REVIEW: AGR2, ERp57/GRP58, and Some Other Human Protein Disulfide Isomerases
S. S. Shishkin*, L. S. Eremina, L. I. Kovalev, and M. A. Kovaleva
Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; E-mail: shishkin@inbi.ras.ru* To whom correspondence should be addressed.
Received July 25, 2013
This review considers the major features of human proteins AGR2 and ERp57/GRP58 and of other members of the protein disulfide isomerase (PDI) family. The ability of both AGR2 and ERp57/GRP58 to catalyze the formation of disulfide bonds in proteins is the parameter most important for assigning them to a PDI family. Moreover, these proteins and also other members of the PDI family have specific structural features (thioredoxin-like domains, special C-terminal motifs characteristic for proteins localized in the endoplasmic reticulum, etc.) that are necessary for their assignment to a PDI family. Data demonstrating the role of these two proteins in carcinogenesis are analyzed. Special attention is given to data indicating the presence of biomarker features in AGR2 and ERp57/GRP58. It is now thought that there is sufficient reason for studies of AGR2 and ERp57/GRP58 for possible use of these proteins in diagnosis of tumors. There are also prospects for studies on AGR2 and ERp57/GRP58 leading to developments in chemotherapy. Thus, we suppose that further studies on different members of the PDI family using modern postgenomic technologies will broaden current concepts about functions of these proteins, and this will be helpful for solution of urgent biomedical problems.
KEY WORDS: protein disulfide isomerases, detection in tumoral cells, markers of cancerDOI: 10.1134/S000629791313004X
Abbreviations: ER, endoplasmic reticulum; NCBI, the USA National Center of Information in Biotechnology; PCR, polymerase chain reaction; PDI, protein disulfide isomerase; SNP’s, single nucleotide polymorphisms.
During the last five years, two proteins of the human protein disulfide
isomerase (PDI) family, which includes about 20 proteins involved in
numerous physiological and pathological processes [1-4], have attracted special
attention. The first of these proteins is the atypical for this family
protein AGR2 (Anterior Gradient homolog 2, also known as AG2, hAG-2,
GOB-4, etc.) (after [5], NCBI Protein,
etc.). The other protein is a typical protein disulfide
isomerase – isoform A3 (PDIA3) or the protein ERp57/GRP58
[6]. This enzyme is characterized by its ability to
catalyze formation, decomposition, and isomerization of disulfide bonds
in protein molecules.
According to NCBI PubMed, more than a hundred papers about each of these two proteins have been published within the last five years. This interest seems to be largely due to the fact that in many of studies both AGR2 and ERp57/GRP58 have been found to participate in molecular processes promoting proliferation of tumor cells. Therefore, it seems reasonable to consider these proteins as potential biomarkers of tumors and as molecular targets for chemotherapy [5-7].
The role of AGR2 and ERp57/GRP58 in various functionally important processes in humans and in other vertebrates – from ontogenetic changes and apoptosis to provision of folding and stress-resistance – is still under active study in many laboratories. Taken together, the accumulated data suggest that these proteins can be considered as being polyfunctional [5-8] and, consequently, as very promising for further studies. The detection of different manifestations of biochemical polymorphism of AGR2 and ERp57/GRP58, including the existence of closely related proteins-members of the PDI family such as AGR3 [9] and also PDI and PDIp (products of the PDIA1 and PDIA2 genes, respectively) [1], is also an argument in favor of this position.
Other members of the PDI family have also been shown to contribute to the control of cell proliferation and to carcinogenesis when it is disordered [1, 10, 11].
Thus, taking into account the great importance of the PDI family members (first of all AGR2 and ERp57/GRP58 proteins) for the solution of many biomedical problems, it seems urgent to consider the main data of recent studies on these proteins and to analyze their possible implications.
GENERAL CHARACTERISTICS OF THE FAMILY OF HUMAN PROTEIN DISULFIDE
ISOMERASES
Most authors refer to the PDI family proteins that significantly differ in size (from 19 to 90 kDa) and in some functional features, in particular, some of them are active disulfide isomerases, although others do not have such activity [1-4, 12]. These proteins have in common an important structural feature, one or several so-called thioredoxin-like domains, and also similar intracellular localization – in the endoplasmic reticulum (ER) and/or in other membrane formations.
These criteria were formulated in one of the first detailed reviews concerning the PDI family [1] and were then used in further publications [4, 12].
In turn, the PDI family is considered as a part of the thioredoxin superfamily, various members of which contain in their polypeptide chains a peculiar structural block or module (~100-120 amino acid residues (a.a.)) similar to a small protein thioredoxin in amino acid sequence including a specific motif of 4 a.a. (Cys-X-X-Cys) [13, 14].
Three-dimensional models of the thioredoxin-like domains indicate that they are the most important modules in the structure of different proteins of the PDI family, usually consisting of five β-layers alternating with four α-helical regions (β-α-β-α-β-α-β-β-α), and the α-helical region on the whole are located in the periphery relative to the centrally located β-layers [13, 15].
Numerous representatives of the thioredoxin superfamily were initially found in prokaryotic organisms but were later also found in eukaryotes including humans. In the human body, these proteins are responsible for many important functions associated with redox reactions (i.e. they are oxidoreductases) [13, 14]. Moreover, many thioredoxin superfamily proteins have features of chaperons that are believed to be just due to the presence of the thioredoxin-like domains in their structure. Thioredoxin itself has been shown to have various functions [16, 17], among which nucleotide reductase activity deserves special attention because it is responsible for the generation of dinucleotides and, consequently, is vital for biosynthesis of DNA. Thus, it is obvious that studies on the thioredoxin superfamily proteins, including those of the PDI family, may be a promising approach for understanding of molecular processes fundamentally important for vital activity and, in particular, leading to production of different functionally active polymers.
To characterize general features of the PDI family taking into account the presence in this family of members with significantly different parameters, it seems reasonable to subdivide the proteins under consideration into three main subgroups and some unusual representatives. General plans of the structure of individual proteins from these subgroups are presented in Fig. 1 (see color insert).
Fig. 1. General schemes of structural organization of proteins from three main subgroups of the human PDI family (PDIA1 as a representative of typical protein disulfide isomerases; ERdj5 as an ER protein; TMX3 as a transmembrane protein), and also unusual members of this family exemplified by ERp18. Calculated values of molecular weight in kDa are given without consideration of signaling peptides. a and a′, thioredoxin-like domains having enzymatic activity; b and b′, thioredoxin-like domains with similar structure but lacking enzymatic activity.
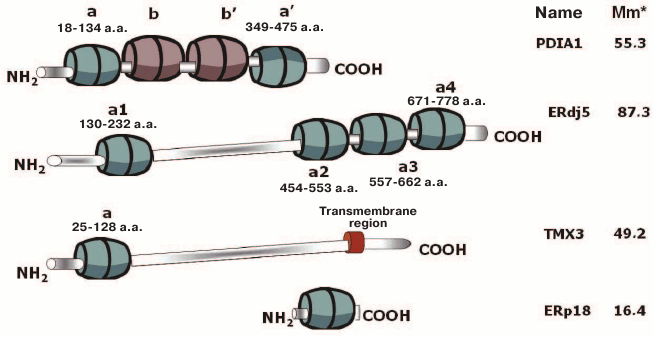
Naturally, the first subgroup is represented by six typical PDIs – proteins with molecular weight in the range of 50-70 kDa, each as a rule containing two thioredoxin-like domains (usually designated as a and a′) and two (b and b′) domains with a similar structure but lacking the enzymatic activity.
Protein disulfide isomerase A1 (also designated as PDI, PDIA1 cellular thyroid hormone-binding protein, prolyl 4-hydroxylase subunit beta, ERBA2L, and p55, after P07237 UniProt) can be considered as a classic example and typical representative of the first PDI family subgroup. This enzyme catalyzes the formation, decomposition, and isomerization of disulfide bonds in protein molecules, has features of a chaperone, acts as a subunit in the prolyl-4-hydroxylase complex, can bind thyroid hormones, and seems to be involved in the transport of triacylglycerols [12, 18, 19]. Its structure is schematically shown in Fig. 1.
Three proteins of this subgroup (PDIp, ERp57/GRP58, and PDILT) have a very similar scheme of structure, whereas each of the proteins ERp72 and PDIr has three thioredoxin-like domains having the enzymatic activity (after [1] and UniProt). And ERp72, similarly to other representatives of this subgroup, also retains two thioredoxin-like domains lacking enzymatic activity. Correspondingly, the molecular weight of ERp72 is about 20% higher than of the other proteins, whereas the PDIr molecular weight is typical because it has only one enzymatically inactive thioredoxin-like domain.
Thus, these PDIs seem to exemplify the “block building” principle, which has provided formation during evolution of a wide variety of proteins in higher eukaryotes.
The second subgroup comprises four ER proteins (ERdj5, ERp5, ERp46, and ERp44) that contain only enzymatically active thioredoxin-like domains [1, 4, 12]. The ERdj5 protein has four such domains (Fig. 1), ERp46 three, ERp5 two, and the ERp44 protein has one such domain.
The third subgroup is represented by four transmembrane proteins (designated TMX and TMX2-4) with structure including one thioredoxin-like domain and a hydrophobic transmembrane region (after [1] and Q9H3N1, Q9Y320, Q96JJ7, Q9H1E5 UniProt). In Fig. 1, the scheme of the TMX3 structure is given as an example.
The proteins ERp27, Erp28 (ERp29), and also ERp18 (thioredoxin domain-containing protein 12) with closely related AGR2 and AGR3 can be considered as unusual members of the PDI family. These proteins have markedly lower molecular weight (<30 kDa) than other PDI family members and contain one thioredoxin-like domain, except for ERp27, which has two functionally inactive such domains (b and b′) [1, 5, 12, 20]. But it should be noted that ERp27 is able to rather specifically bind with ERp57/GRP58.
In humans there are at least two main isoforms of thioredoxin (the cytoplasmic/nuclear Trx1 and mitochondrial Trx2), which are encoded by different genes (TXN and TXN2) [16, 17] and also numerous proteins containing thioredoxin-like domains, including the PDI family members. Their biochemical polymorphism displays various manifestations caused by alternative splicing, amino acid substitutions, and postsynthetic modifications. In particular, TXN gene expression due to alternative splicing is associated with generation of isoforms 1 and 2 (after P10599 UniProt). Examples of the biochemical polymorphism of some proteins of the PDI family will be presented further. Nevertheless, functionally important regions in the thioredoxin-like domains are rather conserved, which is obvious on comparing fragments of amino acid sequences of thioredoxins and thioredoxin-like domains with characteristic motifs inherent in some typical members of the human PDI family (Fig. 2; see color insert).
Fig. 2. Fragments of amino acid sequences from the main human thioredoxins and some members of the PDI family, which include characteristic (Cys-X-X-Cys) or changed motifs consisting of 4 a.a. responsible for enzymatic activity (enclosed in frame and shown by yellow marker). Hydrophobic amino acid residues flanking these motifs from the N-terminal side and shown with gray marker, whereas positively charged lysine and arginine situated on the C-terminal side are shown in blue.

Four proteins under study (PDIA1, ERp57/GRP58, ERp46, and TMX3) from different subgroups but possessing protein disulfide isomerase activity have in the structure of their thioredoxin-like domains characteristic motifs (CGHC) flanked from the N-terminal side with a tryptophan residue (W). Their C-terminal sequence virtually always begins with a positively charged lysine residue, which may be followed by another positive residue (K or R). An exception is ERp46, which in this position has Q followed by positively charged R. Note that on the N-terminal side from the motifs there are blocks of some hydrophobic amino acid residues.
Two unusual members of the PDI family (ERp27 and AGR2) have in the corresponding regions of the amino acid sequences only one cysteine residue. But the motif in the AGR2 sequence (CPHS) was somewhat like the motifs of the functionally active domains in some typical representatives of the PDI family. Thus, protein ERdj5 in the motif of the second functional domain after the obligatory C has P in the second position (after [1] and Q8IXB1 UniProt). The third position is occupied by H in motifs of some typical PDI family proteins (in particular, in those presented in Fig. 2). Finally, S was detected in the fourth position in the motif CRFS of the functionally active domain of the human protein ERp44 ([21] and Q9BS26 UniProt). Note that the CRFS motif is found not only in the human AGR2, but also in sequences of different proteins, PDI family members of other eukaryotes, and these proteins retained the catalytic activity but on a relatively lower level [21, 22]. Moreover, the amino acid sequence of AGR2 from the N-terminal side of the isolated motif CPHS contains a block of hydrophobic amino acid residues, whereas the C-terminal sequence contains the tandem KK. Thus, this similarity can be considered as an argument in favor for AGR2 membership in the PDI family.
HUMAN PROTEIN AGR2 IS AN UNUSUAL REPRESENTATIVE OF THE FAMILY OF
PROTEIN DISULFIDE ISOMERASES: MAIN FEATURES, BIOCHEMICAL
POLYMORPHISM
The protein AGR2 and its close relative AGR3 were assigned to the PDI family later than all other members [22]. However, the most convincing evidences in favor of its belonging to this family were obtained afterwards. Among these data, the fundamental role seems to be played by findings of Park et al. [23]. They succeeded in their studies on mice to detect an unusual catalytic activity of AGR2: it could interact with mucin 2 (MUC2), a large glycoprotein rich in cysteine residues. The cysteine residue in the thioredoxin-like domain of AGR2 was shown to form a heterodisulfide bond with cysteine residues in the MUC2 molecule. This reaction is a prerequisite for secretion of MUC2 by intestinal epithelium cells that allows this protein perform its protective function in the intestine.
Data on AGR2 and AGR3 localization in ER also correlate with their membership in the PDI family [5, 23]. It was recently shown that this feature of AGR2 is determined by a special C-terminal KTEL motif, the complete removal of which or amino acid substitutions in it affecting the protein functions [24]. However, in other members of the PDI family the corresponding motifs (on function retention) have another structure (KDEL, KVEL, etc.) [1]. In total, some NCBI databases designate protein AGR2 (and its gene) as the 17th member of the PDI family (protein disulfide isomerase family A, member 17) and protein AGR3 as the 18th member (protein disulfide isomerase family A, member 18).
Note that studies on the human AGR2 protein were preceded by finding in 1998 of the corresponding gene in the clawed frog Xenopus laevis [25]. It is responsible for normal development of ectodermal cells during early embryogenesis designated by the authors as XAG-2, and its protein product was characterized as a secreted protein. In the same year, a homologous gene (initially named hAG-2) was found in the human genome, and this gene was actively functioning in cultured cells of mammary gland cancer (results of analysis of the corresponding cDNA-library) [26]. Concurrently, in December 1998, information from Zhang and Smith was received by the joined database (EMBL/GenBank/DDBJ databases) about an increased expression of this gene in tumors, in particular, in prostate cancer. This information was not published but was registered (AF115926.1; GI:17998665 GenBank). Thus, this information seems to be the first report about a probable increase in production of the AGR2 protein in prostate cancer.
By now, the gene encoding the human AGR2 protein is characterized in detail (10551 Gene NCBI). It is localized in the 7p21.1 region, occupies about 26 kb, contains seven exons, and is neighboring to the gene encoding AGR3 ([27, 28] and 606358 OMIM NCBI), which allows us to consider these genes (and the encoded proteins) as an example of polylocus polymorphism.
There are data on the possible presence in gene AGR2 of altogether about 500 SNP’s, but in the encoding regions only 12 SNP’s have been revealed, and six of these are non-synonymous (in SNP NCBI). Among the latter, the rs6842 polymorphism has been studied in detail, and it is shown that the substitution of the nucleotide residue T by A results in the amino acid replacement 147N→K (non-synonymous) or the T→C substitution does not lead to amino acid replacement (synonymous). On the whole, just non-synonymous SNP’s and singular amino acid replacements determined by them cause the corresponding biochemical polymorphism of the AGR2 protein (polyallelism).
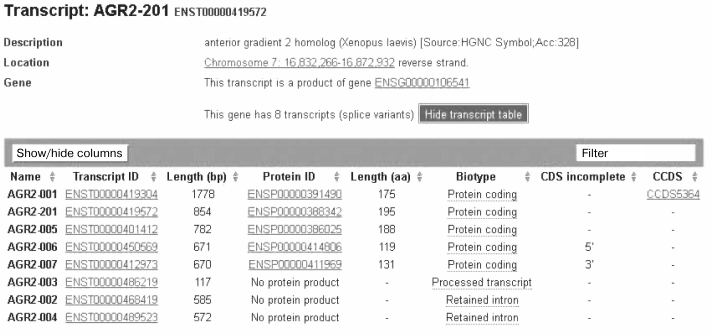
Moreover, The Ensembl Genome project database (after http://www.ensembl.org/Homo sapiens) presents data that expression of the human gene AGR2 due to alternative splicing is associated with production of eight transcripts, and five of them are capable of encoding polypeptide chains with different length, from 195 to 119 a.a (Fig. 3). Thus, alternative splicing significantly contributes to the biochemical polymorphism of AGR2.
Fig. 3. Generalized data on detected transcripts of the human AGR2 gene from The Ensembl Genome project database (after http://www.ensembl.org/Homo sapiens).
It is now known that during post-synthetic modifications the N-terminal signaling polypeptide consisting of 20 a.a. is detached from the newly synthesized AGR2 ([29] and O95994 UniProt). Then the cells secret AGR2, which enters various biological fluids. Thus, AGR2 is secreted into the culture medium during in vitro growing of tumor cells, and the secreted AGR2 retains its ability to stimulate cell proliferation [30]. In patients with some cancers, the blood plasma level of AGR2 was found to be increased [31]. Moreover, an enzyme immunoassay approach was recently developed for determination of AGR2 in urine, allowing detection of this protein in pg/mg of total protein, and then converting to pg/ml of urine [32].
Data of transcriptome and immunochemical analyses indicate that the AGR2 gene is expressed not only in cells of mammary gland and prostate cancers (as mentioned above). This protein has been found in other cancer cells and also in some normal cells (in particular, in cells of intestine, trachea, brain, etc.) ([5], O95994 UniProt and www.proteinatlas.org). Data of studies on AGR2 in tumors will be considered below in more detail.
There are reports about an important role of transcriptional factors of the FOXA family (hepatocyte nuclear factor 3) in the regulation of the AGR2 gene expression. In particular, Foxa1 and Foxa2 were shown to influence the AGR2 gene promoter and increase expression [33, 34]. As a result, the ability of prostate cancer cells for metastasizing and invasion increased. On the contrary, a protein binding ErbB3 (EBP1) inhibited the AGR2 promoter and decreased the stimulatory effects of Foxa1 and Foxa2 [34]. It is now known that EBP1 (designated also as proliferation-associated protein 2G4 and cell cycle protein p38-2G4 homolog) can act as a corepressor of the androgen receptor and bind with very different proteins, including the acetylate histone H1 (after Q9UQ80 UniProt).
Data on the influence of estrogens and androgens on the AGR2 gene expression are different [26, 35-37]. Thus, even in 1998 co-expression of the AGR2 gene and the ER gene encoding the estrogen receptor was found in mammary gland cancer cells [26]. Later, a research group in the USA characterized the AGR2 gene as androgen-inducible [35]. Later, based on data on the co-expression of the AGR2 and ER genes, AGR2 expression was shown to increase in both the hormone-sensitive and hormone-insensitive cells [8, 9]. Finally, in 2013 Bu et al. [36] in some series of experiments directly demonstrated the influence of estrogens and androgens on AGR2 gene (and also of the AGR3 gene) transcription in prostate cancer cells. Concurrently, data appeared indicating involvement of the AGR2 protein in estrogen-induced signaling. These data also suggested that the AGR2 protein could be used as a molecular target in development of chemotherapeutic measures against mammary gland cancer [7, 37]. As a result, many authors now characterize AGR2 as an estrogen- and androgen-sensitive protein with both biosynthesis and subsequent functioning associated with metabolic processes controlled by steroidal sex hormones [7, 35-37].
Thus, there is no doubt that regulation on the transcription level is important for biosynthesis leading to protein AGR2 in health and disease. Moreover, some genotoxic agents (in particular 2,3,7,8-tetrachlorodibenzo-p-dioxin) were shown to influence AGR2 gene expression [38]. It seems that such environmental agents can contribute to deregulation of the AGR2 gene.
Among studies on AGR2 gene expression, special attention should be given to a publication of Hong et al. [39] indicating that AGR2 can also be regulated by the special hypoxia induced factor-1 (HIF-1). HIF-1 consists of several subunits and is a large transcriptional complex termed “master control switch” of genes under the influence of hypoxia [39, 40]. The protein initially induced by hypoxia is the α-subunit of HIF-1. Correspondingly, it is thought that on appearance of hypoxia active production of HIF-1α starts, and then, as a consequence, the master control switch of genes is activated and the whole cell transcription profile changes. Many tumor cells are in the state of hypoxia, which can lead to significant changes in gene expression and also in metabolic processes, and these changes are considered to be a possible cause of tumor cell resistance to the action of chemotherapeutic agents and other damaging factors (after [41]). Thus, the deregulation of AGR2 gene expression under conditions of hypoxia can contribute to phenotypic features of tumor cells.
Functional features of AGR2 are insufficiently studied, but nevertheless it is established that this protein is able to strengthen cell proliferation and mobility and to be favorable for cell survival in culture [30, 42]. Some data suggest that AGR2 can participate in cell transformation. At least, cultured normal mouse fibroblasts (the line NIH3T3) on acquiring after transfection the ability to produce AGR2 also acquire some features of cancer cells, which can be recorded both in vitro and in vivo [42].
There are very interesting reports indicating that AGR2 is probably able to inhibit transcription and/or phosphorylation of protein p53, a very important suppressor of tumor growth [43, 44]. These data were discussed in detail in subsequent reviews and other publications, but the molecular mechanisms of the inhibitory influence of AGR2 on p53 are still not clear [5, 45].
Studies on molecular functions have also revealed that AGR2 can interact with both mucin 2 [23] and mucin 1 [46] and also with other proteins, in particular with α-dystroglycan 1 (DAG1) [28] – a known membrane protein acting as a receptor for some proteins of the extracellular matrix (after Q14118 UniProt). Moreover, AGR2 partners also include a protein termed Ly6/PLAUR domain-containing protein 3 (or GPI-anchored metastasis-associated protein C4.4A homolog) [28], which is involved in cell interactions with the extracellular matrix and in the provision of cell mobility (after O95274 UniProt).
One of the most interesting and comprehensive studies on protein–protein interactions with the involvement of AGR2 was published in 2010 by Maslon et al. [47]. They showed that AGR2 can rather specifically bind with reptin (RuvB-like 2, CAG38538 Protein NCBI) – a protein responsible for some functions in the genetic apparatus of the cell, in particular, for functioning of the histone acetyl transferase complex and its transcription (after Q9Y230 UniProt). The amino acid sequence of AGR2 was also found to have a special motif (FVLLNLVY) responsible for binding with reptin (which also has a special region for this function) and is apparently capable of providing binding with some other proteins. Amino acid substitutions in this motif obtained by directed mutagenesis resulted in a sharp decrease in binding. The functional importance of this motif was also confirmed by the inability to bind reptin of the two AGR2-related proteins (AGR3 and ERp18) with differences in the amino acid sequences of this region.
Using an approach of tandem affinity purification, Yu et al. [48] in 2012 revealed in hepatocellular carcinoma cells 18 AGR2-binding proteins identified as participants of MAPK-signaling and of metabolic pathways controlled by caspases.
Among other studies on protein–protein interactions with involvement of AGR2, special attention should be given to data published in 2013 on the ability of AGR2 to dimerize (or even to oligomerize) [49-52]. At first, Ryu et al. [49] showed that both monomer and dimer of this AGR2 protein appear in large intestine adenocarcinoma cells transfected with a plasmid containing cDNA encoding this protein. These AGR2 forms were revealed using SDS-PAGE and Western blotting, and the authors concluded that the dimerization was due to generation of intermolecular disulfide bonds, similarly to formation by AGR2 of the heterodisulfide bond with cysteine residues in MUC2 molecules [23]. The AGR2 dimerization was also shown to be necessary for its interaction with another ER protein, GRp78 (also termed endoplasmic reticulum lumenal Ca(2+)-binding protein grp78, heat shock 70 kDa protein 5, immunoglobulin heavy chain-binding protein, BiP, etc.). Considering the known role of GRp78 in cellular responses to various stress exposures and chaperone features with formation of multimeric protein complexes (after P11021 UniProt), Ryu et al. [49] suggested that AGR2 dimers could be involved in the functions of GRp78.
On consideration of the molecular mechanisms of so-called endoplasmic reticulum stress (ER stress accompanied by accumulation of proteins with disturbed folding), Kaser et al. [50] virtually in parallel concluded that participation of AGR2 is likely to be its function together with GRp78 in providing normal folding. Disorders in this interaction can lead to some inflammatory diseases of the intestine.
Later, Patel et al. [51] using a balanced analytical ultracentrifugation and some other approaches, including chromatography, found that the usual mature AGR2 (a.a. 21-175) and its shortened form (a.a. 40-175) produce aggregations with molecular weights of 30.6 and 26.0 kDa corresponding to its homo- and heterodimers. Further analysis revealed that for dimerization the region E60-K64 is important. Moreover, studies on polypeptide chains with single amino acid replacements (by directed mutagenesis) showed that no dimers are produced on the 60E→A replacement. And the authors noticed that because the shortened form (a.a. 40-175) participated in the production of dimers, the N-terminal fragment of AGR2 (a.a. 21-40) should remain unstructured under in vitro conditions (and possibly also in vivo). Thus, these authors think that just this region can be responsible for AGR2 functioning in stimulation of cell adhesion and its involvement in metastasis.
The development of a special method for investigation of stability of AGR2 dimers under conditions of treatment with synthetic peptides has been reported [52]. The authors characterized this work as the first step for creating purposeful changes in the properties of AGR2, which is considered as a promising target for chemotherapy of malignant tumors.
Finally, although the main features of human AGR2 are now rather well characterized and indicate its multiple functions, this unusual PDI family representative is still under active studies, and the accumulated data suggest that further studies on AGR2 may be promising for solution of various biomedical problems.
HUMAN PROTEIN ERp57/GRP58 IS A TYPICAL MEMBER OF THE PROTEIN DISULFIDE
FAMILY: GENERAL CHARACTERISTICS AND FUNCTIONAL FEATURES
A human protein that is often called ERp57/GRP58 (from endoplasmic reticulum resident protein 57 and also 58 kDa glucose-regulated protein, endoplasmic reticulum resident protein 60, disulfide isomerase ER-60, ER-60 protease) and thought to be a protein disulfide isomerase A3 (after P30101 UniProt and [1, 6]) has attracted the attention of researchers from the 1990s. This attention is obviously caused by the hypothesis that protein ERp57/GRP58 could be involved in malignant transformation of cells [53].
During the same period, analysis of the amino acid sequence of ERp57/GRP58 calculated from results of the corresponding cDNA sequencing indicated that this protein has two thioredoxin-like domains with characteristic motifs CGHC and also the motif QEDL, suggesting the localization in the ER, and the motif KPKKKKK specific for nuclear localization [54].
Then the ERp57/GRP58 structure was shown to contain, in addition to the two thioredoxin-like functional domains (a and a′), two similar domains (b and b′) but lacking enzymatic activity [55, 56]. And the b and b′ domains were shown to be necessary for production of a stable complex with two lectins that are present in the ER – the soluble calreticulin and membrane-bound calnexin [56, 57]. Moreover, amino acid residues 214K, 274K, and 282R of the b′ domain were shown to play a key role in calnexin binding.
Overall, the general structure of ERp57/GRP58 quite corresponds to the scheme specific for subgroup of traditional protein disulfide isomerases (Fig. 1).
Some data on the human protein ERp57/GRP58 are now summarized in the UniProt database (after P3095) and also in recent reviews [3, 6, 57]. According to the available data, the complete amino acid sequence of ERp57/GRP58 consists of 505 a.a., from which a.a. 1-24 constitute a signaling peptide. It was also shown that within ERp57/GRP58 an intramolecular disulfide bond can be produced (61C-68C within the a domain structure) [58].
The human gene PDIA3 encoding the protein under consideration is also studied rather well. The chromosomal localization of this gene (15q15) is known, as well as the general scheme of the structure of 13 exons (after 602046 OMIM NCBI and GENE NCBI). In the database SNP NCBI, 554 polymorphic variants found in the gene PDIA3 are registered.
Experimental data collected during the last decade leave no doubt that ERp57/GRP58 is a disulfide isomerase, belongs to stress proteins (with synthesis increasing in the case of glucose insufficiency), and is present in the ER and also in other cellular compartments where it performs some important functions [5, 6, 57].
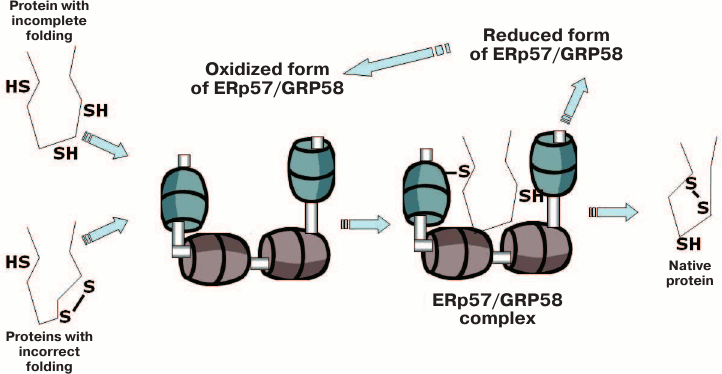
First, ERp57/GRP58 in a complex with calreticulin and calnexin was shown to be responsible for the correct folding of newly synthesized glycoproteins and for control of their quality, which is necessary for the further secretion of such proteins or for insertion into cellular membranes. This function of ERp57/GRP58 seems to be studied most completely. Some recent reviews describing this function in more detail accentuated the important biomedical significance of the correct folding of glycoproteins and the pathogenetic role of accumulated proteins with incorrect folding in various diseases [3, 6, 57]. Schematically, the involvement of ERp57/GRP58 in the correct folding of proteins with incomplete or incorrect folding is presented in Fig. 4 (see color insert).
Fig. 4. Scheme illustrating the participation of ERp57/GRP58 in catalyzing the correct folding of proteins with incomplete or incorrect folding (after [3]).
Second, there are convincing data indicating that ERp57 participates in the assembly of the major histocompatibility complex (MHC class I). Thus, as early as in 1998, Lindquist et al. [59] demonstrated that ERp57 catalyzes formation of disulfide bonds in heavy chains of MHC class I and is a component of this complex. Moreover, this complex was shown to contain calreticulin, calnexin, β2-microglobulin, tapasin, and a special transporter protein TAP (transporter associated with antigen processing, or antigen peptide transporter). These data were repeatedly confirmed and detailed (after [3, 6, 57]). In particular, approximately 10 years later Dong et al. [60] reported that during a stage of MHC class I assembly, heterodimers are produced between ERp57 and tapasin via covalent disulfide bonds with involvement of 57C molecules of ERp57 and 95C molecules of tapasin. Just recently, in 2013, Boyle et al. [61] found an additional component of the major histocompatibility complex – a tapasin-related protein or TAP-binding protein-related protein (TAPBPR). According to their data, MHC class I containing TAPBPR functions inside the Golgi apparatus. Thus, studies on the role of ERp57 in the assembly of the major histocompatibility complex will probably be continued.
Some authors consider the ability of ERp57/GRP58 to interact with nuclear DNA and influence gene expression to be its third principally important function (after [3, 6, 57]). The first reports about the possibility of such interactions of ERp57/GRP58 appeared as early as in the 1990s (after [62]). In 2002, Coppari et al. [63] found ERp57/GRP58 in both ER and cell nuclei. The protein was also shown to interact with DNA molecules through the functionally active thioredoxin domain a′ [64-66]. It was also shown that ERp57/GRP58 is a component of the so-called STAT3-transcriptional complex [66]. The major component of this complex is protein STAT3 (signal transducer and activator of transcription 3) consisting of 770 a.a. and being responsible for the cellular response to interleukins and some growth factors (after P40763 UniProt). STAT3 is a member of the STAT family, which includes six more proteins with highly homologous primary structures and the ability to regulate cell proliferation [67]. In 2010, Coe et al. [73] confirmed the formation of ERp57/GRP58–STAT3 complexes and the ability of ERp57/GRP58 to modulate the signaling triggered by STAT3. They also noted that the death of embryos of knockout mice was probably due just to their inability to synthesize ERp57/GRP58.
The involvement of ERp57/GRP58 in the regulation of gene expression is thought to be associated with the ability of this protein to participate in two other polyprotein complexes that are abbreviated as mTORC1 and mTORC2 (from mammalian target of rapamycin), which also include a special serine/threonine protein kinase (serine/threonine protein kinase mTOR) and some other proteins [68]. This enzyme was also detected in ER, nucleus, and other cellular compartments (after P42345 UniProt), and it is very favorable for generation of complexes with ERp57/GRP58. Both mTORC1 and mTORC2 are involved in various regulatory processes finally resulting in changes in gene expression [69, 70]. Recently, the influence of mTORC1 on STAT3-signaling was found among other effects associated with the regulation of gene expression [70].
According to data of Grillo et al. (2006), ERp57/GRP58 can interact with rather high affinity with protein Ref-1/APE (apurinic-apyrimidinic endonuclease/redox-factor 1), which is involved in DNA repair and also can activate some transcriptional factors [71]. Later, complexes containing Ref-1/APE were shown to immediately bind with DNA molecules and thus to influence the expression of some genes and the activity of transcriptional factors [72, 73]. Note that the above-mentioned HIF-1 and STATS-signaling participants are among the transcriptional factors (and signaling systems) influenced by Ref-1/APE.
Nuclei of human lymphoblasts (Nalm6 line) were also reported to contain a DNA–polyprotein complex with the protein moiety represented by the proteins B1 and B2 from the high mobility group proteins and also by some other proteins including ERp57/GRP58 (denoted ERp60 by the authors) [74]. This complex was capable of binding with synthetic oligodeoxyribonucleotides and could influence gene expression in cultured cells.
Data directly indicating rather specific binding of ERp57/GRP58 with DNA fragments were published in 2007 by Chichiarelli et al. [75]. They studied DNA from HeLa cells, and using chromatin immunoprecipitation isolated 10 fragments binding with ERp57/GRP58. These fragments were cloned and sequenced. Nine of them contained noncoding nucleotide sequences, and seven corresponded to intronic regions of some identified genes. They concluded that some of the detected DNA fragments were hypersensitive to DNase. They suggested that the interaction of ERp57/GRP58 with DNA fragments can lead to changes in the expression of two genes encoding proteins involved in DNA repair.
These studies were continued on DNA from melanoma cells, and the findings published in 2013 [62] indicated that ERp57/GRP58 can recognize DNA sequences in at least the introns of the MSH6, ETS1, and LRBA genes and in the 5′-region of gene TMEM126A, which were attested as target genes. Suppression of ERp57/GRP58 production with interfering RNAs was accompanied by a decrease in the expression of the target genes. The authors paid special attention to the MSH6 gene encoding an important protein involved in the post-replicative repair of DNA (P52701 UniProt). They revealed an interaction of the gene MSH6 fragment recognized by ERp57/GRP58 with protein Ref-1/APE, which also was a participant of the DNA repair systems (see above).
Thus, various indirect and direct experimental data that are available are sufficient for concluding that ERp57/GRP58 is capable of interacting with nuclear DNA, and as a result it influences gene expression.
There are comparatively few data on the other functional features of ERp57/GRP58. Thus, as early as in 1997, Urade et al. [76] considered the presence of CGHC motifs as evidence of proteolytic activity of ERp57/GRP58. Therefore, they named this protein ER-60 protease. Later, this suggestion was supported by works of other authors [77, 78]. In particular, a paper by Rutledge et al. [78] reports that this protein acts as a cysteine proteinase in degradation of apoB100 within liver cells. In work [79], it is reported that human ERp57/GRP58 (ER-60) seems to have transaminase activity responsible for protein–protein cross-linking.
Generalizing the literature data on the structure and functions of human ERp57/GRP58, it should be noted that it is a multifunctional protein involved in very important intracellular processes, and this makes it a very interesting object for various biomedical studies.
ERdj5, TMX, AND SOME OTHER HUMAN PROTEIN DISULFIDE ISOMERASES:
COMMON STRUCTURAL AND FUNCTIONAL FEATURES
This subsection will consider some of the most studied members of a subgroup of the PDI family denoted above as ER proteins (ERdj5, ERp5, ERp46, ERp44) and also subgroups of transmembrane proteins. Virtually all members of these subgroups became known only in the present century, upon the beginning of postgenomic approaches in biochemistry and active use of advanced technologies. In publications presented in the PubMed NCBI database, the main attention among ER proteins is given to ERdj5 (about 25 papers), which has higher molecular weight among the human PDI family members. As a consequence, structural and functional features of ERdj5 will be characterized below as of the representative of the ER subgroup.
A report about ERdj5 was published in 2003 [80], when Cunnea et al. convincingly demonstrated the presence in the ER of human cells of a protein with molecular weight 91 kDa and pI 7.03 that contains four functionally active thioredoxin-like domains (Fig. 1) and also a special so-called DnaJ-domain. The authors assigned this protein as a new representative of protein disulfide isomerases and denoted it as ERdj5 based on specific features of its structure and intracellular localization. In particular, in each thioredoxin-like domain of ERdj5 typical motifs C-X-X-C were found, and in the N-terminal sequence the DnaJ-domain (a.a. 35-100) was revealed.
The presence of the DnaJ-domain in the amino acid sequence of ERdj5 was a reason for inclusion of this protein as a new member of the known superfamily of DnaJ proteins, where it was named DnaJ homolog subfamily C member 10 (after Q8IXB1 UniProt). Members of this superfamily were found in both pro- and eukaryotes and were under active study for more than a decade, which resulted in detection of chaperone features in many of these proteins [81-83].
Even in the first study on human protein ERdj5 [80], both the protein itself and the gene encoding it were characterized. The gene is localized on chromosome 2 (p22.1-p23.1), it contains 23 exons, and its transcription is associated with alternative splicing. Transcripts of the ERdj5 gene were found in all organs studied, but their greatest amount was detected in the pancreas.
The next two publications about human ERdj5 [84, 85] considered this protein in tumor cells. The content of ERdj5 determined immunochemically was threefold higher in hepatocellular carcinoma cells than in the control; therefore, the authors [84] concluded that ERdj5 could be promising as an immunochemical marker for diagnosis of hepatocellular carcinomas. Moreover, in this work results of experiments with interfering RNAs suppressing the ERdj5 synthesis in cultured cells of hepatocellular carcinoma suggested that this protein could contribute the resistance of tumor cells to some chemotherapeutic preparations.
In the study [85], ERdj5, similarly to ERp57/GRP58, was shown to be important for protective reactions developing in the ER in response to oxidative stress. Upon suppressing the synthesis of ERdj5 and ERp57/GRP58 in cultured tumor cells (“knockout” with interfering RNAs), the authors found an increase in apoptosis under the influence of preparations inducing oxidative stress. Therefore, both ERdj5 and ERp57/GRP58 were concluded to be interesting for development of new approaches of antitumor therapy, where these proteins (and/or the genes encoding them) could be used as molecular targets.
Then, in work [86], studies using postgenomic technologies on molecular mechanisms of pathogenesis of chronic pulmonary disease caused by mutations in the gene SFTPC revealed that ERdj5 (and also some other ER proteins) are responsible for normal folding (and also for correcting affected folding) of hydrophobic pulmonary-associated surfactant protein C (SP-C) encoded by this gene and is very important for pulmonary function. Also in 2008 [87] ERdj5 was shown to be a key participant of ER-associated degradation of misfolded or unassembled proteins (ERAD) occurring in the ER. During this degradation (ERAD), ERdj5 functions within a special multiprotein complex. The authors also determined that another important participant of ERAD was a protein called ER-resident chaperone BiP (synonyms: heat shock 70 kDa protein 5, 78-kDa glucose-regulated protein, HSPA5, etc.) (after P11021 UniProt).
In a series of subsequent studies, the important role of ERdj5 during ERAD was confirmed and detailed, and other protein participants of ERAD were also determined [88-91].
In particular, in 2009 Riemer et al. [88] found that, in addition to ERdj5 and BiP, a special flavoprotein was involved in the ERAD process, and detection of this flavoprotein began from analysis of genomic information in silico. Among DNA regions earlier characterized as containing an open reading frame, the authors noted one such potential gene (RefSeq: NP 079231, gene name: FOXRED2) localized on chromosome 22 (after 613777 OMIM NCBI). Basing on the nucleotide sequence, this gene was supposed to encode an amino acid sequence consisting of 684 a.a., which included an N-terminal signaling peptide (26 a.a.), a dinucleotide-binding domain, and the C-terminal motif KEEL specific for soluble proteins of the ER.
With this in mind, the same authors [88] isolated the hypothetical protein from human cultured cells, characterized its features (which essentially corresponded to the calculated ones), and showed that it was a protein capable of binding with ERdj5 and also with some other known participants of ERAD. This flavoprotein was named ERFAD (ER flavoprotein associated with degradation). It is now also called by another name – FAD-dependent oxidoreductase domain-containing protein 2 (after Q8IWF2 UniProt).
Moreover, Riemer et al. [88] found some more proteins interacting with ERdj5 and involved in ERAD, in particular a protein designated SEL1L (from the term protein sel-1 homolog 1; synonym – suppressor of lin-12-like protein 1) (after Q9UBV2 UniProt). SEL1L is also known to be a participant of the system of an ubiquitin-dependent system of degrading misfolded proteins. Another protein under study, OS-9 (or amplified in osteosarcoma 9), was found to be a lectin involved in ERAD of nonglycosylated misfolded proteins (after Q13438 UniProt).
Concurrently, Thomas and Spyrou [89] found that in neuroblastoma cells ERdj5 is involved in development of apoptosis induced by some chemotherapeutic preparations. Therefore, ERdj5 is interesting as a potential molecular target for antitumor preparations.
The contribution of ERdj5 to folding of secreted proteins was reported by Hosoda et al. [90]. Their study was performed on a model – in particular, ERAD processes were analyzed in salivary glands of knockout laboratory mice. Note that mice with ERdj5 knockout were characterized by the authors as viable and healthy.
The main results of works performed during the first decade of this century on ER components (including ERdj5) involved in recognizing proteins with incomplete or incorrect folding and in subsequent ERAD were generalized in a review by Maattanen et al. [91]. They presented a scheme of ERAD demonstrating interrelations between ERdj5, BiP, ERFAD, SEL1L, and OS-9, and also with the other participant of ERAD – and an ER degradation enhancing α-mannosidase-like ER protein (EDEM) [92].
In the beginning of the new decade, Tamura et al. [93] reported that ERdj5 not only interacts with the factor EDEM1, but it also contributes to its maturation. And the interaction with ERdj5 leads to detachment from EDEM1 of a signaling peptide and transition of this factor into soluble form. The ratio of the soluble and membrane forms of EDEM1 was evaluated as an important parameter of functioning of the overall ERAD system.
Among publications of 2011, a work of Hagiwara et al. [94] should be noted. They carefully studied the structure of mouse ERdj5 and indicated that in addition to four functionally active thioredoxin-like domains, the protein also contains two inactive domains, b1 and b2. However, similar variants of the structure were not found in human ERdj5 by other authors.
In four publications of 2013 [95-98], attention was mainly given to some biomedical aspects of human ERdj5. Thus, Diamanti et al. [95] studied the influence of 2-hydroxyethylmetacrylate (a monomer used to prepare hydrophilic polymers for biomedical purposes) on primary cultures of human pulp cells and found a significant increase in the synthesis of ERdj5 (as well as of BiP), most likely due to development of ER stress. The ER stress is thought to be mainly caused by the accumulation of proteins with incomplete or incorrect folding.
Williams et al. [96] reported that Rdj5 and protein Sel1L, which is believed to have an adaptor function, were shown to produce a complex participating (together with BiP) in transfer of the cholera toxin catalytic subunit (CTA1). These data contribute to modern concepts about molecular mechanisms responsible for the toxic effect of CTA1. Data obtained on a model system [97] suggested that ERdj5 could be involved in and play a protective role in pathogenesis of some neurodegenerative diseases (Alzheimer’s disease, etc.). It was also found [98] that ERdj5, due to its reducing activity, can break incorrect disulfide bonds and correct the folding of a low density lipoprotein receptor – a protein playing a key role in development of atherosclerosis.
Thus, all data now available on features and functions of ERdj5 indicate that this member of the PDI family is involved in reactions protecting cells against accumulation of proteins with incomplete or incorrect folding. Because ER stress caused by accumulation of such proteins is found in various diseases, there is no doubt that further studies on ERdj5 are of interest.
Important information has already been obtained concerning the functional characteristics of other members of the human PDI family proteins of the ER subgroup (ERp5, ERp46, ERp44). Thus, ERp5 is known to participate in regulatory processes in platelets [99]. Moreover, as a component of membranes of leukemia cells, ERp5 acts as a receptor for so-called tumor-associated NKG2D-ligands and, therefore, is considered to be a participant of cell malignant transformation in leukemias [100, 101]. Thus, studies on ERp5 have a definite biomedical orientation, and this protein is even considered as a potential molecular target for chemotherapeutic agents [100].
The discovery of a subgroup of transmembrane proteins of the PDI family and initial studies on them have been performed mainly by Japanese researchers. In 2001, Matsuo et al. [102] reported that among genes that responded to treatment with regulatory proteins of the TGF-β family in human adenocarcinoma A549 cells, there is a gene encoding a new protein consisting of 280 a.a. The amino acid sequence of this protein contains a thioredoxin-like domain with a Cys-Pro-Ala-Cys motif, which allowed the authors to assign this protein to the thioredoxin superfamily. They also recorded in its amino acid sequence a potential transmembrane domain (a.a. 183-203). The newly discovered protein was named thioredoxin-related transmembrane protein (TMX).
Matsuo et al. [102] also found that the recombinant TMX protein had a disulfide reductase activity. Northern blotting revealed expression of the gene encoding TMX in all tested human tissues – from skeletal muscles and heart, where minimal levels were recorded, to the maximal activity in spleen and liver. Results of immunoblotting with anti-TMX antibodies showed that the protein was present mainly in the microsomal fraction, less in plasma membranes, and was not found in nuclei. Such intracellular distribution of TMX was also confirmed by other methods, so preferential localization of TMX in ER was concluded.
In a subsequent work [103], Matsuo et al. demonstrated that the earlier described TMX has protein disulfide isomerase activity and probably acts as a regulator of protein folding.
In 2009, the same authors reported that in human cells (strains A549 and 293 obtained, respectively, from lung carcinoma and embryonal kidney), TMX was mainly in the reduced state [104]. It was also shown that TMX could interact with calnexin, but not with calreticulin, and that for its functioning a heterodisulfide bond should be produced with molecules of heavy chains of the major histocompatibility complex (MHC class I). But, as differentiated from ERp57/GRP58, TMX was not necessary for correct assembly of the MHC class I, but under conditions of ER stress this protein prevented transport of misfolded heavy chains from the ER into the cytoplasm and their subsequent proteasomal degradation. Finally, it was concluded that TMX performed in ER a specific function associated with control of disorders in protein folding.
Considering the above-listed features, including the localization in the ER, Ellgaard and Ruddock [1] and later also other authors [3] began to attribute TMX to the PDI family. Some other proteins containing thioredoxin-like domains and transmembrane motifs (TMX2-4) are also considered members of this family [3].
Now it is acknowledged that TMX (also called transmembrane Trx-related protein and TMX1) is involved in various redox reactions associated with reversible oxidation of two thiol groups within the active center of this protein (after Q9H3N1 UniProt). Rather complete data (obtained on the protein level) have been reported about the primary structure of TMX and also of its post-synthetic modifications, including phosphorylation of three serine residues.
Moreover, recent studies on membranes associated with mitochondria (considered as a special part of ER) [105] revealed that TMX molecules are covalently bound with palmitic acid. It was concluded that this binding is through a.a. 205C and 207C and provided correct inclusion of the protein into membranes, as well as its active functioning.
Finally, in 2013 it was reported that in liver cells TMX has a protective function during inflammations and the associated oxidative stress [106].
There are at least three more known members of the PDI family belonging to the subgroup of membrane proteins: TMX2, TMX3, and TMX4 [1, 3] (in some reviews [3, 107] TMX5 was also mentioned, but we failed to find other information about it). According to the available data, these proteins have protein disulfide isomerase (oxidoreductase) activity and the C-terminal motif indicating localization within the ER (after Q9Y320, Q96JJ7 UniProt and [108]). There are still few data about these functions, but it seems that these proteins will be studied more carefully in the nearest future.
STUDIES ON AGR2 AND ERp57/GRP58 IN TUMOR AND OTHER ACTIVELY
PROLIFERATING CELLS
From the first decade of this century, AGR2 and ERp57/GRP58 in human cancer cells have been studied in many laboratories in the USA, Western Europe, China, and also in Russia. Dozens of papers have been published about these subjects, and these proteins are still given great attention.
In fact, all studies on human AGR2 were initiated by the above-mentioned work of Thomson and Weigel [26] performed in 1998 just on tumor cells. In a culture of mammary gland cancer cells, they detected co-expression of the gene encoding AGR2 and the gene of estrogen receptor. Based on this finding, they supposed that AGR2 could be involved in pathogenesis of highly differentiated hormone-sensitive mammary gland cancer.
Then, until 2005 single papers reported that AGR2 was present in mammary gland cancer cells and was involved in some regulatory processes. It seems that Fletcher et al. [28] were the first to directly detect immunohistochemically the AGR2 protein in mammary gland cancer cells. Based on determination of the corresponding transcripts, they concluded that there is a direct correlation of AGR2 with estrogen receptors and an inverse correlation with epidermal growth factor.
Using proteomic analysis of proteins in cells of hormone-resistant and hormone-sensitive lines of mammary gland cancer cells, Huber et al. [109] identified AGR2 in two-dimensional electrophoregrams. In the hormone resistant cells the AGR2 level was lower than in the hormone-sensitive cells. Results of a parallel study on transcripts (by hybridization on DNA chips) were similar to the proteomic analysis data and also confirmed the presence of a correlation between AGR2 and estrogen receptors. They concluded that AGR2 could be involved in intracellular processes in the hormone-sensitive cells of mammary gland cancer.
Attention to the role of AGR2 in tumors significantly increased from 2005, after appearance of three early publications [35, 110, 111] about increased level of this protein (and/or expression of its gene) in prostate cancer. Thus, Zhang et al. [35] found significantly increased levels of AGR2 gene transcripts in the majority of tissue specimens of prostate cancer and in the model cell line LNCaP. Moreover, immunochemical determination of AGR2 revealed a high content of this protein in prostate cancer (and in so-called prostatic intraepithelial neoplasia) and also in the LNCaP cells. In summary, the authors concluded that AGR2 is interesting as a potential biomarker of prostate cancer and a possible target for chemotherapy.
In the study [110], increased expression of the AGR2 gene was shown in the overwhelming majority of tissue specimens of prostate cancer (89%). In work [111], active expression of the AGR2 gene was found in circulating tumor cells in patients with prostate cancer metastases (and also in metastases of mammary cancer, etc.), and thus it could be considered as a marker of the beginning of metastasis.
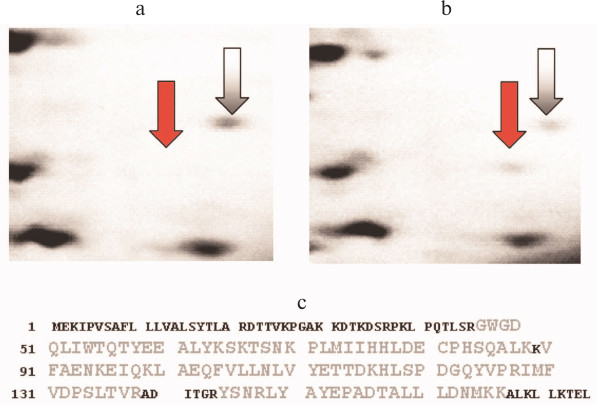
In 2006, Kovalev et al. [112], using proteomic methods (two-dimensional electrophoresis and mass-spectrometry), identified AGR2 in malignant prostate tumors, and they noted that this protein could be interesting as a biomarker of such tumors because it was not detected in benign hyperplasia (Fig. 5; see color insert).
Fig. 5. Results of proteomic identification of AGR2 protein in prostate tissues (after [112]). a) Fragment of two-dimensional electrophoregram of a benign tumor specimen; the gray arrow shows a marker protein (identified as peptidyl prolyl cis-trans-isomerase B), the red arrow shows the supposed position of the AGR2 protein. b) A similar fragment of two-dimensional electrophoregram of a cancer tissue specimen; designations are the same as in Fig. 5a. c) Results of mass-spectrometry of the fraction indicated by the red arrow in Fig. 5b. In the amino acid sequence of the AGR2 protein, the sequence regions corresponding to the detected tryptic peptides (coverage 64%) are shown in larger and lighter letters.
During the subsequent period, more than a dozen publications have reported the presence of AGR2 in prostate cancer, and there is virtually no doubt that this protein has features of a biomarker of this pathology. Some information about the main studies on this subject published during the period of 2007-2013 is summarized in the table.
Main publications during 2007-2013 concerning AGR2 in prostate cancer
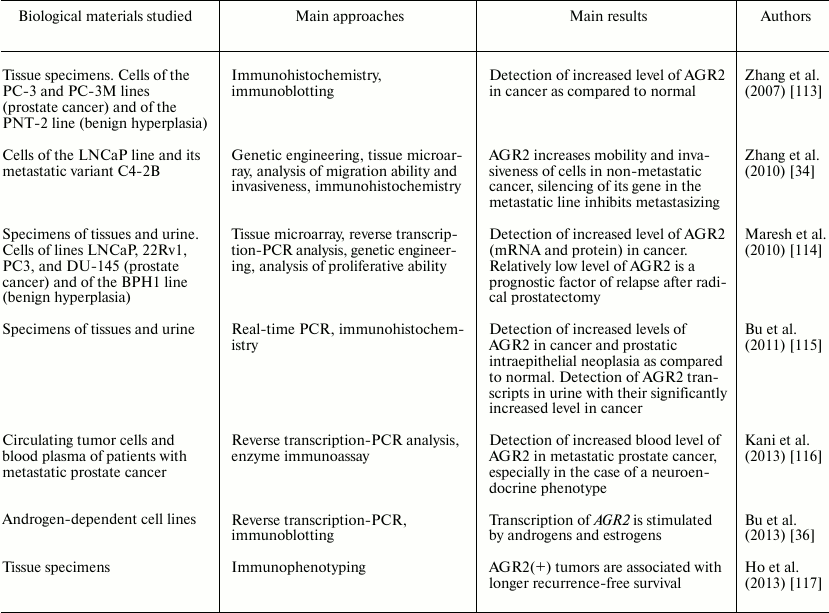
During the period from 2007 to 2013, dozens of papers were also published reporting increased level of AGR2 in tissues and cell lines of mammary gland primary cancer, for example, works [118, 119]. High levels of AGR2 (and/or its transcripts) were also found in malignant tumors of other localizations, in particular in cancer of ovary [120], esophagus [121], stomach [122], pancreas [30], and lungs [123].
Many authors have pointed to presence of a correlation between the high level of AGR2 and increased mobility of tumor cells [120] and their ability to metastasize [51, 124]. It should be noted that AGR2 seems to promote mobility (and dissemination) not only of tumor cells. Thus, Hapangama et al. reported about a contribution of AGR2 to the increased invasiveness of the endometrium cells in endometriosis and subsequent ectopia of these cells [125].
In generalizing these results obtained by many different researchers, we conclude that AGR2, being a secreted protein, can be considered a rather common biomarker of adenocarcinomas with different localization. Also, separate pilot studies performed with limited groups of patients with tumors [126] indicate the reasonability of clinical testing for validation of AGR2 as a diagnostically significant protein marker.
However, it must be taken into consideration that changes in AGR2 level can also occur in some pre-cancer diseases. In any case, increased level of AGR2 (estimated by the content of mRNA and/or protein) was observed in prostatic intraepithelial neoplasia [35, 115], intestinal metaplasia of esophagus [127], and in pancreatic intraepithelial neoplasia [128]. However, the AGR2 level determined by transcripts was decreased in ulcerative colitis, which is believed to be a facultative pre-cancer [33].
Pointing to the possibility of using AGR2 as a biomarker of adenocarcinoma, it must be remembered that this protein is responsible for important functions in embryogenesis not only of Xenopus (as mentioned above), but also in mammals. Gupta et al. [129] showed that during stomach formation in embryogenesis in rats, AGR2 was responsible for the balance between the cell proliferation and differentiation. Thus, AGR2 promoted the differentiation and specialization of gastric epithelium cells and concurrently inhibited the proliferative activity of stem cells.
Finally, it should be emphasized that individual papers reported a decrease in AGR2 level in some forms of cancer. In particular, decreased level of AGR2 was found in urothelial carcinoma of urinary bladder [130] and in planocellular cancer of the oral cavity [131].
The current interest in the role of AGR2 in malignancies is also confirmed by results of bibliometric analysis of the number of publications presented in the PubMed NCBI database. Seventy-five percent of 128 papers about AGR2 contain findings obtained on malignant tumors and/or cultured tumor cells. And 90% of all studies on AGR2 in cancer were published in 2005 or later. Moreover, during the period 2005-2009, four to six papers per year were published, whereas during 2010-2011 there were 16 such papers per year and already 25 papers in 2012.
Along with AGR2 considered as an unusual member of the PDI family, a typical protein disulfide isomerase, ERp57/GRP58 protein, also attracts the attention of researchers investigating specific features of metabolism in malignant tumor cells. It mentioned above, as early as in the middle of 1990s a possible involvement of this protein in oncogenic transformation was suggested in some reports [53]. During this period, some authors [132] indicated that ERp57/GRP58 (ERp61 is a synonym) could be secreted by tumor cells into the culture medium, although most of this protein remained within the cells.
In 2002, Guo et al. [133] reported that ERp57/GRP58 interacts with some participants of so-called STAT signaling (in particular with STAT3), which was already known to play an important role in malignant transformation of cells (reviewed in [134]).
Based on Northern blotting results, Celli and Jaiswal [135] reported in 2003 that the content of transcripts encoding ERp57/GRP58 was higher in tumors of mammary gland, uterus, lungs, and stomach than in the corresponding normal tissues. Convincing data on the contribution of ERp57/GRP58 to the cytotoxic effect of the antitumor preparation mitomycin C were presented in the same work. In particular, due to its oxidoreductase activity, ERp57/GRP58 promoted mitomycin-induced generation of crosslinking in DNA molecules.
These data on the involvement of ERp57/GRP58 in carcinogenesis and in action mechanisms of some antitumor preparations were later confirmed and detailed in works of other researchers.
Thus, Eufemi et al. [66] reported in 2004 about the presence of ERp57/GRP58 in the regulatory complex STAT3–DNA, and Adikesavan and Jaiswal [136] established in 2007 that just functionally active thioredoxin-like domains of ERp57/GRP58 were necessary for generation of the mitomycin-induced DNA crosslinking. The main results of studies on ERp57/GRP58 during the first decade of this century are considered in review [57].
During 2011-2012, about a dozen works were published expanding ideas about the role and mechanisms of ERp57/GRP58 functioning in tumor cells. Thus, Grindel et al. [137] noted dysregulated expression of the gene encoding ERp57/GRP58 and showed that the treatment of hepatocellular carcinoma cells with tumor necrosis factor (TNF-α) resulted in a change in intracellular distribution of ERp57/GRP58 – the protein began to be actively transferred from the cytoplasm into the nucleus. Liao et al. [138] demonstrated the ability of ERp57/GRP58 to modulate the invasiveness of uterine cervix cancer cells and proposed the use of this protein as a prognostic biomarker. In work [139], ERp57/GRP58 was studied (together with HSP70 and calreticulin) in tumor cells subjected to so-called photodynamic therapy.
Finally, in 2013 Elisa et al. [140] reported that in mammary gland cancer cells ERp57/GRP58 is involved in cascade processes triggered by the epidermal growth factor receptor, the role of which in carcinogenesis was characterized in many experimental works and reviews (e.g. [141]). Also in 2013, Santana-Codina et al. [142], upon analyzing protein–protein interactions in mammary gland cancer cells including a specific osteotrophic subclone, concluded that ERp57/GRP58 functions as a molecular center in the protein network in tumor cells and promotes metastasizing into bones.
Thus, the available data clearly suggest that ERp57/GRP58 is involved in carcinogenesis and that its features are to be taken into account in studies on malignant tumors, including the prediction of metastasizing and the development of new chemotherapeutic agents.
So, it is clear that both AGR2 and ERp57/GRP58 will remain very attractive objects for various biomedical studies directed to solution of urgent problems in molecular oncology.
During a relatively short period – less than 20 years – human proteins called AGR2 and ERp57/GRP58 have been actively studied, and this has resulted in numerous data on their structure, functions, and biochemical polymorphism. Transcriptomic, proteomic, and other postgenomic technologies have played a determining role in these studies.
The established features allowed researchers to assign these proteins to the rather large family of protein disulfide isomerases. Naturally, a specific feature determining the membership in the PDI family is the ability to catalyze formation of disulfide bonds in proteins. This ability was found in both AGR2 and ERp57/GRP58. The presence of thioredoxin-like domains in their amino acid sequences, as well as of special C-terminal motifs characteristic for proteins localized in the endoplasmic reticulum, are also considered important reasons for assigning them to the PDI family.
The main functions of the ERp57/GRP58 protein in normal cells seem to be sufficiently characterized, whereas the role of AGR2 protein in human in norm is studied insufficiently. There are few data about other members of the PDI family, so the field for further studies is still large.
Results of studies on AGR2 (a small protein with a single thioredoxin-like domain with a special catalytic motif) allow researchers to consider it as an unusual member of the PDI family. In contrast, ERp57/GRP58 has properties of a typical protein disulfide isomerase. Nevertheless, both proteins are found to be participants in carcinogenesis, and this attracted and continues to attract the attention of many researchers in different countries.
Among the results of studies on AGR2 and ERp57/GRP58 in malignant tumor specimens and in cultured cancer cells, especially interesting are data suggesting that these proteins possess features of biomarkers. As a consequence, it is reasonable to think that there is reason to begin works to validate AGR2 and ERp57/GRP58, and in the case of success these proteins will be promising for being used in diagnosis of oncological diseases. There are also other prospects for continuation of biomedical studies on AGR2 and ERp57/GRP58 proteins associated, in particular, with works in the field of chemotherapy.
In conclusion, one can hope that further studies on different members of the PDI family with postgenomic technologies will enhance the modern knowledge about functions of these proteins in the human body that in turn will be of help for solution of urgent biomedical problems.
This work was supported by the State Contract No. 14.740.11.0762.
REFERENCES
1.Ellgaard, L., and Ruddock, L. W. (2005) EMBO
Rep., 6, 28-32.
2.Feige, M. J., and Hendershot, L. M. (2011) Curr.
Opin. Cell Biol., 23, 167-175.
3.Andreu, C. I., Woehlbier, U., Torres, M., and Hetz,
C. (2012) FEBS Lett., 586, 2826-2834.
4.Laurindo, F. R., Pescatore, L. A., and de
Fernandes, C. (2012) Free Radic. Biol. Med., 52,
1954-1969.
5.Brychtova, V., Vojtesek, B., and Hrstka, R. (2011)
Cancer Lett., 304, 1-7.
6.Turano, C., Gaucci, E., Grillo, C., and
Chichiarelli, S. (2011) Cell Mol. Biol. Lett., 16,
539-563.
7.Salmans, M. L., Zhao, F., and Andersen, B. (2013)
Breast Cancer Res., 15, 204; [Epub ahead of print]
23635006 [PubMed – as supplied by publisher].
8.Verma, S., Salmans, M. L., Geyfman, M., Wang, H.,
Yu, Z., Lu, Z, Zhao, F., Lipkin, S. M., and Andersen, B. (2012) Dev.
Biol., 369, 249-260.
9.Gray, T. A., MacLaine, N. J., Michie, C. O.,
Bouchalova, P., Murray, E., Howie, J., Hrstka, R., Maslon, M. M.,
Nenutil, R., Vojtesek, B., Langdon, S., Hayward, L., Gourley, C., and
Hupp, T. R. (2012) J. Immunol. Meth., 378, 20-32.
10.Zhang, D., and Richardson, D. R. (2011) Int.
J. Biochem. Cell. Biol., 43, 33-36.
11.Benham, A. M. (2012) Antioxid. Redox.
Signal., 16, 781-789.
12.Imaoka, S. (2011) Int. Rev. Cell. Mol.
Biol., 290, 121-166.
13.Carvalho, A. P., Fernandes, P. A., and Ramos, M.
J. (2006) Prog. Biophys. Mol. Biol., 91, 229-248.
14.Pan, J. L., and Bardwell, J. C. (2006) Protein
Sci., 15, 2217-2227.
15.Gulerez, I. E., Kozlov, G., Rosenauer, A., and
Gehring, K. (2012) Acta Crystallogr. Sect. F Struct. Biol. Cryst.
Commun., 68, 378-381.
16.Watson, W. H., Yang, X., Choi, Y. E., Jones, D.
P., and Kehrer, J. P. (2004) Toxicol. Sci., 78, 3-16.
17.Myers, C. R., Myers, J. M., Kufahl, T. D.,
Forbes, R., and Szadkowski, A. (2011) Mol. Nutr. Food Res.,
55, 1361-1374.
18.Lumb, R. A., and Bulleid, N. J. (2002) EMBO
J., 21, 6763-6770.
19.Schwaller, M., Wilkinson, B., and Gilbert, H. F.
(2003) J. Biol. Chem., 278, 7154-7159.
20.Alanen, H. I., Williamson, R. A., Howard, M. J.,
Hatahet, F. S., Salo, K. E., Kauppila, A., Kellokumpu, S., and Ruddock,
L. W. (2006) J. Biol. Chem., 281, 33727-33738.
21.Anelli, T., Alessio, M., Mezghrani, A., Simmen,
T., Talamo, F., Bachi, A., and Sitia, R. (2002) EMBO J.,
21, 835-844.
22.Persson, S., Rosenquist, M., Knoblach, B.,
Khosravi-Far, R., Sommarin, M., and Michalak, M. (2005) Mol.
Phylogenet. Evol., 36, 734-740.
23.Park, S. W., Zhen, G., Verhaeghe, C., Nakagami,
Y., Nguyenvu, L. T., Barczak, A. J., Killeen, N., and Erle, D. J.
(2009) Proc. Natl. Acad. Sci. USA, 106, 6950-6955.
24.Gupta, A., Dong, A., and Lowe, A. W. (2012) J.
Biol. Chem., 287, 4773-4782.
25.Aberger, F., Weidinger, G., Grunz, H., and
Richter, K. (1998) Mech. Dev., 72, 115-130.
26.Thompson, D. A., and Weigel, R. J. (1998)
Biochem. Biophys. Res. Commun., 251, 111-116.
27.Petek, E., Windpassinger, C., Egger, H., Kroisel,
P. M., and Wagner, K. (2000) Cytogenet. Cell. Genet., 89,
141-142.
28.Fletcher, G. C., Patel, S., Tyson, K., Adam, P.
J., Schenker, M., Loader, J. A., Daviet, L., Legrain, P., Parekh, R.,
Harris, A. L., and Terrett, J. A. (2003) Br. J. Cancer,
88, 579-585.
29.Zhang, Z., and Henzel, W. J. (2004) Protein
Sci., 13, 2819-2824.
30.Ramachandran, V., Arumugam, T., Wang, H., and
Logsdon, C. D. (2008) Cancer Res., 68, 7811-7818.
31.Chung, K., Nishiyama, N., Yamano, S., Komatsu,
H., Hanada, S., Wei, M., Wanibuchi, H., Suehiro, S., and Kakehashi, A.
(2011-2012) Cancer Biomark., 10, 101-107.
32.Wayner, E. A., Quek, S. I., Ahmad, R., Ho, M. E.,
Loprieno, M. A., Zhou, Y., Ellis, W. J., True, L. D., and Liu, A. Y.
(2012) Prostate, 72, 1023-1034.
33.Zheng, W., Rosenstiel, P., Huse, K., Sina, C.,
Valentonyte, R., Mah, N., Zeitlmann, L., Grosse, J., Ruf, N., Nurnberg,
P., Costello, C. M., Onnie, C., Mathew, C., Platzer, M., Schreiber, S.,
and Hampe, J. (2006) Genes Immun., 7, 11-18.
34.Zhang, Y., Ali, T. Z., Zhou, H., D’Souza,
D. R., Lu, Y., Jaffe, J., Liu, Z., Passaniti, A., and Hamburger, A. W.
(2010) Cancer Res., 70, 240-248.
35.Zhang, J. S., Gong, A., Cheville, J. C., Smith,
D. I., and Young, C. Y. (2005) Genes Chromosomes Cancer,
43, 249-259.
36.Bu, H., Schweiger, M. R., Manke, T., Wunderlich,
A., Timmermann, B., Kerick, M., Pasqualini, L., Shehu, E., Fuchsberger,
C., Cato, A. C., and Klocker, H. (2013) FEBS J., 280,
1249-1266.
37.Vanderlaag, K. E., Hudak, S., Bald, L.,
Fayadat-Dilman, L., Sathe, M., