transferred into 50-ml Erlenmeyer flasks containing 10 ml BBM medium with 1.0 mg/liter atrazine. This step was repeated when the alga grew into logarithmic phase again with atrazine concentration increasing to 0.5 and 1.0 mg/liter sequentially. The algal lines that grew quickly in the process were selected. Their genomic DNA was extracted by the hexadecyltrimethylammonium bromide (CTAB) method, and the mutated genes were amplified using primer pairs of atzA-F and atzA-R and then ligated with pMD19-T Simple vector (Takara Biotechnology) for sequencing.
Expression and purification of mutated AtzA in E. coli. After sequencing confirmation, the mutated atzA genes were restricted with SacI/NotI from the respective pMD19-T-atzA and ligated with pET41a(+) that was also restricted with the same endonucleases. The resulting plasmids were then transformed into BL21(DE3). A single colony was incubated in LB medium containing 50 μg/ml kanamycin at 37°C. When A600 reached 0.8, isopropyl-β-D-thiogalactopyranoside (IPTG) was added into the medium to final concentration of 1 mM, and the cells were cultivated for a further 10 h. The cells were then harvested and lysed by ultrasonication. Protein from the precipitate and the supernatant, representing insoluble and soluble fractions, were analyzed by SDS-PAGE in 12.5% polyacrylamide gel.
Inclusion bodies were separated from 50 ml cell cultures and washed extensively with 10 ml of buffer A (9.5 mM NaH2PO4, 40.5 mM Na2HPO4, 0.5 M NaCl, 1 M urea, pH 7.4), and incubated for 30 min at room temperature. The lysate was centrifuged for 15 min at 12,000g, and the precipitate was resuspended in buffer B (9.5 mM NaH2PO4, 40.5 mM Na2HPO4, 0.5 M NaCl, 2% Triton X-100, and 20 mM EDTA, pH 7.4). After incubation at room temperature for another 30 min and centrifugation for 15 min at 12,000g, the precipitate was resuspended in solubilization buffer (9.5 mM NaH2PO4, 40.5 mM Na2HPO4, 0.5 M NaCl, 5 mM imidazole, 8 M urea, pH 7.4) until fully dissolved. The dissolved solution was stirred for at least 6 h at room temperature and filtered through a 0.45 μm pore membrane. The filtered lysate was incubated with 2 ml nickel-nitrilotriacetic acid (Ni-NTA) agarose FF (Weishibohui Science and Technology Co., Ltd., China) and equilibrated in solubilization buffer for 30 min at 20°C. Then the column was washed with urea-containing buffers (9.5 mM NaH2PO4, 40.5 mM Na2HPO4, 0.5 M NaCl and 8, 6, 4 or 2 M urea, pH 7.4) at 20°C. The Ni-NTA column was washed with buffer C (9.5 mM NaH2PO4, 40.5 mM Na2HPO4, 0.5 M NaCl, 40 mM imidazole, pH 7.4) to elute other proteins. Finally, the refolded AtzA was eluted from the Ni-NTA by using buffer D (9.5 mM NaH2PO4, 40.5 mM Na2HPO4, 0.5 M NaCl, 0.4 M imidazole, pH 7.4) at 20°C. The concentration of refolded protein was measured by the Bradford method [24]. The enzymes were incubated with 0.2 mM FeSO4 for 30 min at 25°C before activity assays.
