REVIEW: Chemistry Enters Nucleic Acids Biology: Enzymatic Mechanisms of RNA Modification
S. Boschi-Muller and Y. Motorin*
Université de Lorraine, Laboratoire IMoPA, UMR 7365 CNRS-UL, Faculté de Médecine de Nancy, 9 rue de la forêt de Haye, BP 184, 54505 Vandoeuvre les Nancy, France; fax: 33 (0) 383-685-509; E-mail: Yuri.Motorin@univ-lorraine.fr* To whom correspondence should be addressed.
Received August 1, 2013
Modified nucleotides are universally conserved in all living kingdoms and are present in almost all types of cellular RNAs, including tRNA, rRNA, sn(sno)RNA, and mRNA and in recently discovered regulatory RNAs. Altogether, over 110 chemically distinct RNA modifications have been characterized and localized in RNA by various analytical methods. However, this impressive list of known modified nucleotides is certainly incomplete, mainly due to difficulties in identification and characterization of these particular residues in low abundance cellular RNAs. In DNA, modified residues are formed by both enzymatic reactions (like DNA methylations, for example) and by spontaneous chemical reactions resulting from oxidative damage. In contrast, all modified residues characterized in cellular RNA molecules are formed by specific action of dedicated RNA-modification enzymes, which recognize their RNA substrate with high specificity. These RNA-modification enzymes display a great diversity in terms of the chemical reaction and use various low molecular weight cofactors (or co-substrates) in enzymatic catalysis. Depending on the nature of the target base and of the co-substrate, precise chemical mechanisms are used for appropriate activation of the base and the co-substrate in the enzyme active site. In this review, we give an extended summary of the enzymatic mechanisms involved in formation of different methylated nucleotides in RNA, as well as pseudouridine residues, which are almost universally conserved in all living organisms. Other interesting mechanisms include thiolation of uridine residues by ThiI and the reaction of guanine exchange catalyzed by TGT. The latter implies the reversible cleavage of the N-glycosidic bond in order to replace the initially encoded guanine by an aza-guanosine base. Despite the extensive studies of RNA modification and RNA-modification machinery during the last 20 years, our knowledge on the exact chemical steps involved in catalysis of RNA modification remains very limited. Recent discoveries of radical mechanisms involved in base methylation clearly demonstrate that numerous possibilities are used in Nature for these difficult reactions. Future studies are certainly required for better understanding of the enzymatic mechanisms of RNA modification, and this knowledge is crucial not only for basic research, but also for development of new therapeutic molecules.
KEY WORDS: RNA-modification, methylation, pseudouridine, catalytic mechanismsDOI: 10.1134/S0006297913130026
Abbreviations: MTase, methyltransferase; SAM/AdoMet, S-adenosyl-L-methionine.
After DNA transcription by RNA polymerase(s), the majority of primary
RNA transcripts in the cell undergo additional steps of RNA maturation
to give rise to mature cellular RNA species. Depending on the organism,
these steps consist of 5′- and 3′-trimming, intron excision
[1, 2], as well as chemical
modification of parental A, U, C, and G nucleotides. This particular
RNA-modification step is insured by numerous specific enzymatic
activities called RNA-modification enzymes. Depending on the chemical
structure of the resulting modified nucleotide, low molecular weight
metabolic co-substrates (also commonly called cofactors) are frequently
required for these reactions (e.g. AdoMet/SAM for methylations). Over
110 chemically different modified nucleotides have been characterized
to date in RNAs from various living organisms [3].
The majority of these modifications represent nucleotide methylations,
but other reactions are also allowed, like isomerization, thiolation,
reduction, etc. In some instances the whole enzymatic cascade is
necessary for complete synthesis of a complex modified nucleotide
(so-called hypermodified nucleotides in RNA, or RNA hypermodification).
Some examples of modified nucleotides discussed in this review are
shown in Fig. 1.
Fig. 1. Chemical structures of some modified nucleotides.
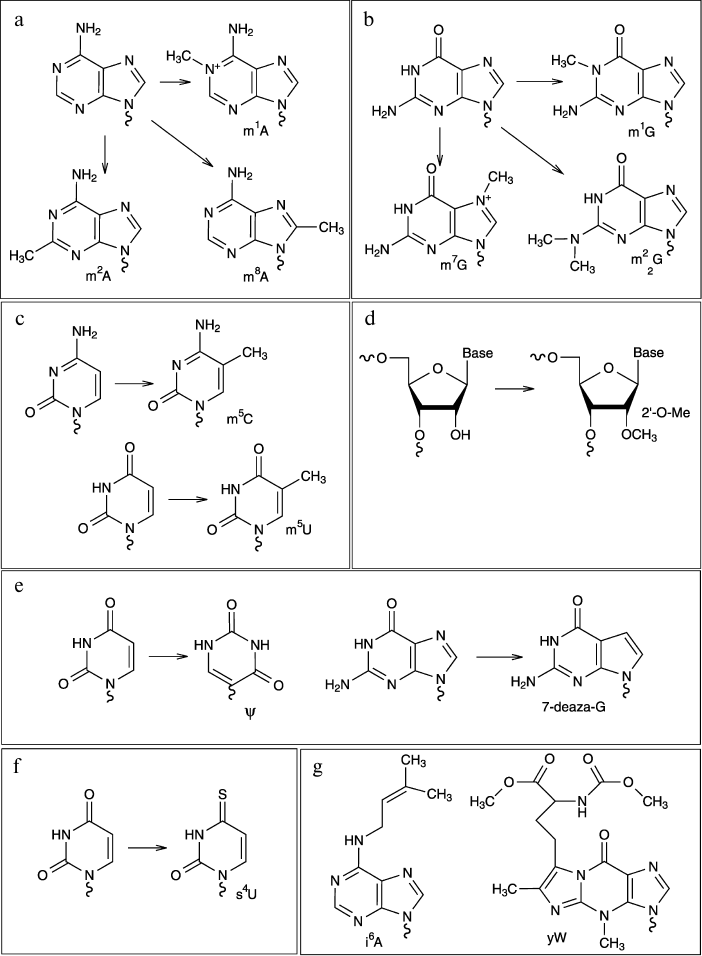
Modified RNA residues are found in all studied organisms, and many of them are highly conserved in a given RNA species. The best examples of such conservation are the so-called TψC- and D-loops present in almost any tRNA species; these loops contain highly conserved characteristic modified residues ribothymidine (T = m5U), pseudouridine (ψ), and dihydrouridine (D). There are many other examples of high evolutionary conservation of both modified nucleotides in RNA and also the respective RNA-modification machineries responsible for their biosynthesis.
Numerous studies have pointed out the crucial importance of modified nucleotides for RNA folding, 2D and 3D structure [4], thermal and conformational stability, and also interactions with other RNA and protein partners in the cell. These aspects are reviewed elsewhere [5, 6].
RNA-MODIFICATION ENZYMES. BRIEF OVERVIEW OF THE RNA MODIFICATION
PROCESS IN THE CELL
Since all known modified nucleotides in RNA result from enzymatic modification, several classes of specific RNA-modification enzymes have been characterized [7]. The most evident classification is thus based on the chemical nature of the resulting modified nucleotide and, in some instances, on the nature of the RNA substrate recognized by the protein. Most common RNA-modification enzymes are RNA:methyltransferases (RNA:MTase) [5] (methylation represents about 70% of known modified residues in RNA) and RNA:pseudouridine-(ψ)-synthases. An important number of other RNA-modification enzymes is now characterized, and biochemical pathways for biosynthesis of these particular nucleotides are now uncovered (see MODOMICS database for further information) [3]. However, despite this outstanding progress in genomic characterization of the RNA-modification machinery, the exact chemical and kinetic mechanism of RNA modification frequently remains elusive. In this review, we will consider in detail only some major classes of RNA-modification enzymes, namely RNA:MTases, RNA:ψ-synthases, guanine transglycosylases TGT, and tRNA-thiolases.
Major Classes of RNA-Modification Enzymes
RNA:methyltransferases. As mentioned above, the RNA:MTases represent a good part of known RNA-modification enzymes. These enzymes belong to a vast group of methyltransferases that catalyze the transfer of a CH3-group (Me-group) from a methyl donor to a biomolecule [8, 9]. Depending on the presence of characteristic sequence motifs and on the global organization of the protein fold, the MTases are placed into five distinct classes. Most DNA: and RNA:MTases belong to classes I (the most represented) [8] and IV (mostly SpoU type 2′-O-RNA:MTases and m1G-tRNA:MTases) [10, 11]. From the kinetic and mechanistic point of view, RNA:MTases can be compared to other methyltransferases acting on DNA, proteins, and even on small molecules [12]. The almost universal methyl donor for all Me-transfer reactions in living organisms is S-adenosyl-L-methionine (SAM or AdoMet); only a few MTases use other Me-donors, like N5,N10-methylenetetrahydrofolate (CH2H4-folate) [13].
Analysis of numerous known AdoMet (SAM)-dependent MTases pointed out the existence and relatively strong conservation of protein motifs involved in AdoMet binding. These motifs have been identified in almost any MTase, independently from its substrate specificity, and are numbered from I to X. Some of these known motifs stabilize AdoMet in the enzyme active site, but motifs IV, V, and VI are also frequently associated with the pure catalytic step of the Me-transfer reaction [8, 9]. In addition, a different catalytic mechanism for RNA methylation was recently discovered. These unusual RNA:MTases also use SAM as a source of the Me-group, but they belong to the so-called radical SAM family. This family includes numerous enzymes catalyzing radical reactions using SAM as a substrate, and some of these proteins are able to transfer a Me-group to a biological substrate. Only a few radical SAM MTases acting on RNA are known, the best-characterized examples being m2A- and m8A-generating enzymes RlmN and Cfr from antibiotic-resistant bacteria [14-16]. One other RNA:modification enzyme, TYW1, which is not an RNA:MTase but uses the same radical SAM reaction mechanism, participates in the early biosynthetic steps of the hypermodified base wybutosine in eukaryotic tRNA [17].
Pseudouridine synthases. RNA:ψ-synthases families. The modified nucleotide pseudouridine is the most abundant modification in cellular RNAs and was even initially considered as the fifth nucleotide in RNAs. However, later on, its post-transcriptional origin become clear, and pseudouridine is now one of the founding members of the modified RNA nucleotides family. With rare exceptions, pseudouridine residues have been found in all analyzed RNA species, including tRNA, rRNA, snRNA, snoRNA, and others. Like all other modified nucleotides, pseudouridine is incorporated post-transcriptionally, by specific action of RNA:ψ-synthases [18-20].
All known RNA:ψ-synthases are grouped into six distinct families depending on their sequence and structure conservation. These families are named after founding protein members characterized in E. coli (families I to V) or, if absent in eubacteria, after an archaeal protein (Pus10 family). The great diversity of RNA:ψ-synthases implies only a low conservation of the catalytic amino acid residues involved in catalysis. Only one catalytic residue (generally Asp) is conserved in all classes of the characterized enzymes, and in some cases it is associated to a basic residue like Lys or Arg (see Fig. 2) [21].
Fig. 2. Conserved sequence motif in RNA-pseudouridine synthases families. Only representative members of each family are depicted.

Despite important differences in size, sequence, and overall protein folding, all these enzymes catalyze the same chemical reaction (isomerization of an incorporated U residue in RNA into pseudouridine). Even if the reaction product is identical, the RNA substrate for this class of RNA-modification enzymes differs very considerably in size and 3D structure. Many RNA:ψ-synthases specifically recognize tRNAs, while others act on snRNA, rRNA, etc. In some instances the same enzyme acts on both tRNA and other RNAs, since the recognition unit comprises only a limited structural domain [22, 23].
tRNA-guanine transglycosylases. The reaction catalyzed by tRNA-guanine transglycosylases (TGT) was first discovered fortuitously by the observation that the radioactively labeled G base is incorporated into tRNA fraction upon incubation in cell-free extract. The understanding of the biological meaning of this phenomenon came much later, when deaza-guanosine derivatives (modified nucleotides of the queuosine family) were discovered in the anticodon loop of both bacterial and higher eukaryotic tRNA molecules. Indeed, the enzymes of the TGT family catalyze the exchange of initially incorporated G residue by a deaza-guanosine base, which is synthesized by a separate metabolic pathway. This incorporated queuosine Q (or pre-queuosine, preQ1) base is further modified to become Q, ManQ, or GalQ [3]. A very similar reaction was also found in Archaea, where the exchanged archaeosine residue is located at position 15 in the D-loop, except that the G residue is replaced by preQ0 and further converted to archaeosine.
To achieve the base replacement, TGT enzymes use the precise cleavage of the C–N glycosidic bond (like do RNA:ψ-synthases) followed by formation of a covalent reactive intermediate between the RNA substrate and enzyme. This reaction intermediate is further used to incorporate an external 7-deaza-guanosine derivative.
The crystal structure of the Z. mobilis TGT complexed with substrate RNA minihelix demonstrated the existence of the covalent reaction intermediate between the catalytic Asp residue and the 1′ carbon of the ribosyl moiety of the replaced G nucleotide [24-27].
tRNA thiolases. Biosynthesis of thiolated nucleotides in tRNA is catalyzed by a group of sulfurtransferases that are responsible for synthesis of s2U, s4U, s2C, and also ms2A and its derivatives. These thiolated nucleotides are important both for mRNA decoding during protein synthesis and, in addition, are implicated in bacterial photosensitivity to UV light [28]. The best-studied example of tRNA-sulfurtransferase is the enzyme ThiI responsible for s4U formation at position 8 in tRNA [29, 30]. In contrast, the enzymes (MTTases) responsible for ms2-group (methyl-thio) formation belong to the radical SAM enzyme family [31].
Other enzymes. The above-cited examples demonstrate important variability of catalytic strategies developed by Nature for RNA modification. However, some general principles are conserved from one species/reaction to another. For example, the catalytic Cys residue is used for activation of the U base in cmnm5U synthesis [32-34], while the activation of the exocyclic NH2 group in i6A synthesis takes place using an approach similar to exocyclic NH2-methylation [35]. As mentioned above, the [Fe-S] cluster protein TYW1 implicated in wybutosine biosynthesis uses the radical SAM catalytic reaction for formation of the cyclic intermediate imG-14 from initially formed m1G at position 37 in the tRNA anticodon loop [17]. The catalytic mechanisms of many other RNA-modification enzymes still remain to be elucidated in detail.
Major Reaction Steps in the RNA-Modification Process
Independently from the exact chemical structure of the modified residue, at the molecular level the whole enzymatic reaction of RNA modification can be tentatively split into at least four elementary steps:
– RNA substrate and cofactor (co-substrate) binding;
– conformational change of both RNA and protein to enter the target nucleotide into the active site;
– the “chemical” catalytic step;
– product release.
At every such elementary step, the amino acids of the enzyme enter in contact with the RNA chain via electrostatic, H-bond, and van der Waals interactions. Electrostatic interactions between negatively charged phosphate moieties in RNA and Lys and Arg residues in the protein are known to stabilize the transient RNA–protein complex and favor the subsequent conformational change of the RNA and enzyme. During this mutual structural adaptation between macromolecules, the target base is frequently stabilized in the active site by stacking interaction with an aromatic Phe or Tyr residue. During the catalytic step of the reaction, the amino acids of the enzyme active site contribute to changes in the distribution of electronic density and thus to activation of a given atom or a group for the modification reaction. In the last step, the modified RNA chain has to be released from the enzyme, and this dissociation step can represent a rate-limiting step in the reaction.
RNA Substrate Stabilization
Interaction between the RNA-modification enzyme and its cognate RNA substrate takes place at multiple point contacts required for efficient substrate binding and modification. These interactions can be nonspecific (e.g. strong electrostatic interactions with RNA phosphate residues) and specific, allowing the precise recognition of the RNA sequence. The specific interactions generally rely on the contacts between the base and polar amino acids in the active site via H-bonds. As mentioned above, the target base enters the enzyme active site due to important RNA conformational change and is stabilized via stacking interaction with the appropriate aromatic amino acid. Moreover, this stacking stabilization may also contribute to reactivity of the base during the catalytic step.
ENZYMATIC MECHANISMS OF RNA MODIFICATION
In the following paragraph, only the chemical aspects of mechanisms and catalysis are described, when they are known. These mechanisms have been discovered for the majority during the past decade via comprehensive mutational analysis and high-resolution crystal structures, which are reviewed elsewhere and will not be described here (for examples see [36-41]).
Chemical Properties of Nucleotides in an RNA Chain
An unmodified RNA transcript is composed of parental nucleotides A, C, G, and U incorporated during transcription. Phosphate residues in RNA molecules are not modified enzymatically, even if such possibility exists with strong alkylating reagents. The ribose moiety is naturally modified only at position of the 2′-OH group, and the vast majority of RNA-modification reactions touch different atoms (C, N, and O) in nucleobases (see Fig. 3). Depending on the exact chemical structure of the nucleobase, the relative reactivity of different cyclic and exocyclic positions can vary; however, some reactions are common for pyrimidine (C and U) or purine (A and G) bases. For example, the C5=C6 bond in pyrimidine bases exhibits similar reactivity to nucleophilic reagents, or the N7 position of A and G is a subject for alkylation [42].
Fig. 3. Chemical reactivity of nucleotides. Only the highly reactive centers, as shown by the use of chemical reagents, are represented. The nucleophilic centers are indicated by black circles, the electrophilic ones by stars, and those subject to oxidant by triangles.
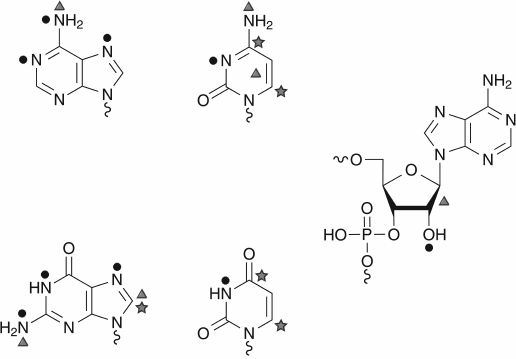
Carbon atoms at position 6 in the pyrimidine ring are the most reactive spots for nucleophilic reagents due to electronic density displacement towards carbonyl groups (O= at position 4 in U or position 2 in C). The most common nucleophilic reagents used in nucleic acid chemistry are bisulfite as well as hydrazine and hydroxylamine derivatives. In contrast, the C5 carbon is known to react with strong electrophilic reagents, like Br2, ICl (halogenation reactions) or Hg(OAc)2 (mercuration) [42].
Nitrogen atoms in pyrimidine rings (N3 in pyrimidines, N1 and N7 in purines) are common targets for chemical alkylation by electrophilic reagents or for addition reactions with an activated C=C bond (carbodiimides, acrylonitrile).
Exocyclic nitrogen atoms in A and C show important reactivity with aldehydes (e.g. formaldehyde), glyoxal, or nitrous acid (HNO2). Many of these reactive target atoms are used also for enzymatic modification of RNA molecules.
Another weak point in RNA (and DNA) is the N-glycosidic bond between the base and ribose (deoxyribose). At low pH, the N-glycosidic bonds are rapidly cleaved due to protonation of the base at N3 (in pyrimidines) and N7 in purines [42].
Mechanism of RNA-Methylation by Methyltransferases
All RNA:MTases proceed with direct or indirect transfer of a methyl group to a target atom resulting in C-methylation, O-methylation, or N-methylation. In the great majority of cases, the methyl group is derived from S-adenosyl-L-methionine (AdoMet/SAM) (Fig. 4b), and the reaction looks like an SN2 displacement mechanism involving attack of a nucleophile from the RNA substrate on the electrophilic methyl group of SAM with concomitant release of S-adenosyl-L-homocysteine (AdoHcy). A wide variety of mechanisms have evolved to activate the catalytic nucleophile atom dependent on its chemical properties. On one hand, due to their intrinsic nucleophilic character, methylations at N and O atoms involve a direct transfer of the methyl group, which can require a proton removal before, concurrent with, or after methyl transfer. The AdoMet conformation is also crucial to the mechanism of methyl transfer, which necessarily involves the precise juxtaposition of the electrophilic methyl group with a nucleophilic target atom. On the other hand, the sp2-hybridized carbons display electrophilic rather than nucleophilic character, preventing a direct transfer mechanism for C-methylations. In these cases, two strategies have evolved: either a two electron transfer reaction via a two-state mechanism with formation of a covalent enzyme–RNA intermediate, or a one electron transfer mechanism involving intermediate methylation of a cysteine residue.
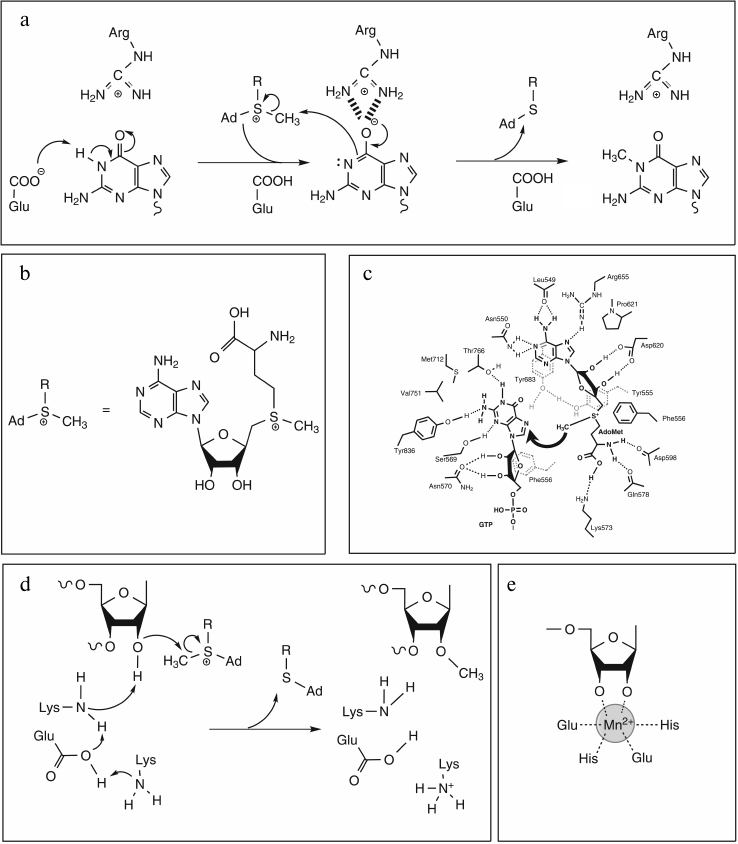
Fig. 4. Proposed mechanisms for N- and O-methylation reactions. a) N-methylation of guanine catalyzed by Trm5 (adapted from [43]). Proton abstraction by glutamate residue acting as general base yields deprotonated guanine with electron density shifted to O6 stabilized by the positively charged arginine residue. Nucleophilic attack on AdoMet by deprotonated N1 then leads to product formation. b) Chemical structure of AdoMet and its schematic representation. c) Schematic diagram of the active site of vaccinia virus mRNA cap N7-MTase (adapted from [45]) with putative interactions with AdoMet and cap (GTP) during methyl transfer reaction. Dashed lines indicate potential hydrogen bonds. Residues Tyr555 and Phe556 are located in the N-terminal lid peptide. d) 2′-O-methylation: proposed catalytic mechanism of nsp10 (adapted from [47]). The ε-amino group of the catalytic Lys, deprotonated by the concerted action of Glu and Lys residues, activates the 2′-OH group, which in turn can attack the electrophilic AdoMet methyl group. e) Chemical structure of the metal ion coordinated by the two OH groups of RNA substrate and four amino acid side chains in Hen1 active site (adapted from [51]).
Catalysis Strategies
These few examples highlight the great diversity of catalytic scenarios.
N-Methylation. Even if the mechanism of N-methylation seems to be universal, the catalysis does not appear to be restricted to one model. Indeed, the chemical mechanism being presumed to entail an in-line SN2-type displacement mechanism with the incoming nitrogen nucleophile and AdoHcy leaving group occupying apical positions in a putative pentavalent transition state of the methyl carbon. In this reaction, catalysis may intervene at different levels, not necessarily independent of each other: chemical catalysis, substrate proximity, orientation or transition-state stabilization.
One of the best studied examples is chemical catalysis used by Trm5 enzyme to methylate the N1 position of guanine. It has been shown by site-directed mutagenesis and kinetic approaches that a Glu residue located in the active site plays a major role in catalysis, probably as a general base. Another key residue, an Arg also located in the active site, is proposed to stabilize the oxyanion at O6 position of G (Fig. 4a) [43].
The N-MTases can also operate at the N7 atom of guanine. In these cases, methylation does not require the removal of a proton before methyl transfer, and consequently the developed catalytic strategy is different. Indeed, from site-directed mutagenesis studies on the vaccinia virus cap mRNA-modifying enzyme, catalysis has been proposed to be achieved via stabilization of the transition state by different interactions between amino acid side chains from the N-terminal lid peptide specific for these MTases and the substrates. These interactions implicate in particular two aromatic residues (one Tyr and one Phe), which provide an electron-rich environment surrounding the two cationic centers (N7 and Sγ) in the predicted SN2 transition state [44, 45] (Fig. 4c).
Moreover, a similar catalytic strategy seems to be developed by the ErmC′ enzyme, which catalyzes the N-methylation of the N6 atom of adenine despite the requirement for a proton abstraction in the reaction. Indeed, from mutagenesis approaches, it has been shown that only an active site Tyr residue implicated in base stabilization is indispensable [46].
O-Methylation. The catalysis is essentially devoted to facilitating the removal of a proton from the RNA OH-group, an essential step for this SN2-type displacement mechanism, via a general base catalytic mechanism, as is the case for nsp10 and VP39 enzymes, for examples. From structural and site-directed mutagenesis studies, it has been proposed than a Lys residue activates the 2′-OH group. The Lys ε-amino group is proposed to be itself activated by a concerted mechanism involving Glu and Lys (nsp10) or Asp and Arg (VP39) residues. Catalysis of nsp10 should also implicate an Asp residue probably involved in the transition state stabilization via electrostatic interactions [47, 48] (Fig. 4d).
Nevertheless, as often is the case, evolution has found a different way to achieve the specific 2′-O-methylation at the 3′-terminal nucleotide in RNA chain. This mechanism necessitates the presence of an essential metal cation, which can be Mg2+ or Mn2+, and it represents a unique case in which the catalytic strategy directs substrate specificity. Indeed, the metal has to be coordinated by the two OH groups of the ribose substrate (2′- and 3′-) and four strictly conserved residues (two Glu and two His) (Fig. 4e). The fact that metal coordination implicates the 3′-OH explains the specificity of Hen1 enzyme for methylation only at 3′-terminal nucleotides [49-51]. The metal-dependent mechanism of 2′-O-methylation remains to be elucidated.
C-Methylation. Methylations at C5 to produce 5-methyluridine (ribothymidine) and 5-methylcytosine implicate Michael addition at the C6 ring position by a nucleophilic sulfhydryl group of the enzyme to generate a covalent intermediate. Formation of this covalent Michael adduct leads to activated C5 of the nucleotide and results in methyl transfer from the SAM cofactor. Then base abstraction of a proton from C5 leads to formation of the product. In terms of catalysis, these reactions use a general base to facilitate the β-elimination, and this base has been shown to be a carboxylic acid group for m5U (Glu residue) or a thiolate for m5C (Cys residue) (Fig. 5a) [36, 52-54].
Fig. 5. Proposed mechanisms for C-methylation reactions. a) Mechanism and catalysis of m5C formation (adapted from [52]). The reaction proceeds through the nucleophilic attack at C6 of cytosine by the thiolate group of a catalytic Cys residue. Formation of the covalent Michael adduct leads to activated C5 of the nucleoside and results in methyl transfer from the reactive methylthiol of the AdoMet cofactor. Another Cys residue acts as a general base in the β-elimination step. b) Mechanism of m8C formation (adapted from [56]). The reaction consumes two AdoMet equivalents to support both methyl transfer to catalytic Cys residue (step 1) and subsequently generation of the 5′-deoxyadenosine (5′dA, step 2).
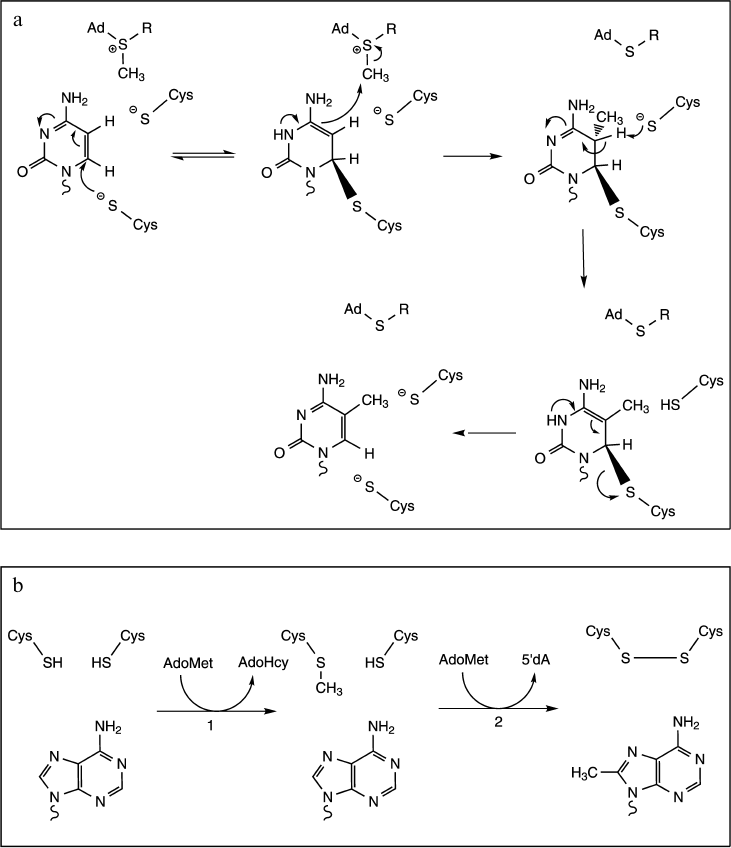
It is noteworthy that in the case of C5 methylation by m5U-MTase TrmFO, the methyl group is transferred from 2,10-methylene-tetrahydrofolate and not from SAM. However, the mechanism is quite similar to that with SAM, with: (1) a covalent catalysis via formation of a thiolate covalent adduct on C6 to activate C5 in the nucleobase and to facilitate its methylation, and (2) a general base catalysis by another Cys residue to facilitate β-elimination [13, 55].
Finally, methylations at purine carbon atoms C2 and C8 have recently been shown to be catalyzed by radical SAM enzymes RlmN and Cfr. In both cases, the methyl group is transferred by a ping-pong mechanism involving intermediate methylation of a catalytic Cys residue. Indeed, each turnover requires two molecules of AdoMet. The first molecule is used to methylate a Cys residue via a SN2 mechanism, while the second one reacts with the [4Fe-4S] cluster to generate a 5′-deoxyadenosine radical. Finally, the 5′-deoxyadenosine radical reacts with the methyl cysteine thioether to generate an oxidative radical intermediate that facilitates methyl transfer to the nonreactive C8 or C2 of adenine (Fig. 5b) [16, 56].
Mechanism of Pseudouridine Synthases
The conversion of uridine to pseudouridine is catalyzed by different families of RNA:-ψ-synthases; they all employ the same mechanism involving an essential Asp residue as a nucleophile [21, 57]. This mechanism implies the break of the N-glycosidic bond between the uracil ring and the ribose leading to the formation of a covalent intermediate on the Asp residue, followed by a rotation of the retained base in the enzyme active site and further re-attachment of the same base via a C–C bond to ribose. These enzymes have been identified and extensively studied for a long time, however the exact molecular mechanism of the isomerization reaction is still poorly understood. Indeed, two alternative mechanisms, differing notably in the nature of the covalent intermediate formed after the nucleophilic attack of the catalytic carboxylate group, are still debated [58]. In the first one, the carboxylate group attacks the C6 of the uracil ring leading to the formation of a Michael adduct intermediate and to the subsequent break of the glycosidic bond (Fig. 6a). The second one involves a nucleophilic attack on C1′ of the ribose to liberate the anion of uracil and generate an acyl intermediate (Fig. 6b). In both cases, the generation of the pseudouridine requires a final deprotonation step of C5 and protonation at N1. It is noteworthy that a third alternative mechanism implying a glycal intermediate could exist, at least for the reaction of TruB enzyme with the 5-fluorouridine analog [59].
Fig. 6. Proposed mechanisms for cleavage of the C–N glycosidic bond. a) ψ-Generation via formation of a Michael adduct by nucleophilic attack of the active site Asp residue on C6 of U (adapted from [20]). The glycosidic bond breaks and the tethered uracil rotates around the ester bond, and the new C–C bond forms. The aspartate then departs as a leaving group, the C5 is deprotonated, and the N1 is protonated. b) ψ-Generation via formation of an acylal intermediate by nucleophilic attack of the active site Asp residue at C1′ of U (adapted from [20]). The tightly bound uracilate anion then rotates around an axis perpendicular to the ring, and the new C–C bond forms with the aspartate as the leaving group. The subsequent deprotonation of C5 and protonation of N1 yield ψ. c) The TGT reaction proceeds through a nucleophilic attack of the catalytic aspartyl carboxylate at the C1′ of the ribose to give a covalent TGT–RNA intermediate. After guanine exchange for the modified base, nucleophilic attack of the base N9 at the glycosidic carbon generates the modified product.
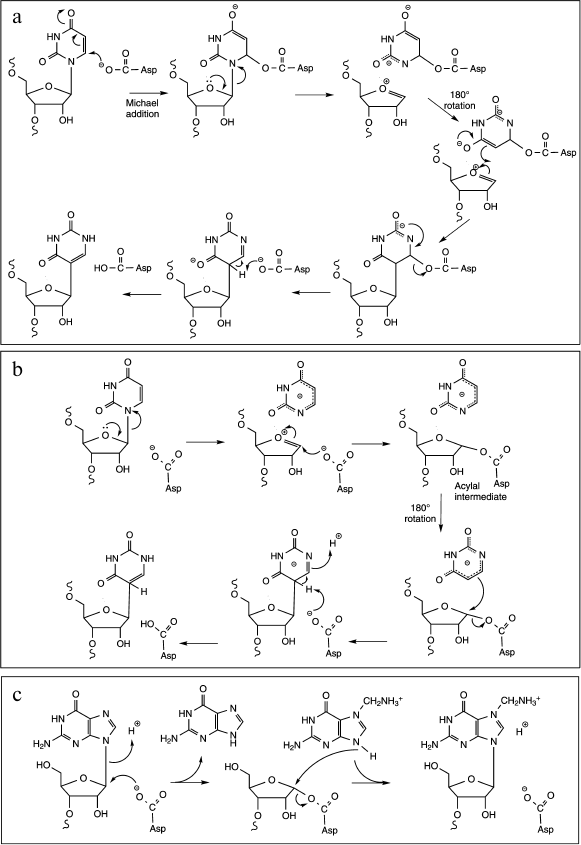
In term of catalysis, the conversion of a uridine residue to pseudouridine, the resulting product is thermodynamically more stable than uridine, thus the reaction does not consume energy. However, the activation of the C–N bond for cleavage requires an important energy input coming from protein conformational change during a catalytic (or pre-catalytic step). In some aspects the first step of U-to-ψ isomerization may resemble cleavage of the C–N glycosidic bond by DNA-glycosylases during the initial steps of DNA repair or to base exchange reaction catalyzed by TGT (tRNA-guanine transglycosylases, see below). Detailed studies of the kinetics of U-to-ψ conversion were performed on various RNA:ψ-synthases, but it cannot be excluded that the exact catalytic mechanism varies from one class to another, and thus the data cannot be easily extrapolated from one enzyme to another. Moreover, the essential Asp residue has been proposed to play the role of general acid/base catalysis involved in C5 deprotonation, while the specific amino acid for protonation of N1 is still unknown. More work is required to reach a conclusion [60, 61].
Mechanism of tRNA-Guanine Transglycosylases
The tRNA-guanine transglycosylases (TGT) catalyze a base-exchange reaction that involves the cleavage of a guanosine N-glycosidic bond and utilize a double-displacement mechanism. The TGT reaction proceeds through a nucleophilic attack of the catalytic aspartyl carboxylate at the C1′ of the ribose to give a covalent TGT–RNA intermediate. After guanine exchange for the modified base, nucleophilic attack of the base N9 at the glycosidic carbon generates the modified product (Fig. 6c) [24, 62]. In terms of catalysis, the TGT mechanism requires the protonation of the displaced guanine and the deprotonation of the incoming heterocyclic base. This role seems to be fulfilled by a second Asp residue acting via a water molecule proton relay as a general acid-base [25]. Moreover, recent kinetic data support a role of the 2′-OH group in stabilizing the growing negative charge on the nucleophilic aspartate as the glycosidic bond is broken during the breakdown of the covalent complex [63].
Mechanism of s4U-Thiolase ThiI
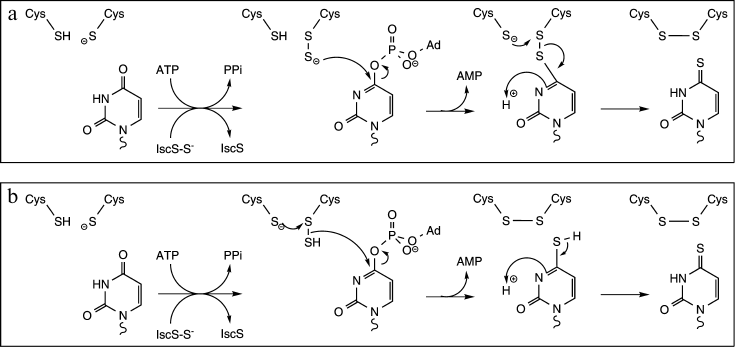
ThiI catalyzes the transfer of a sulfur atom from a donor molecule to position 4 of uridine through the formation of a persulfide intermediate (Fig. 7) [29]. In most bacteria, the donor is the persulfide form of the cysteine desulfurase IscS [40]. IscS is a pyridoxal-5′-phosphate-dependent cysteine desulfurase that forms a persulfide group on an active site Cys at the expense of free Cys. The terminal sulfur is then transferred to ThiI, where it resides on the catalytically essential Cys residue [30]. In methanogenic archaea, IscS is not essential for thiouridine synthesis and S2– has been shown to be a physiologically relevant sulfur donor [64]. The reaction also requires the presence of activated uridine, which is also generated by ThiI at the expense of ATP [65]. The last part of the reaction consists of the transfer of sulfur from the ThiI persulfide intermediate to the activated uridine residue leading to the formation of thiouridine and of a disulfide bond on ThiI. Reduction of that disulfide bond regenerates ThiI for another round of catalysis [30].
