SkBQ – Prooxidant Addressed to Mitochondria
M. Y. Vyssokikh1*, B. V. Chernyak1, L. V. Domnina1, D. S. Esipov1, O. Y. Ivanova1, G. A. Korshunova1, R. A. Symonyan1, M. V. Skulachev1,2, T. V. Zinevich1, and V. P. Skulachev1
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Vorob’evy Gory 1/40, 119992 Moscow, Russia; fax: +7 (495) 939-0338; E-mail: mikhail.vyssokikh@gmail.com; mike@genebee.msu.su; korsh@genebee.msu.ru2Institute of Mitoengineering, Vorob’evy Gory 1, 119992 Moscow, Russia; E-mail: info@skq-project.ru
* To whom correspondence should be addressed.
Received September 24, 2013
Oxidative stress and mitochondrial dysfunction are the key links in the chain of development of pathologies associated with the violation of cellular energy metabolism. Development of mitochondria-addressed compounds highly specific for chemical processes is one of the most promising ways to develop approaches to the treatment of inherited and age-related diseases with mitochondrial etiology. Correlation of structure and chemical activity of the test compounds from a class of lipophilic cations revealed the key role of substituents in the aromatic ring of 1,4-benzoquinones in the manifestation of high antioxidant properties. In this work, it is shown that a synthesized benzoquinone derivative conjugated in position 6 with membrane-penetrating cation of decyltriphenylphosphonium and with substituents at position 2, 3, and 5 (SkBQ) has much lower antioxidant and significantly higher prooxidant activity in comparison with similar derivatives of plasto- and toluquinone SkQ1 and SkQT1 in experiments on isolated mitochondria. At the same time, SkBQ, like SkQ1 and SkQT1, can be reduced by the respiratory chain in the center i of complex III and decrease the mitochondrial membrane potential. In cell cultures of human fibroblasts, it was revealed that SkBQ does not protect cells from apoptosis induced by hydrogen peroxide. Under the same conditions, SkQ1 and SkQT1 exhibit a powerful protective effect. Thus, SkBQ can be seen as a mitochondria-addressed prooxidant. The possibility of using SkBQ as an anticancer drug for the treatment of cancers such as prostate cancer whose cells are sensitive to mitochondrial reactive oxygen species is discussed.
KEY WORDS: mitochondria, reactive oxygen species, mitochondria-addressed anti- and prooxidantsDOI: 10.1134/S0006297913120079
Quinones, one of the major classes of natural compounds, have the ability to enter redox reactions, including performing a unique function of electron transfer inside energy-transforming cell membranes. Quinones are the key cofactors of enzymatic reactions matching the processes of electron transfer in the oxidation of respiratory substrates to form a transmembrane electrochemical potential of hydrogen ions that cells use for the phosphorylation of ATP during respiration and photosynthesis [1].
In addition to participating in electron transfer between redox centers of the respiratory chain in mitochondria, partially reduced quinone interacts with oxygen in center (i0) and complex III under certain conditions, resulting in the formation of superoxide-precursor of reactive oxygen species (ROS) [2]. To prevent oxidation of such labile macromolecules as nucleic acids, proteins, and compounds containing unsaturated bonds, antioxidant protective system cells support the level of superoxide and other ROS below the critical value. These systems are non-proteinaceous components such as glutathione and a series of enzymes utilizing radicals such as superoxide dismutase, catalase, glutathione peroxidase, and others. The disbalance of production and utilization rates of ROS in the cell give rise an increase in oxidative damage and the development of the phenomenon of oxidative stress, leading to the eventual elimination of cells exposed to it along the path of programmed cell death [3, 4].
Increase of oxidative stress is largely due to the accumulation of damage in the poorly protected mitochondrial DNA (mtDNA), localized close to the inner membrane, where ROS can be produced. The accumulation of oxidative damage leading to increasing respiratory chain dysfunction and development of pathologies associated primarily with insufficient energy metabolism of cells have the collective name of “mitochondrial diseases” [5, 6].
In recent years, the attention of researchers was attracted by an opportunity to reduce the level of oxidative damage of biological macromolecules by means of antioxidants [7, 8]. Application of both types of conventional antioxidants – soluble (like N-acetylcysteine, glutathione, etc.) and lipophilic (such as vitamin K, derivatives of vitamin E, carotenoids, ubiquinone, etc.) – has resulted in limited success due to a combination of low affinity and selectivity of the compounds used. However, several studies have shown some therapeutic effect of ubiquinone and its analog decylubiquinone (Q10) and of idebenone and polyphenols [9, 10].
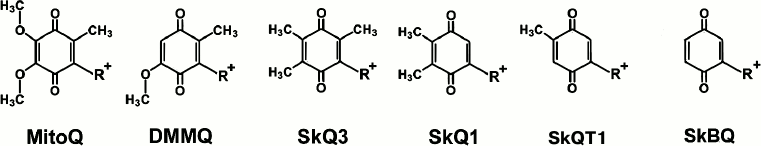
Recently, a new approach to the development of a possible therapy of pathologies with mitochondrial oxidative origin was proposed. It is based on the use of the ability of mitochondria to accumulate penetrating cations, since the mitochondrion is a unique intracellular organelle charged negatively. A family of compounds was created based on the molecule of a conjugate between penetrating decyltriphenylphosphonium cation and substituted quinone. The antioxidant capacity and pronounced cell structure protective effect against induced or endogenous oxidative stress were shown for the compounds [11-14]. It turned out that the change in the number and structure of the substituents on the aromatic ring of these compounds (Fig. 1) dramatically increases antioxidant properties of the test substances. However, along with inductive effect decline and the number of substituents in aromatic ring core reduction, the reactivity of the quinone increases. To determine the properties of quinone completely devoid of substituents in positions 2, 3, and 5, 1,4-benzoquinone was conjugated with decyltriphenylphosphonium and studied as a model molecule in experiments on isolated rat heart mitochondria and cell culture of human dermal fibroblasts.
Fig. 1. Mitochondria-addressed cation derivatives of quinone. MitoQ, 10-(2,3-dimethoxy-5-methyl-1,4-benzoquinonyl-6)decyltriphenylphosphonium; DMMQ, 3-demethoxy-mitoQ; SkQ3, 10-(methylplastoquinonyl-6)decyltriphenylphosphonium; SkQ1, 10-(plastoquinonyl-6)decyltriphenylphosphonium; SkQT1, 10-(p-toluquinonyl)decyltriphenylphosphonium; SkBQ, 10-(p-benzoquinonyl)decyltriphenylphosphonium. R+, decyltriphenylphosphonium cation.
MATERIALS AND METHODS
SkBQ – 10-(p-benzoquinonyl)decyltriphenylphosphonium bromide – was synthesized according to the following scheme:
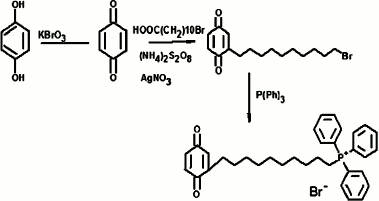
Progress of synthesis: p-benzoquinone was obtained by oxidation of hydroquinones with potassium bromate in an aqueous medium in the presence of sulfuric acid by heating to 50°C. The resulting p-benzoquinone was alkylated by bromoundecanoic acid in a radical substitution reaction with simultaneous decarboxylation catalyzed by silver nitrate in the presence of ammonium persulfate. The p-benzoquinone and 11-bromundecanoic acid were taken in equimolar amounts, and the reaction was carried out for 3 h in a mixture of acetonitrile and water (3 : 4) at the reaction temperature of 65°C. The reactions were monitored by TLC on silica gel in chloroform. The completion of the reaction was judged by disappearance of the initial quinone spot. Upon completion, the reaction mixture was diluted with water, and 10-(p-benzoquinonyl)decyl bromide was extracted with diethyl ether. After evaporation of the ether, the residue was applied to a silica gel column with chloroform as eluent. Fractions containing the individual compound with the same chromatographic mobility value, Rf (chloroform) = 0.57 were combined and evaporated on a rotary evaporator. The resulting material (yield 51%) was introduced into the next reaction without further purification. Equimolar amounts of 10-(p-benzoquinonyl)decyl bromide and triphenylphosphine ([P(Ph)3]) in a solution of 96% ethanol were heated for 72 h at 75-80°C in a sealed flask. After the reaction mixture was evaporated, the residue was repeatedly re-sedimented from dichloromethane with diethyl ether. Further purification of the compound was achieved by column chromatography on silica gel using a mixture of dichloromethane and ethanol (5 : 1) as eluent. Crude yield was 64%. The final purification of the target compound was carried out by reverse phase HPLC. A Luna CN 100A column (Phenomenex, USA) was used with a gradient of ethanol from 50 to 70% over 17 min. The resulting drug SkBQ was of 93% purity. Calculated mass [M + 1] = 510, found [M] = 509.8. The structure of the compound was confirmed by its NMR spectrum.
Mitochondria were isolated from rat hearts at 4°C. The rats were decapitated, and the hearts were removed and washed in cooled isolation medium (250 mM sucrose, 10 mM Mops-KOH, pH 7.4, 1 mM EGTA, and 0.1% bovine serum albumin). The heart was minced with scissors in a Petri dish, and then the slurry was weighed and suspended in a chilled glass beaker with isolation medium in a ratio of 10 ml/g of cardiac tissue. A 0.5-ml sample of nagarse (type XXIV; Sigma, USA) containing 2.5 mg of protein in 1 ml of isolation medium was added to the suspension. The suspension was then homogenized in a glass Potter homogenizer for 1-2 min. The homogenate was diluted with the isolation medium to the ratio of 20 ml/g of original tissue and centrifuged for 10 min at 600g. The supernatant was collected and centrifuged 10 min at 8500g. The precipitate was resuspended in a minimum volume, homogenized, and transferred into a centrifuge cup, diluted in 20 ml of isolation medium, and then centrifuged again for 10 min at 8500g. The supernatant was discarded, and the pellet was resuspended in 300 µl of isolation medium.
Mitochondrial membrane potential was measured using Safranin O as an indicator [15]. The measuring medium contained 250 mM sucrose, 10 mM MOPS-KOH, pH 7.4, 0.1 mM EGTA, 5 mM succinate, 2 µM rotenone, and 15 µM Safranin O. The concentration was 0.7 mg of mitochondrial protein/ml. Light absorption was measured at wavelengths of 555 and 523 nm on an Aminco DW2000 spectrophotometer in the dual-wavelength mode.
The rate of mitochondrial respiration was measured using a Clark electrode at 25°C. The incubation medium contained 250 mM sucrose, 10 mM HEPES, 1 mM EGTA, 2 mM MgCl2, 2 mM KH2PO4, 5 mM succinate, and 2 µM rotenone.
The level of lipid peroxidation in mitochondria was determined by the accumulation of malondialdehyde (MDA) reacting with thiobarbituric acid (TBA); the method was described previously by Yagi et al. [16] with modifications described in the work of Jentzsch et al. [17]. The content of MDA and TBA adduct was determined spectrophotometrically with the Aminco DW200 in dual-beam mode at 535 nm and expressed as nmol of MDA/mg of mitochondrial protein. To obtain the calibration curve for dependence of absorbance at 535 nm on the content of adduct of MDA and TBA, standard 50 µM solution of MDA was prepared by hydrolysis of 10 mM 1,1,4,4-tetraethoxypropane by 10 mM hydrochloric acid for 10 min at room temperature.
Formation of H2O2 in the mitochondrial suspensions was determined fluorimetrically by oxidation of Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine; Invitrogen, USA). Mitochondria (0.25 mg protein/ml) in isolation medium without albumin were placed into a plastic fluorometer cuvette (volume 0.5 ml), horseradish peroxidase (final content of 1 IU/ml), and 4 µl of 10 mM solution of Amplex Red in dimethyl sulfoxide was added. Formation of hydrogen peroxide was induced by adding of 1 µM antimycin A to the suspension. The fluorescence was measured at 595 nm with excitation at 550 nm on a Hitachi MPF4 spectrofluorimeter at 25°C.
The degree of SkBQ reduction was determined spectrophotometrically in dual-beam mode on the Aminco DW2000 (within the range of 250-350 nm) by the change in absorbance at 276 nm with subtraction of the absorbance at the isosbestic point at 285 nm. The experiment began by adding 5 mM succinate and 2 µM rotenone to the medium containing 0.5 mg of mitochondrial protein at 25°C. Registration time was 45 min, and spectra were recorded every 3 min, 1 nm step, rate 1 nm/s, and the decline of optical density was recorded in the absence or in presence of 1 μM antimycin A.
Cell death was determined by nuclear morphology after staining with Hoechst 33342 (1 µg/ml); cells with condensed and fragmented chromatin are assumed to be apoptotic. Preparations were analyzed using a fluorescence microscope at excitation wavelength of 365 nm.
RESULTS AND DISCUSSION
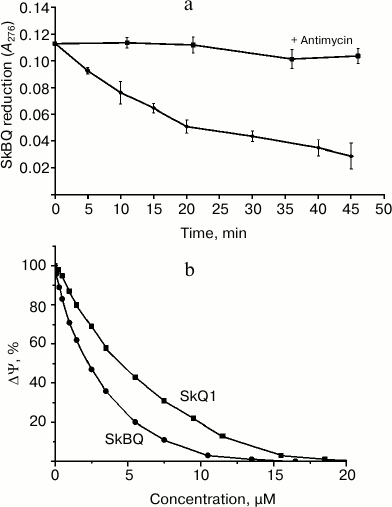
Interaction of SkBQ with the mitochondrial respiratory chain at the level of complex III was demonstrated by its reduction in the medium containing the energized mitochondria (Fig. 2a). In the presence of antimycin A reduction does not occur, allowing us to locate the site of interaction SkBQ with the respiratory chain as center i of complex III. The kinetics of SkBQ reduction is similar to that of conjugated substituted quinones such as SkQ1 [18]. Figure 2b shows the effect of SkBQ on the membrane potential (ΔΨ) of mitochondria energized with succinate in the presence of rotenone and oligomycin. The compound decreases ΔΨ at somewhat lower concentrations than SkQ1.
Fig. 2. Interaction of SkBQ with mitochondria respiration chain of rat heart. a) Reduction of 1,4-benzoquinone conjugated with decyltriphenylphosphonium by mitochondria respiratory chain energized by succinate with rotenone in the presence (upper curve) and in the absence (lower curve) of antimycin A (1 μM); b) effect of SkBQ and SkQ1 on transmembrane potential.
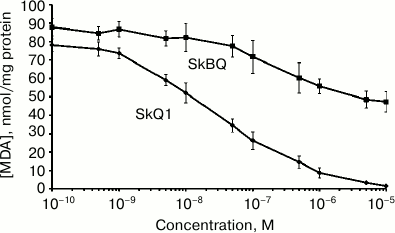
The antioxidant activity of SkBQ was investigated on the model system where oxidative stress is induced in mitochondria. It was found that SkBQ has significantly less pronounced protective action for mitochondria than SkQ1 upon induction of lipid peroxidation by the Fenton reaction (Fig. 3).
Fig. 3. SkBQ fails to inhibit lipid peroxidation caused by exogenous oxidative stress in mitochondria. SkBQ and SkQ1 (0.1 nM-10 µM) were added to mitochondria energized with succinate in the presence of rotenone. For induction of the Fenton reaction, 10 μM FeSO4 and 100 μM H2O2 were added to the mitochondria, and the suspension was incubated for 45 min at 25°C with constant stirring. MDA was extracted as described in “Materials and Methods”.
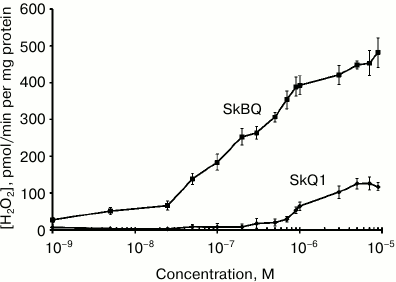
When measuring hydrogen peroxide produced by mitochondria in the presence of antimycin A (Fig. 4), it appears that submicromolar concentrations of SkBQ stimulate H2O2 production to a much greater extent than SkQ1.
Fig. 4. SkBQ stimulates hydrogen peroxide production in rat heart mitochondria in the presence of 1 μM antimycin A.
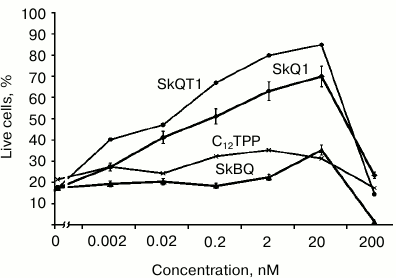
In experiments with cultured human fibroblasts, it has been shown that SkBQ practically does not protect the cells from apoptosis induced by hydrogen peroxide. Under the same conditions, SkQ1 and SkQT1 showed effective protection, while C12TPP (analog of SkQ without quinone) was generally ineffective (Fig. 5).
Fig. 5. Effect of penetrating cations on H2O2-induced apoptosis of skin fibroblasts. Human skin fibroblasts were preincubated for 24 h with SkQ1, SkBQ, SkQT1, or C12TPP. Then H2O2 was added to the incubation medium to 0.5 mM final concentration. After 24 h of incubation with H2O2, dead and live cells were counted on the microscope.
Thus, the compound SkBQ, having no substituents in the quinone ring at positions 1-5, proved to have very weak antioxidant activity in experiments on isolated mitochondria. In cells, it shows no protective effect in oxidative stress conditions and does not protect them from the loss in the induction of apoptosis by hydrogen peroxide. At the same time, SkBQ at submicromolar concentrations demonstrates remarkable prooxidant effect on isolated mitochondria. It seems promising to study the possibility of using SkBQ as an anticancer drug on the types of cancers that can be treated by chemotherapeutic prooxidation. Thus, activation of mitochondrial ROS generation in prostate cancer cells leads to the death of these cells [19]. This effect is opposite to the action of antioxidants such as SkQs (SkQ1, SkQR1, SkQT1) on cells of different types of sarcoma. In this case, according to our data, reducing the generation of mitochondrial ROS kills cancer cells by inhibiting the cell cycle after mitosis, which activates apoptosis [14].
REFERENCES
1.Mitchell, P. (1966) Biol. Rev., 41,
445-502.
2.Kramer, D. M., Roberts, A. G., Muller, F., Cape,
J., and Bowman, M. K. (2004) Methods Enzymol., 382,
21-45.
3.Amstad, P., Moret, R., and Cerutti, P. (1994) J.
Biol. Chem., 269, 1606-1609.
4.Hockenbery, D. M., Oltvai, Z. N., Yin, X.-M.,
Milliman, C. L., and Korsmeyer, S. J. (1993) Cell, 75,
241-251.
5.Richter, C., Gogvadze, V., Laffranchi, R.,
Schlapbach, R., Schweizer, M., Suter, M., Walter, P., and
Yaffee, M. (1995) Biochim. Biophys. Acta, 24, 67-74.
6.Wallace, D. C. (2001) Novartis Found. Symp.,
235, 247-263, discussion 263-266.
7.Skulachev, V. P., Bogachev, A. V., and Kasparinsky,
F. O. (2010) Membrane Bioenergetics [in Russian], Moscow State
University Publishers, Moscow.
8.Halliwell, B. (2001) Drugs Aging, 18,
685-716.
9.Lenaz, G., Bovina, C., D’Aurelio, M.,
Fato, R., Formiggini, G., Genova, M. L., Giuliano, G., Merlo
Pich, M., Paolucci, U., Parenti Castelli, G., and Ventura, B.
(2002) Ann. N.Y. Acad. Sci., 959, 199-213.
10.Garcia-Gimenez, J. L., Gimeno, A., Gonzalez-Cabo,
P., Dasi, F., Bolinches-Amoros, A., Molla, B., Palau, F., and Pallardo,
F. V. (2011) PLoSOne, 6, e20666.
11.Kelso, G. F., Porteous, C. M., Coulter, C. V.,
Hughes, G., Porteous, W. K., Ledgerwood, E. C., Smith, R. A., and
Murphy, M. P. (2001) J. Biol. Chem., 276, 4588-4596.
12.Skulachev, V. P. (2005) IUBMB Life,
57, 305-310.
13.Ross, M. F., Da Ros, T., Blaikie, F. H., Prime,
T. A., Porteous, C. M., Severina, I. I., Skulachev, V. P., Kjaergaard,
H. G., Smith, R. A., and Murphy, M. P. (2006) Biochem. J.,
400, 199-208.
14.Severina, I. I., Severin, F. F., Korshunova, G.
A., Sumbatyan, N. V., Ilyasova, T. M., Simonyan, R. A., Rogov, A. G.,
Trendeleva, T. A., Zvyagilskaya, R. A., Dugina, V. B., Domnina, L. V.,
Fetisova, E. K., Lyamzaev, K. G., Vyssokikh, M. Y., Chernyak, B. V.,
Skulachev, M. V., Skulachev, V. P., and Sadovnichii, V. A. (2013)
FEBS Lett., 587, 2018-2024.
15.Akerman, K. E. O., and Wikstrom, M. K. F. (1976)
FEBS Lett., 68, 191-197.
16.Yagi, K., Nishigaki, I., and Ohama, H. (1968)
Vitamins, 37, 105-112.
17.Jentzsch, A. M., Bachmann, H., Furst, P., and
Biesalski, H. K. (1996) Free Radic. Biol. Med., 20,
251-256.
18.Skulachev, V. P., Antonenko, Y. N., Cherepanov,
D. A., Chernyak, B. V., Izyumov, D. S., Khailova, L. S., Klishin, S.
S., Korshunova, G. A., Lyamzaev, K. G., Pletjushkina, O. Y., Roginsky,
V. A., Rokitskaya, T. I., Severin, F. F., Severina, I. I., Simonyan, R.
A., Skulachev, M. V., Sumbatyan, N. V., Sukhanova, E. I., Tashlitsky,
V. N., Trendeleva, T. A., Vyssokikh, M. Y., and Zvyagilskaya, R. A.
(2010) Biochim. Biophys. Acta, 1797, 878-889.
19.Rico-Bautista, E., Zhu, W., Kitada, S.,
Ganapathy, S., Lau, E., Krajewski, S., Ramirez, J., Bush, J. A., Yuan,
Z., and Wolf, D. A. (2013) Oncotarget., 4, 1212-1229.