Specific activity and kinetics of the variants. AtzA activity was assayed based on its ability to convert atrazine into hydroxyatrazine, and initial rates of atrazine disappearance and hydroxyatrazine formation were estimated by high-performance liquid chromatography (HPLC). Briefly, activity assays were performed in 0.4 ml 100 mM Tris-HCl buffer (pH 7.2) with approximately 20 µg/ml enzyme and 46 μM atrazine. The enzyme kinetic values were determined in the same reaction system as described above, with atrazine concentration ranging from 4.6 to 140 μM and enzyme concentrations of approximately 10 µg/ml. The reaction mixture was incubated at 25°C for 10 min. After the incubation, residual atrazine was extracted by 0.4 ml dichloromethane, and the samples were centrifuged at 18,514g for 10 min. The supernatant was filtered through a 0.2-μm filter and then subjected to CoM 6000 HPLC system (CoMetro Technology, USA) analysis on an analytical C18 (5 μm, 250 × 4.6 mm) reverse-phase column at 30°C with linear gradients as follows: 0 to 6 min, 10 to 25% acetonitrile; 6 to 21 min, 25 to 65% acetonitrile; 21 to 23 min, 65 to 100% acetonitrile; and 23 to 25 min, 100% acetonitrile [8] with monitoring at wavelength 228 nm. The activity unit was defined as the amount of enzyme that converts 1 µmol of atrazine into hydroxyatrazine per minute at room temperature. The enzyme kinetic parameters were determined using Lineweaver–Burk plots. Four independent experiments were conducted.
Data analysis. The data were analyzed with SPSS 11.0 software (SPSS Inc., USA). The t-test for independent samples was performed to assess the differences between wild-type AtzA and the variants. Results are given as means with 95% confidence intervals.
Homology modeling of AtzA. The amino acid sequence of wild-type AtzA was submitted to the Phyre2 server (http://www.sbg.bio.ic.ac.uk/phyre2) for homology modeling in the intensive mode [25], which is based on a profile–profile alignment algorithm. Six templates were selected based on heuristics to maximize confidence, percentage identity, and alignment coverage. These proteins, their PDB code, and sequence identity with AtzA were described as following: amidohydrolase from an environmental sample of Sargasso sea (3hpa, 30%), adenosine deaminase from Xanthomonas campestris (4dzh, 28%), adenosine deaminase homolog from Chromobacterium violaceum (4f0r, 25%), adenosine deaminase from Pseudomonas aeruginosa (4dyk, 25%), atrazine chlorohydrolase TrzN from Arthrobacter aurescens (3lsb, 23%), and amidohydrolase family protein from Oleispira antarctica (3lnp, 22%).
The AtzA amino acid sequence was submitted to the 3DLigandSite server (http://www.sbg.bio.ic.ac.uk/3dligandsite/) [26] for potential ligand position and binding site prediction based on superimposition of homology models. The conserved and active sites of AtzA were analyzed using the NCBI Conserved Domains Search (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) through multiple sequences alignment.
RESULTS
Library construction and high-throughput screening. Haematococcus pluvialis is a unicellular freshwater species of Chlorophyta from the family Haematococcaceae. When the algal cells were cultured in a medium containing 0.1 mg/liter or higher atrazine, their growth was completely inhibited (data not shown). Thus, 0.1 mg/liter atrazine was considered as the lethal concentration inhibiting the algal cell growth.
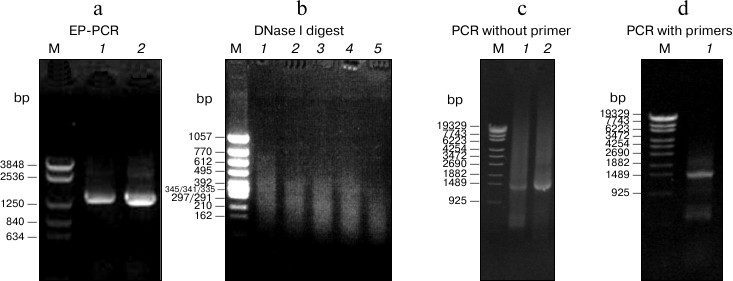
To obtain beneficial mutations, EP-PCR and DNA shuffling were combined to generate a random mutagenesis library of atzA. Two mutagens, NaNO2 and CoCl2, were used in EP-PCR (Fig. 1a). In the DNA shuffling procedure, the digestion condition of DNase I was determined as 20 min at 15°C, and 50-200-bp digested products were collected for reassembly (Fig. 1b). The concentrations of digested fragments and reassembled fragments in primerless PCR and conventional PCR were both confirmed as 40 ng/μl (Fig. 1, c and d). The library contained over 1.5·105 colonies. It was then introduced into H. pluvialis and screened on BBM plates with 0.5 mg/liter atrazine. The transformants that contained improved enzymes grew vigorously and developed into a single colony, whereas the ones with no or unprofitable mutation died. Visible single algal colonies were observed after 3-week growth on 5-fold lethal concentration of atrazine (0.5 mg/liter) on plates. With several rounds of repeated screening in liquid medium, 15 variants were selected for further sequencing.
