Internal Initiation of Polyuridylic Acid Translation in Bacterial Cell-Free System
E. A. Sogorin, S. Ch. Agalarov, and A. S. Spirin*
Institute of Protein Research, Russian Academy of Sciences, ul. Institutskaya 4, 142290 Pushchino, Moscow Region, Russia; fax: +7 (495) 514-0218; E-mail: spirin@vega.protres.ru; sultan@vega.protres.ru* To whom correspondence should be addressed.
Received September 4, 2013
The task of the present work was to answer the question: is the free 5′-end needed for effective translation of a model polyribonucleotide template – polyuridylic acid – in a bacterial (E. coli) cell-free system? For this purpose, the template activities of the original polyuridylic acid with its free 5′-end and the polyuridylic acid with blocked 5′-end were compared in the bacterial cell-free translation system. To block the 5′-end, the cytidylic oligodeoxyribonucleotide with fluorescein residue at its 5′-end and uridylic oligoribonucleotide sequence at its 3′-end, schematically described as FAM(dC)10(rU)50, was covalently attached (ligated) to the 5′-end of the template polyuridylic acid. It was shown that the efficiency of polyphenylalanine synthesis on the 5′-blocked template and on the polyuridylic acid with free 5′-end was virtually the same. It was concluded that bacterial ribosomes are capable of effectively initiating translation at the polyuridylic sequence independently of the 5′-end of template polyribonucleotide, i.e. via an internal initiation mechanism, in the absence of a Shine–Dalgarno sequence and AUG start codon.
KEY WORDS: polyuridylic acid, translation initiation, polyphenylalanine synthesis, T4 RNA ligase, cell-free translationDOI: 10.1134/S0006297913120055
Abbreviations: FAM, carboxyfluorescein; Hepes, N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid; polyU, polyuridylic acid; TBE, Tris-borate buffer; TCA, trichloroacetic acid; Tris, tris(hydroxymethyl)methylamine.
The canonical translation initiation in prokaryotes begins with the
binding of the small ribosomal subunit with the purine-rich
Shine–Dalgarno (SD) sequence, located near the start AUG-codon
[1]. This sequence is partially complementary to
the pyrimidine-rich 3′-proximal portion of the 16S rRNA sequence,
which is called “anti-Shine–Dalgarno” (ASD). The
correct recognition of the start codon depends on the base pairing of
these sequences in the region several nucleotides upstream of the
AUG-codon. The SD-sequence can thus be regarded as an analog of the
internal ribosomal entry site (IRES) in eukaryotes. It should be noted,
however, that translation initiation in prokaryotes is possible in
cases when the mRNA does not contain the Shine–Dalgarno sequence.
In some cases the prokaryotic ribosome binds to the leader sequences of
the mRNA of some plant viruses [2], which do not
contain a site of the complementary pairing with the ASD. Another
example of a noncanonical translation initiation mechanism is
initiation on the leaderless mRNA [3]; in this case
the ribosome binds directly with the 5′-terminal AUG codon, from
which the open reading frame begins. At last, the most known
effectively translated model mRNA – polyuridylic acid
(polyU) – contains neither the Shine–Dalgarno sequence
nor the AUG codon at the 5′-end. In the latter case, one can
assume two possible mechanisms of translation initiation: either the
5′-end of the polyuridylic template is necessary for translation
initiation, stringing itself to the ribosomal particle (just as the tip
of a thread enters into a needle eye), or the inner portion of the
polyuridylic acid is an analog of the eukaryotic site of the internal
initiation (the so-called IRES element in RNA of eukaryotic viruses),
and, therefore, the prokaryotic ribosome is capable of effectively
initiating translation without the Shine–Dalgarno sequence,
without the AUG codon, and independently of the 5′-end of the
mRNA.
Since the pioneering work on deciphering of the genetic code [4], polyuridylic acid (polyU) has been widely used in studies of the fundamental mechanisms of translation as a model of the polyribonucleotide template for polypeptide (polyphenylalanine) synthesis in bacterial cell-free translation systems. Polyuridylic acid is very well translated, and, therefore, the initiation of translation (a stage largely determining the rate of protein synthesis) in this template proceeds effectively. In an early work [5] on the interaction of ribosomes with polyU, electron microscopic data were obtained that were interpreted as indicating that the ribosome reveals a particular affinity to one of the ends of the polyribonucleotide. However, the methodical correctness of this work raised great doubts (see “Results and Discussion”). Until now, there is no direct experimental evidence of requirement of the free 5′-end for translation of the polyuridylic template by prokaryotic ribosomes. Thus, the question has remained unanswered whether the free 5′-end is required for effective translation initiation of the polyuridylic acid.