Fig. 7. Proposed mechanism for thiouridine formation catalyzed by ThiI (adapted from [30]). ThiI catalyzes the transfer of the terminal sulfur atom of a persulfide form of IscS (IscS–S–) to the catalytically essential Cys residue leading to the formation of the ThiI persulfide intermediate. ThiI displays also an ATP pyrophosphatase activity for the adenylation of uridine. Two mechanisms have been proposed for the last part of the reaction: a) the terminal sulfur of the ThiI persulfide intermediate directly attacks the C4 of activated uridine, or (b) ThiI generates via intradisulfide bond formation hydrogen sulfide, which subsequently serves as a nucleophile to displace the activated oxygen of uridine.
CONCLUSION
RNA modifications represent a complex and important field of research regarding the vast number of enzymes, chemistry, and catalytic mechanisms. Regarding the involved chemical mechanisms, their fine characterization remains often partial despite extensive development of mutagenesis and chemical and structural methods, and further work is required to complete their description. Moreover, with the exception of a few cases, the kinetic characterization, including identification of the rate-limiting step, remains limited, and this may constitute an obstacle to the understanding of the mechanisms. For example, analysis of rate-limiting steps of U-to-ψ conversion was performed using diverse bacterial and archaeal enzymes by steady-state and pre-steady state experiments, and in all cases the rate-limiting step has been shown to be associated with the catalysis step [24]. But this is not always the case. Indeed, pre-steady-state analysis of the N-methylation catalyzed by two analogous Trm5 and TrmD has shown that they display distinct rate-limiting steps, which can be correlated with observed structural differences [66]. At least, the simple observation of a covalent complex is not sufficient to prove intermediacy. To be a true intermediate, a complex must be both chemically and kinetically competent, as it was shown in the case of the TGT [63]. Another aspect, which has not been extensively studied, concerns the efficiency of the different enzymes in vitro and in vivo. Despite the fact that an enzyme is able to catalyze a specific reaction in vitro, its relevance in vivo could depend on its efficiency.
REFERENCES
1.Hocine, S., Singer, R. H., and Grunwald, D. (2010)
Cold Spring Harbor Perspect. Biol., 2, a000752.
2.Lamond, A. I. (1991) Curr. Opin. Cell Biol.,
3, 493-501.
3.Machnicka, M. A., Milanowska, K., Osman Oglou, O.,
Purta, E., Kurkowska, M., Olchowik, A., Januszewski, W., Kalinowski,
S., Dunin-Horkawicz, S., Rother, K. M., et al. (2013) Nucleic Acids
Res., 41, D262-267.
4.Helm, M. (2006) Nucleic Acids Res.,
34, 721-733.
5.Motorin, Y., and Helm, M. (2011) Wiley
Interdiscip. Rev. RNA, 2, 611-631.
6.Motorin, Y., and Helm, M. (2010)
Biochemistry, 49, 4934-4944.
7.Ansmant, I., and Motorin, I. (2001) Mol.
Biol., 35, 248-267.
8.Schubert, H. L., Blumenthal, R. M., and Cheng, X.
(2003) Trends Biochem. Sci., 28, 329-335.
9.Kozbial, P. Z., and Mushegian, A. R. (2005) BMC
Struct. Biol., 5, 19.
10.Watanabe, K. (2005) J. Biol. Chem.,
280, 10368-10377.
11.Watanabe, K., Nureki, O., Fukai, S., Endo, Y.,
and Hori, H. (2006) J. Biol. Chem., 281, 34630-34639.
12.Cheng, X., and Roberts, R. J. (2001) Nucleic
Acids Res., 29, 3784-3795.
13.Hamdane, D., Argentini, M., Cornu, D.,
Myllykallio, H., Skouloubris, S., Hui-Bon-Hoa, G., and
Golinelli-Pimpaneau, B. (2011) J. Biol. Chem., 286,
36268-36280.
14.Hutcheson, R. U., and Broderick, J. B. (2012)
Metallomics, 4, 1149-1154.
15.Vey, J. L., and Drennan, C. L. (2011) Chem.
Rev., 111, 2487-2506.
16.Grove, T. L., Benner, J. S., Radle, M. I., Ahlum,
J. H., Landgraf, B. J., Krebs, C., and Booker, S. J. (2011)
Science, 332, 604-607.
17.Noma, A., Kirino, Y., Ikeuchi, Y., and Suzuki, T.
(2006) EMBO J., 25, 2142-2154.
18.Hamma, T., and Ferre-D’Amare, A. R. (2006)
Chem. Biol., 13, 1125-1135.
19.Charette, M., and Gray, M. W. (2000) IUBMB
Life, 49, 341-351.
20.Mueller, E. G., and Ferre-D’Amare, A. R.
(2009) in DNA and RNA Modification Enzymes: Structure, Mechanism,
Function and Evolution, Landes Biosciences, Austin, USA, pp.
363-376.
21.Ramamurthy, V., Swann, S. L., Paulson, J. L.,
Spedaliere, C. J., and Mueller, E. G. (1999) J. Biol. Chem.,
274, 22225-22230.
22.Motorin, Y., Keith, G., Simon, C., Foiret, D.,
Simos, G., Hurt, E., and Grosjean, H. (1998) RNA, 4,
856-869.
23.Behm-Ansmant, I., Urban, A., Ma, X., Yu, Y.-T.,
Motorin, Y., and Branlant, C. (2003) RNA, 9,
1371-1382.
24.Kittendorf, J. D. (2003) J. Biol. Chem.,
278, 42369-42376.
25.Kittendorf, J. D., Barcomb, L. M., Nonekowski, S.
T., and Garcia, G. A. (2001) Biochemistry, 40,
14123-14133.
26.Chervin, S. M., Kittendorf, J. D., and Garcia, G.
A. (2007) Methods Enzymol., 425, 121-137.
27.Stengl, B., Reuter, K., and Klebe, G. (2005)
Chembiochem, 6, 1926-1939.
28.Rajakovich, L. J., Tomlinson, J., and Dos Santos,
P. C. (2012) J. Bacteriol., 194, 4933-4940.
29.Palenchar, P. M., Buck, C. J., Cheng, H., Larson,
T. J., and Mueller, E. G. (2000) J. Biol. Chem., 275,
8283-8286.
30.Mueller, E. G., Palenchar, P. M., and Buck, C. J.
(2001) J. Biol. Chem., 276, 33588-33595.
31.Arragain, S., Handelman, S. K., Forouhar, F.,
Wei, F.-Y., Tomizawa, K., Hunt, J. F., Douki, T., Fontecave, M.,
Mulliez, E., and Atta, M. (2010) J. Biol. Chem., 285,
28425-28433.
32.Meyer, S., Scrima, A., Versees, W., and
Wittinghofer, A. (2008) J. Mol. Biol., 380, 532-547.
33.Armengod, M.-E., Moukadiri, I., Prado, S.,
Ruiz-Partida, R., Benitez-Paez, A., Villarroya, M., Lomas, R., Garzon,
M. J., Martinez-Zamora, A., Meseguer, S., et al. (2012)
Biochimie, 94, 1510-1520.
34.Osawa, T., Ito, K., Inanaga, H., Nureki, O.,
Tomita, K., and Numata, T. (2009) Structure, 17,
713-724.
35.Zhou, C., and Huang, R. H. (2008) Proc. Natl.
Acad. Sci. USA, 105, 16142-16147.
36.Hou, Y.-M., and Perona, J. J. (2010) FEBS
Lett., 584, 278-286.
37.Guelorget, A., and Golinelli-Pimpaneau, B. (2011)
Structure, 19, 282-291.
38.Iwata-Reuyl, D. (2003) Bioorg. Chem.,
31, 24-43.
39.Spedaliere, C. J., Ginter, J. M., Johnston, M.
V., and Mueller, E. G. (2004) J. Am. Chem. Soc., 126,
12758-12759.
40.Mueller, E. G. (2006) Nat. Chem. Biol.,
2, 185-194.
41.Motorin, Y., Lyko, F., and Helm, M. (2010)
Nucleic Acids Res., 38, 1415-1430.
42.Behm-Ansmant, I., Helm, M., and Motorin, Y.
(2011) J. Nucleic Acids, 2011, 408053.
43.Christian, T., Lahoud, G., Liu, C., Hoffmann, K.,
Perona, J. J., and Hou, Y.-M. (2010) RNA, 16,
2484-2492.
44.Zheng, S., and Shuman, S. (2008) RNA,
14, 2297-2304.
45.De la Pena, M., Kyrieleis, O. J. P., and Cusack,
S. (2007) EMBO J., 26, 4913-4925.
46.Maravic, G., Feder, M., Pongor, S., Flogel, M.,
and Bujnicki, J. M. (2003) J. Mol. Biol., 332,
99-109.
47.Decroly, E., Debarnot, C., Ferron, F., Bouvet,
M., Coutard, B., Imbert, I., Gluais, L., Papageorgiou, N., Sharff, A.,
Bricogne, G., et al. (2011) PLoS Pathog., 7,
e1002059.
48.Hodel, A. E., Gershon, P. D., and Quiocho, F. A.
(1998) Mol. Cell, 1, 443-447.
49.Chan, C. M., Zhou, C., Brunzelle, J. S., and
Huang, R. H. (2009) Proc. Natl. Acad. Sci. USA,
106, 17699-17704.
50.Tkaczuk, K. L., Obarska, A., and Bujnicki, J. M.
(2006) BMC Evol. Biol., 6, 6.
51.Huang, R. H. (2012) Biochemistry,
51, 4087-4095.
52.Foster, P. G., Nunes, C. R., Greene, P.,
Moustakas, D., and Stroud, R. M. (2003) Structure, 11,
1609-1620.
53.Lee, T. T., Agarwalla, S., and Stroud, R. M.
(2005) Cell, 120, 599-611.
54.Lee, T. T., Agarwalla, S., and Stroud, R. M.
(2004) Structure, 12, 397-407.
55.Nishimasu, H., Ishitani, R., Yamashita, K.,
Iwashita, C., Hirata, A., Hori, H., and Nureki, O. (2009) Proc.
Natl. Acad. Sci. USA, 106, 8180-8185.
56.Challand, M. R., Salvadori, E., Driesener, R. C.,
Kay, C. W. M., Roach, P. L., and Spencer, J. (2013) PloS One,
8, e67979.
57.Huang, L., Pookanjanatavip, M., Gu, X., and
Santi, D. V. (1998) Biochemistry, 37, 344-351.
58.Gu, X., Liu, Y., and Santi, D. V. (1999) Proc.
Natl. Acad. Sci. USA, 96, 14270-14275.
59.Miracco, E. J., and Mueller, E. G. (2011) J.
Am. Chem. Soc., 133, 11826-11829.
60.Hamilton, C. S., Spedaliere, C. J., Ginter, J.
M., Johnston, M. V., and Mueller, E. G. (2005) Arch. Biochem.
Biophys., 433, 322-334.
61.Phannachet, K., Elias, Y., and Huang, R. H.
(2005) Biochemistry, 44, 15488-15494.
62.Romier, C., Reuter, K., Suck, D., and Ficner, R.
(1996) Biochemistry, 35, 15734-15739.
63.Garcia, G. A., Chervin, S. M., and Kittendorf, J.
D. (2009) Biochemistry, 48, 11243-11251.
64.Liu, Y., Zhu, X., Nakamura, A., Orlando, R.,
Soll, D., and Whitman, W. B. (2012) J. Biol. Chem., 287,
36683-36692.
65.You, D., Xu, T., Yao, F., Zhou, X., and Deng, Z.
(2008) Chembiochem, 9, 1879-1882.
66.Zhou, J., Lv, C., Liang, B., Chen, M., Yang, W.,
and Li, H. (2010) J. Mol. Biol., 401, 690-695.
67.Wright, J. R., Keffer-Wilkes, L. C., Dobing, S.
R., and Kothe, U. (2011) RNA, 17, 2074-2084.