Fig. 1. Mutagenesis library construction by EP-PCR and DNA shuffling. a) Electrophoresis profile of atzA after EP-PCR. Lanes: 1) EP-PCR product induced by CoCl2; 2) EP-PCR product induced by NaNO2. b) Electrophoresis profile of mixed EP-PCR products digested with DNase I at 15°C. Lanes 1-5) DNase I digestion for 4, 8, 12, 16, or 20 min, respectively. Approximately 50-200-bp random fragments after 20 min digestion (lane 5) were selected as template for reassembly. c) Reassembly profile of PCR without primers. Lanes: 1, 2) reassembly product using 20 or 40 ng/µl digested fragment as template, respectively. The fragment concentration 40 ng/µl was selected as the optimal condition. d) Amplification profile of PCR with primers using 40 ng/µl reassembled fragments as template.
Sequence analysis of eight variants and their specific activities. Sequencing results revealed that eight different variants were obtained, while the other seven were repeated variants. Their host algae lines grew vigorously in medium containing 0.5 mg/liter atrazine, in which wild type was completely inhibited (Fig. 2; see color insert). The amino acid sequence information is shown in the table. Among these variants, there were two single-site substitution variants (10-7, 21-1), one dual-site substitution variant (22-4), and two triple-site substitution variants (16-1, 15-1). Besides, there were 4, 6, and 7 substitutions in 7-10, 48-6, and 32-2, respectively. Variants 10-7, 16-1, 15-1, 22-4, and 21-1 had completely different substitutions without overlapping at any site, whereas 7-10, 48-6, and 32-2 had four common substitutions (M315I, H399Q, N429S, V466A). The occurrence of the four substitutions in the improved variants indicated that these substitution sites are probably important for AtzA activity. Compared with 7-10, 48-6 had two additional (D30G, R389S) substitutions, while 32-2 had three additional (D30G, Q71R, R389S) substitutions, with only one addition (Q71R) over 48-6.
Fig. 2. Screening for AtzA variants with enhanced activity in H. pluvialis. Growth status of H. pluvialis transformants harboring wild-type and improved AtzA enzymes cultivated in liquid medium with 1 mg/liter atrazine for 5 weeks.

Mutation sites, specific activity, and kinetic parameters of the
variants relative to wild type
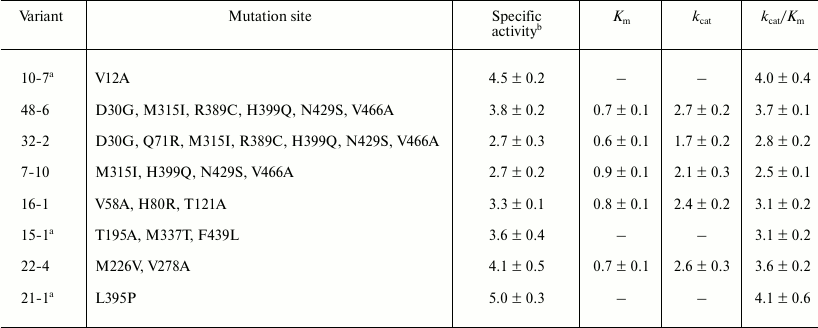
Note: The specific activity, Km,
kcat, and kcat/Km
of the wild-type AtzA-AD1 were set as 1 and they were 2.6 ± 0.1
U/mg, 146 ± 10 µM, 1.6 ± 0.1
s–1, and 1.1·104 ±
0.1·104
s–1·M–1, respectively. The
data are expressed as mean ± SE (n = 4).
a As the Km value of this variant exceeded
the maximum solubility of atrazine in water (153 µM), its
precise Km and kcat values could
not be estimated.
b The specific activity was determined with 46 μM
atrazine.