To answer the question, we have constructed a polyuridylic acid with blocked 5′-end. The blocking was achieved by covalent attachment of ten cytidylic deoxyribonucleotides and fluorescein to the 5′-end of the polyuridylic acid, which excludes initiation directly from the 5′-end of the polyribonucleotide template. This template has been tested in a cell-free translation system. It was found that the efficiency of polypeptide (polyphenylalanine) synthesis on the 5′-blocked template and on the polyuridylic acid with the free 5′-end was virtually the same. Thus, for the first time, direct evidence has been obtained that translation initiation of the polyuridylic template by prokaryotic ribosomes can begin with its internal site.
MATERIALS AND METHODS
Reagents. In this work reagents and materials of the following companies were used: ribonuclease S1, T4 RNA ligase, RNase inhibitor RiboLock, and length marker RiboRulerTM Low Range RNA ladder (Fermentas, Lithuania); polyU (Sigma, USA); [14C]phenylalanine (Amersham, England); 3MM Chr filters (Whatman, England); Minisolve 1 liquid scintillator (Genzyme, USA).
Fragmentation of high molecular weight polyuridylic acid by limited hydrolysis with ribonuclease S1. Partial cleavage of the commercial preparation of high molecular weight polyuridylic acid (polyU) was performed in a reaction mixture (total volume 50 µl) containing acetate buffer (40 mM CH3COONa, pH 4.5, 0.3 mM NaCl, 2 mM ZnSO4), 1 mg polyU, and 2 units of nuclease S1. The mixture was incubated for 10 min at 37°C. The reaction was stopped by adding an equal volume of phenol. Nucleic acids were precipitated by adding sodium acetate (pH 5.5) up to 300 mM and 2.5 volumes of 96% ethanol. The precipitated nucleic acids were collected by centrifugation for 15 min in the cold, and the precipitates were washed with 80% ethanol, air dried, and dissolved in 30 µl of deionized water.
The fragmented polyU was subjected to periodate oxidation in the presence of 20 mM sodium periodate and 65 mM sodium acetate (pH 4.0) for 1.5 h at 16°C. After incubation, an equal volume of 90% formamide in TBE buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA) was added to the solution, and the mixture was heated at 95°C for 1 min.
Fractionation and purification of polyU fragments. After the periodate reaction, the polyU fragments were electrophoretically separated in 6% denaturing polyacrylamide gel containing 7 M urea. The size of the fragments was estimated in relation to the commercial preparation of RNA length marker RiboRulerTM Low Range RNA ladder (100-1000 nt). The fragments were visualized in the gel by staining with toluidine blue. The area of the gel containing polyU fragments with length of ~100 and ~150 nt was excised with a sterile needle and transferred into a 1.5 ml mini-tube. Phenol (400 µl) and a solution containing 500 mM CH3COONa (pH 5.0) with 2 mM EDTA (600 µl) were added to the gel, and the mixture was shaken vigorously for 30 min. Then, the gel was precipitated in a MiniSpin centrifuge (15 min, 12,100g). The upper (aqueous) phase containing polyU was collected and concentrated by repeated extraction of water with butanol. The polyU was ethanol precipitated as described above and dissolved in water.
Ligation of the fragments of polyuridylic acid with fluorescein-labeled 5′-blocking oligonucleotide. For the purpose of blocking the 5′-polyU end, we have developed a design of the special blocking oligonucleotide FAM(dC)10(rU)50, which contains a fluorescent group (FAM) at the 5′-end, and is further followed by 10 deoxyribocytidylic and 50 ribouridylic nucleotides. By our request, the synthesis of this chimerical oligonucleotide was carried out by the Sintol (Russia). The scheme of blocking of the 5′-end of the polyU is given below:
FAM(dC)10(rU)50 + (rU)~100 = FAM(dC)10(rU)~150.
The polyU fragment of ~100 nt and the chimeric oligonucleotide FAM(dC)10(rU)50 was ligated using T4 RNA ligase. The reaction mixture was prepared in a buffer of 50 mM Tris-HCl (pH 7.5) with 10 mM MgCl2 and 10 mM DTT and contained 50 µg of RNA polyU, 20 µg FAM(dC)10(rU)50, 0.2 unit/µl of T4 RNA ligase, 1 mM ATP, 0.5 unit/µl RNase inhibitor RiboLock, and 20% PEG-4000. The mixture was incubated for 15 h at 25°C. The reaction mixture was deproteinized with an equal volume of phenol, the upper aqueous phase was collected, and the polynucleotide was ethanol precipitated as described above and dissolved in water. The product of the ligation reaction (FAM(dC)10(rU)~150) was purified by gel electrophoresis as described in the section “Fractionation and Purification of polyU Fragments”.