The recombinant proteins of the wild-type and mutated enzymes were purified using Ni2+-affinity columns. As shown in the table, all variants showed significant specific activity improvement over the wild type, as expected. Of them, six variants (21-1, 10-7, 22-4, 48-6, 15-1, 16-1) had more than 3-fold improvement relative to the wild type. The two single-site substitution variants, 21-1 and 10-7, exhibited the most significant improvement, indicating that these amino acid substitutions (L395P, V12A) are important for AtzA activity. Two amino acid substitutions (M226V, V278A) in 22-4 contributed to 4.1-fold increase relative to the wild type. Three substitutions (T195A, M337T, F439L) in 15-1 enhanced the activity 3.6-fold. Four substitutions (M315I, H399Q, N429S, V466A) in 7-10 improved its specific activity 2.7-fold. Interestingly, compared with 7-10, two extra substitutions (D30G, R389C) in 48-6 increased the specific activity from 2.7- to 3.8-fold relative to wild type. However, compared with 48-6, one additional substitution (Q71R) in 32-2 decreased the activity from 3.8- to 2.7-fold, indicating Q71R has a negative role. Remarkably, these variants (48-6, 32-2, 7-10) carrying the four consensus substitutions (M315I, H399Q, N429S, V466A) exhibited more than 2.7-fold increase in specific activity, strongly suggesting that one or several of these mutations are very important to the dechlorinating activity of AtzA.
Kinetic analysis of variants with enhanced activity. The kinetic parameters of eight variants are also shown in the table. As the Km values of 10-7, 15-1, and 21-1 exceeded the maximum solubility of atrazine in water, their precise kcat and Km values could not be estimated. Variant 32-2 displayed the lowest Km value (0.6-fold of wild type), while 48-6 showed the largest improvement in catalytic activity (2.7-fold of wild type). The most enhanced catalytic efficiency (kcat/Km value) was found in 21-1, which exhibited 4.1-fold of the wild type. Overall, kcat/Km values of these variants increased 2.5- to 4.1-fold relative to the wild type.
As for Km value, 32-2 harboring seven substitutions had the lowest Km value. And 7-10, which had the same four substitutions (M315I, H399Q, N429S, V466A) as 32-2 and 48-6, decreased the Km to 0.9-fold of wild type. Variant 48-6, which had two (D30G, R389C) extra substitutions compared with 7-10, had a considerable reduction in Km value to 0.7-fold of wild type. Variant 32-2, which had one (Q71R) extra substitutions compared with 48-6, further decreased the Km value to 0.6-fold of wild type, indicating that Q71R has a beneficial role in decreasing Km value. Variant 16-1 containing three amino acid substitutions (V58A, H80R, T121A) and 22-4 containing two substitutions (M226V, V278A) also had considerable reduction in Km value. As for kcat value, 7-10 increased the value to 2.1-fold of wild type, whereas variant 48-6, which had extra D30G and R389C compared with 7-10, further increased the value to 2.7-fold. Compared with 48-6, 32-2 had a significantly decreased kcat value, indicating that the additional substitution Q71R in 32-2 reduced the catalytic activity of AtzA significantly. Variants 22-4 and 16-1 harboring two or three substitutions also enhanced catalytic activity to 2.6- and 2.4-fold of that of wild type, respectively. Notably, although the precise kcat and Km values of the two single-site substitution variants (21-1 and 10-7) were not obtained, they showed 4.1- and 4.0-fold kcat/Km value of wild type, indicating that substitution of V12A and L395P had great impact on the catalytic efficiency.
Structure modeling and distribution of substitution sites. The model of AtzA is shown in Fig. 3a (see color insert). Six templates were involved in AtzA modeling construction. Four hundred and fifty residues (95% of total 474 residues) were modeled at >90% accuracy. The alignment coverage accounted for nearly all the sequences of the six templates, and the confidence value between AtzA and each template was 100%. The MAMMOTH-lnE score, which reflects the similarity of modeled structure with its templates, reached as high as 37.7. The upper part of Fig. 3a is a typical (β/α)8 domain of the amidohydrolase superfamily, with an eight β-strands barrel core coated by eight α-helices. The lower part is a dual β-sheet domain, which is comprised of two parallel β-sheets (Fig. 3b). The one near the (β/α)8 domain consists of five β-strands, while the other has four. Hydrogen bond analysis demonstrated that several important residues (Val12, Val58, Leu395) in the (β/α)8-neighbouring β-sheet are involved in hydrogen bonds to stabilize the β-sheet structure (Fig. 3b). Ligand-binding residue analysis revealed that an iron ion is perched in the β-barrel core in the (β/α)8 domain, while five residues (His66, His68, His243, His276, Asp327) surrounding the iron ion were found involved in ligand binding (Fig. 3c). Conserved and active residue analysis showed these five residues, as well as Glu246, are highly conserved.
Fig. 3. Homology modeling of AtzA. a) Overall diagram of the AtzA monomer. The substitution sites observed in this study are displayed as red spheres. The predicted active center and substrate-binding site are enclosed in a cyan dotted rectangle. b) Dual β-sheet domain and hydrogen bond analysis of Val12 and Leu395. The hydrogen bonds are displayed as white dotted lines. c) Close view of AtzA active center. The iron ion is displayed as a blue sphere. The residues involved in ligand or substrate binding, which were predicted in this study, are displayed in magenta or yellow color.
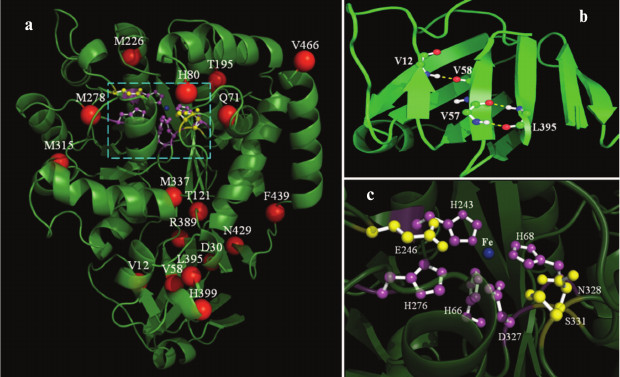
The substituted amino acid residues observed in this study are also displayed in Fig. 3a. They are mainly distributed in two regions. One is the peripheral region of (β/α)8 domain, and the other is the region in and around (β/α)8-neighboring β-sheet. Gln71 (32-2), His80 (16-1), Thr195 (15-1), Met226 (22-4), Val278 (22-4), Met315 (48-6, 32-2, 7-10), Met337 (15-1), and Val466 (48-6, 32-2, 7-10) are located randomly in the former region, while Val12 (10-7), Asp30 (48-6, 32-2), Val58 (16-1), Arg389 (48-6, 32-2), Leu395 (21-1), His399 (48-6, 32-2, 7-10), and Asn429 (48-6, 32-2, 7-10) are in the later one. Besides, Thr121 (16-1) and Phe439 (15-1) are in the loop region that links the (β/α)8 with the dual β-sheet domain.
DISCUSSION
Several researchers have modified AtzA by different strategies previously. Using an AtzA–atrazine complex model, a combinatorial limited site-saturation mutagenesis was performed on five amino acid residues of the putative substrate-binding pocket [17], which is the only research aimed at improving the catalytic efficiency of AtzA so far. In this study, directed evolution was performed to improve the catalytic efficiency of AtzA. As this is a random mutagenesis technology, it is crucial to combine it with a simple and reliable high-throughput screening method. Haematococcus pluvialis was selected as the receptor of a screening system because of its innate advantages. (i) It propagates very quickly with a doubling time of several hours. (ii) Its cultivation method is simple and convenient. Visible single algae will appear on a BBM plate after three weeks. (iii) It is sensitive to atrazine, and the lethal concentration is just 0.1 mg/liter. (iv) It can be easily transformed by electroporation and has high transformation efficiency. Obviously, the H. pluvialis screening system using atrazine as screening pressure is a typical positive high-throughput screening system.
Only eight variants were successfully obtained from the random mutagenesis library in the study. The relatively small number of variants obtained may be due to the conservative feature of AtzA. Although the mutagenesis library was not very large (approximately 1.5·105), it contained abundant mutation types. Before screening, four fifths were single or multiple nucleotide substitution variants, while one fifth were deletion or insertion variants (data not shown). After several rounds of screening, 15 variants with enhanced vitality were obtained. Among them, four variants had the identical amino acid sequence as 48-6, three as 16-1, and two as 10-7 and 22-4, respectively. The presence of duplicate variants indicated the saturation of the screening process and the conservatism of AtzA. The conservative feature of AtzA also accounted for the slowness of AtzA in natural evolution during the evolutionary process [27].
Overexpression of recombinant proteins in E. coli is widely used to produce proteins for laboratory study. However, approximately 70% of recombinant proteins were overexpressed as insoluble inclusion bodies [28]. This was also the case in our study. To obtain soluble active proteins, the inclusion bodies need to be first made soluble in a denaturant, and then this is followed by a refolding process. This procedure has been used for more than 20 years and works quite well for many inclusion body proteins [29]. In a recent study, 76% of 88 proteins were refolded with an average of >75% yield of soluble proteins using a two-step-denaturing and refolding method [28]. In this study, we generated refolded AtzA proteins from inclusion bodies to determine the specific activity and kinetics of the wild type and the variants. Comparing the kcat/Km of wild type with that published previously, the value determined in this study was slightly lower, 1.1·104 vs. 1.4·104 s–1·M–1 [8, 17]. This may be caused by the efficiency of protein refolding and experimental differences. Despite these factors existing, our data still indicated most of the protein refolded into its active form, and determination of enzyme characteristic parameters through the protocol was feasible.
To learn the effect of amino acid residues on AtzA function, the specific activity and kinetic parameters (Km, kcat, kcat/Km) were also determined. Unfortunately, a relatively large number of amino acid substitutions within one variant made it difficult to assess the individual contribution of each substitution to the enzyme activity. However, some substitutions can still be clearly described. Besides V12A (10-7) and L395P (21-1), M226V in dual-site substitution variant 22-4 is also very important to AtzA, as site-directed mutagenesis at Val278 had very limited impact on AtzA activity [30]. Moreover, Q71R had a negative impact on AtzA activity but a positive role in decreased Km value according to the comparison between 48-6 and 32-2. Besides, the high frequency of substitutions at the four sites M315, H399, N429, and V466 in the improved variants also suggested that these residues were involved in enzyme function, separately or interactively. None of the substitution sites except Gln71 [17] observed in our study were characterized previously. Thus, our work provides new clues concerning the structure–function relationship of AtzA.
Although AtzA is known to be a metalloenzyme of the amidohydrolase superfamily [9], its crystal structure has not been solved. In this study, six structure-known proteins having the highest sequence identities with AtzA were selected as templates for homology modeling [31]. Although they have low sequence identity with AtzA (22-30%), the model had a high confidence match (>90% confidence), which qualified for building a correct model [25]. The modeled AtzA had 100% confidence with the respective templates, demonstrating that they had true homology. The considerably higher MAMMOTH-lnE score (37.7) than a cutoff (4.5) [32] also confirmed that the AtzA model is reliable and the ligand-binding site prediction is accurate. Although reliable, inadequacies still existed. For example, during homology modeling, 22 residues (5%) were modeled by an unreliable ab initio method, which may affect the model accuracy to some extent. However, as they are located in N-terminal, C-terminal, and linker region of the protein, the affect may be relatively limited. In this model, the iron ion was found perched in the β-barrel core of the (β/α)8 domain, while five residues (His66, His68, His243, His276, Asp327) were predicted to be involved in ligand binding, in accordance with the “enzyme–substrate” model of Scott et al. [17]. Besides, Asn328, Ser331, and Glu246, which were considered to relate with substrate binding [17, 18], were found close to five ligand-binding residues in the β-barrel core. Thus, it is concluded that the active center and substrate-binding site are probably located in the β-barrel core of the (β/α)8 domain, which fits well the characteristics of the amidohydrolase superfamily described by Seibert and Raushel [10].
In this study, nearly half of the amino acid substitutions observed were in the peripheral region of the (β/α)8 domain, while no substitution occurred in the β-barrel core, indicating that residues in the β-barrel core had relatively low substitutability indices. As evolutionarily conserved residues always show low substitutability indices [33], this demonstrates from another perspective that the active center and substrate-binding residues are located in the β-barrel core of the (β/α)8 domain.
As the two most important residues (Val12, Leu395) identified in this study were situated in the same β-sheet of the dual β-sheet domain, this (β/α)8-neighboring β-sheet definitely plays an important role in enzyme function. In addition, the β-sheet is another concentrated region where amino acid substitution occurred in the study, which further confirmed our conclusion. Hydrogen bond analysis revealed that Val12, Val58, and Leu395 are involved in hydrogen bond formation between β-strands in (β/α)8-neighboring β-sheet to stabilize the structure. Substitution of these residues may cause changes in stability of the structure. Taken together, substitutions in the (β/α)8-neighboring β-sheet significantly improve AtzA activity, probably by affecting the overall structural stability of AtzA.
In summary, a mutagenesis library of atzA was constructed by combining EP-PCR and DNA shuffling, and eight AtzA variants with enhanced enzyme activity were obtained using the H. pluvialis expression system. Characterization of the improved variants and the modeled AtzA structure not only contribute to understanding of the structure–function relationship of AtzA, but it can also serve as the basis for future evolution experiments.
The authors are grateful to Dr. Jia Liu (Department of Chemistry and Biochemistry, University of Maryland) for critically reading the manuscript.
This work was supported by a grant of the National Natural Science Foundation of China (No. 31070717), the Key Program of the Natural Science Foundation of Tianjin (Grant No. 12YFJZJC01700), Tianjin International Science and Technology Cooperation Project (No. 09ZCGHHZ00500), and the Key Technologies R & D Program of Tianjin (No. 11ZCGYNC01000).
REFERENCES
1.Gavrilescu, M. (2005) Eng. Life
Sci., 5, 497-526.
2.Van der Meer, J. R. (2006) Front.
Ecol. Environ., 4, 35-42.
3.Rhine, E. D., Fuhrmann, J. J., and Radosevich, M.
(2003) Microb. Ecol., 46, 145-160.
4.Erickson, L. E., Lee, K., and Sumner, D. D. (1989)
Crit. Rev. Environ. Control, 19, 1-14.
5.Macias-Flores, A., Tafoya-Garnica, A., Ruiz-Ordaz,
N., Salmeron-Alcocer, A., Juarez-Ramirez, C., Ahuatzi-Chacon, D.,
Mondragon-Parada, M. E., and Galindez-Mayer, J. (2009) World J.
Microbiol. Biotechnol., 25, 2195-2204.
6.Zhang, Y., Jiang, Z., Cao, B., Hu, M., Wang, Z.,
and Dong, X. (2011) Int. Biodeter. Biodegr.,
65, 1140-1141.
7.De Souza, M. L., Wackett, L. P., Boundy-Mills, K.
L., Mandelbaum, R. T., and Sadowsky, M. J. (1995) Appl.
Environ. Microbiol., 61, 3373-3378.
8.De Souza, M. L., Sadowsky, M. J., and Wackett, L.
P. (1996) J. Bacteriol., 178, 4894-4900.
9.Seffernick, J. L., McTavish, H., Osborne, J. P., de
Souza, M. L., Sadowsky, M. J., and Wackett, L. P. (2002)
Biochemistry, 41, 14430-14437.
10.Seibert, C. M., and Raushel, F. M. (2005)
Biochemistry, 44, 6383-6391.
11.Rousseaux, S., Hartmann, A., Lagacherie, B.,
Piutti, S., Andreux, F., and Soulas, G. (2003) Chemosphere,
51, 569-576.
12.Strong, L. C., McTavish, H., Sadowsky, M. J., and
Wackett, L. P. (2000) Environ. Microbiol., 2,
91-98.
13.Wang, L., Samac, D. A., Shapir, N., Wackett, L.
P., Vance, C. P., Olszewski, N. E., and Sadowsky, M. J. (2005) Plant
Biotechnol. J., 3, 475-486.
14.Wang, H., Chen, X., Xing, X., Hao, X., and Chen,
D. (2010) Plant Cell Rep., 29, 1391-1399.
15.Chen, K., and Arnold, F. H. (1993) Proc.
Natl. Acad. Sci. USA, 90,
5618-5622.
16.Turner, N. J. (2009) Nat. Chem.
Biol., 5, 567-573.
17.Scott, C., Jackson, C. J., Coppin, C. W.,
Mourant, R. G., Hilton, M. E., Sutherland, T. D., Russell, R. J., and
Oakeshott, J. G. (2009) Appl. Environ. Microbiol.,
75, 2184-2191.
18.Raillard, S., Krebber, A., Chen, Y., Ness, J. E.,
Bermudez, E., Trinidad, R., Fullem, R., Davis, C., Welch, M.,
Seffernick, J., Wackett, L. P., Stemmer, W. P. C., and Minshull, J.
(2001) Chem. Biol., 8, 891-898.
19.Seffernick, J. L., and Wackett, L. P. (2001)
Biochemistry, 40, 12747-12753.
20.Bischoff, H. W., and Bold, H. C. (1963)
Phycological Studies IV, The University of Texas publication
(6318), Austin, pp. 1-95.
21.Jones, A., Lamsa, M., Frandsen, T. P., Spendler,
T., Harris, P., Sloma, A., Xu, F., Nielsen, J. B., and Cherry, J. R.
(2008) J. Biotechnol., 134, 325-333.
22.Stemmer, W. P. (1994) Nature, 370,
389-391.
23.Brown, L. E., Sprecher, S. L., and Keller, L. R.
(1991) Mol. Cell. Biol., 11, 2328-2332.
24.Bradford, M. M. (1976) Anal.
Biochem., 72, 248-254.
25.Kelley, L. A., and Sternberg, M. J. E. (2009)
Nat. Protoc., 4, 363-371.
26.Wass, M. N., Kelley, L. A., and Sternberg, M. J.
E. (2010) Nucleic Acids Res., 38, W469-W473.
27.Hernandez, M., Villalobos, P., Morgante, V.,
Gonzalez, M., Reiff, C., Moore, E., and Seeger, M. (2008) FEMS
Microbiol. Lett., 286, 184-190.
28.Liu, X., and Parales, R. E. (2009) Appl.
Environ. Microbiol., 75, 5481-5488.
29.Yang, Z., Zhang, L., Zhang, Y., Zhang, T., Feng,
Y., Lu, X., Lan, W., Wang, J., Wu, H., Cao, C., and Wang, X. (2011)
PLoS One, 6, e22981.
30.Burgess, R. R. (2009) Methods
Enzymol., 463, 259-282.
31.Chen, D., Chen, X., and Cai, B. (2004) Acta
Scientiarum Naturalium Universitatis Nankaiensis, 37,
109-114 [in Chinese].
32.Bordoli, L., Kiefer, F., Arnold, K., Benkert, P.,
Battey, J., and Schwede, T. (2009) Nat. Protoc.,
4, 1-13.
33.Ortiz, A. R., Strauss, C. E. M., and Olmea, O.
(2002) Protein Sci., 11, 2606-2621.
34.Guo, H. H., Choe, J., and Loeb, L. A. (2004)
Proc. Natl. Acad. Sci. USA,
101, 9205-9210.
35.Baker, D. (2000) Nature, 405,
39-42.