Escherichia coli cell-free translation system. The S30 extract from E. coli was prepared by the procedure described in [6]. The translational mixture contained 26 mM Hepes-KOH, pH 7.6, 0.03 mM folic acid, 1.2 mM ATP, 0.8 mM GTP, 1.7 mM DTT, 0.18 mM total tRNA E. coli, 13.4 mM Mg(OAc)2, 187.5 mM KOAc, 0.08 mg/ml mRNA, 0.25 mg/ml creatine phosphokinase, 80 mM phosphocreatine, 4% PEG-8000, 25% S30 extract E. coli, 0.5 unit/µl RNase inhibitor RiboLock, and 0.005 mM [14C]phenylalanine ([14C]Phe, 19 GBq/mmol). The reaction was carried out at 25°C. Aliquots of the reaction mixture were applied to 1-cm square pieces of 3MM Chr paper, which were then boiled in 10% trichloroacetic acid (TCA) for 5 min and re-boiled in fresh 10% TCA for 2 min. The paper was washed with acetone and dried at room temperature. Measurements of radioactivity in the samples to determine the intensity of the incorporation of [14C]phenylalanine into a polypeptide were performed on a LS 6500 Multi-Purpose Scintillation Counter (Beckman Coulter, USA) in 2 ml of Minisolve 1 liquid scintillator.
RESULTS AND DISCUSSION
Preparation of 5′-blocked polyU: synthesis of the preparation FAM(dC)10(rU)~150. Our task was to attach a polyU fragment of a certain length to the 3′-end of the labeled synthetic deoxyribo-ribonucleotide FAM(dC)10(rU)50 by their ligation using T4 RNA ligase. The length of the attached polyU fragment was chosen to be about 100 nt for the following reasons. On one hand, we sought to achieve the maximum length of the polyU fragment to increase the yield of synthesized polyphenylalanine. On the other hand, the full length of the reaction product should not be excessive to ensure its clear separation from the reaction substrates in the course of electrophoretic purification. The analysis of electrophoretic separation of polyU fragments of different lengths in polyacrylamide gel revealed an optimal length of the attached polyU fragment of about 100 nt. As stated in “Materials and Methods”, to obtain such a polyU fragment, a commercial polyU preparation was subjected to limited hydrolysis with nuclease S1. This nuclease leaves a monophosphate group at the 5′-end of the cleaved RNA, which is necessary for successful subsequent ligation.
Figure 1 shows the results of electrophoretic analysis of the polyU preparations used at all stages of fragmentation, modification, and purification in comparison with RNA markers of different lengths. Figure 1a (lane 1) shows the electrophoretic distribution of the polyU chains in the original commercial preparation, and adjacent lane 2 represents the result of fragmentation of this preparation by nuclease S1. Lane 3 of the same plate (Fig. 1a) shows the distribution of marker RNAs, on the basis of which to isolate the desired fragment polyU, an area of the gel corresponding to the length of the marker RNA of about 100 nt was chosen. Electrophoresis of the purified polyU fragment corresponding to the RNA marker of about 100 nt in length is shown in Fig. 1a, lane 4.
Fig. 1. Analysis of the polyU preparations by electrophoresis on a 6% polyacrylamide gel under denaturing conditions (7 M urea). a) Partial digestion and preparative isolation of the polyU fraction: 1) commercial polyU preparation; 2) commercial polyU preparation after treatment with nuclease S1; 3) RNA markers; 4) purified preparation polyU~100. b) Effect of periodate oxidation of polyU~100 on its ability to form oligomeric and circular forms in the ligation reaction: 1) RNA markers; 2) polyU~100 after periodate oxidation and incubation under ligation reaction conditions; 3) polyU~100 and its oligomeric forms after incubation under ligation reaction conditions. c) Obtaining of the 5′-blocked polyU: 1) RNA markers; 2) the result of the ligation reaction of the FAM(dC)10(rU)50 and polyU~100 (upper band corresponds to the product of the reaction – FAM(dC)10(rU)~150); 3) purified blocked polyU (FAM(dC)10(rU)~150); 4) purified preparation polyU~150. RNA markers: 100, 200, 300, 400, 600, 800, and 1000 nt.
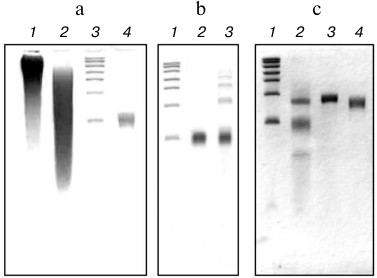
The next stage of our experiment was ligation of the isolated fragment polyU~100 with synthetic chimeric oligodeoxyribo-ribonucleotide FAM(dC)10(rU)50 using RNA ligase (see “Materials and Methods”). Figure 1b demonstrates the need for a preliminary procedure of periodate oxidation of polyU fragments to avoid the formation of oligomeric and circular forms of the polyU chains because of crosslinking their free 5′-ends with the 3′-ends of other or their own chains in the ligation reaction. Indeed, in the case of polyU without pretreatment with sodium periodate (compare lanes 2 and 3, Fig. 1b), the appearance of the oligomeric products characterized by the appearance of bands with slower electrophoretic double and triple chain lengths was observed.
It should be noted that this oligomerization during the ligation reaction was excluded in the case of oligonucleotide FAM(dC)10(rU)50, as its 5′-end is blocked by fluorescein. All this predetermined the selective ligation reaction involved in only the 3′-hydroxyl group of the 3′-end of FAM(dC)10(rU)50 and the 5′-phosphate of the 5′-end of the polyU~100. Thus, the only possible reaction product to be ligated should be FAM(dC)10(rU)~150. Indeed, as can be seen from Fig. 1c, lane 2, the product of the ligation reaction had an electrophoretic mobility expected for FAM(dC)10(rU)~150, and well separated from the reaction substrates. The fluorescence of the corresponding band confirmed its identification as FAM(dC)10(rU)~150.
The product of the ligation reaction was purified from the reaction substrates by preparative electrophoresis. The results of the purification are shown in Fig. 1c, lane 3. Figure 1c (lane 4) also shows the preparation of the polyU fragment about 150 nt in length, produced and purified as well as polyU~100. This preparation was used as a control in the translation experiment.
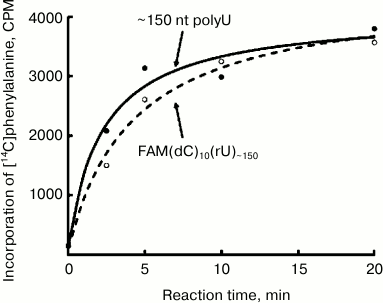
Translation of FAM(dC)10(rU)~150 and (rU)~150 in the E. coli cell-free system. Figure 2 shows the time courses of polyphenylalanine synthesis for the 5′-blocked and control polyU preparations. As can be seen, both the rate of translation, and the yield of the product for both preparations are virtually the same. It is known that DNA in normal conditions cannot serve as a template for translation [7, 8]. Therefore, the translation initiation at the template blocked by 5′-fluorescein and polydeoxyribonucleotide should begin with some polyU region. Thus, in this case there is an internal (independent of the free 5′-end) initiation.
Fig. 2. Time course of polyphenylalanine synthesis on the polyU template. Solid curve (black circles), ~150 nt polyU; dashed curve (open circles), 5′-blocked polyU: the preparation FAM(dC)10(rU)~150.
As mentioned in the introductory section, in their early work Matthaei et al. [5] reported on the trend of the ribosomal particles to attach to one of the polyU ends. This conclusion was based on electron microscopic images of a mixture of the 30S, 50S subunits, and polyU. However, the authors of that paper made a number of methodological flaws. In particular, the isolation of the ribosomal subunits and the experiment on their binding to polyU were performed in the complete absence of monovalent cations, which are absolutely necessary for the functioning of ribosomes.
From the experiment described in the present study, we can conclude that the prokaryotic ribosome is essentially capable to initiate translation at polyuridylic sequence, regardless of the 5′-end of the template and in the absence of the Shine–Dalgarno sequence and the AUG start codon.
The authors are grateful to A. S. Sokolov for assistance in some experiments.
This work was supported by grants from the Russian Foundation for Basic Research (Nos. 12-04-01179-a and 13-04-40213-N) as well as a grant from the Program “Molecular and Cell Biology” of the Presidium of the Russian Academy of Sciences.
REFERENCES
1.Shine, J., and Dalgarno, L. (1974) Proc. Natl.
Acad. Sci. USA, 71, 1342-1346.
2.Gallie, D. R., Sleat, D. E., Watts, J. W., Turner,
P. C., and Wilson, T. M. A. (1987) Nucleic Acids Res.,
15, 3257-3273.
3.Murray, I. A., Gil, J. A., Hopwood,
D. A., and Shaw, W. V. (1989) Gene, 85,
283-291.
4.Nirenberg, M. W., and Mattei, J. H. (1961) Proc.
Natl. Acad. Sci. USA, 47, 1588-1602.
5.Matthaei, H., Amelunxen, F., Eckert, K., and
Heller, G. (1964) Ber. Bunsengesellschaft Phys. Chemie,
68, 735.
6.Shirokov, V. A., Kommer, A., Kolb, V. A., and
Spirin, A. S. (2007) Methods Mol. Biol., 375, 19-55.
7.McCarthy, B. J., and Holland, J. J. (1965) Proc.
Natl. Acad. Sci. USA, 54, 880.
8.Morgan, A. R., Wells, R.
D., and Khorana, H. G. (1967) J. Mol. Biol., 26,
477.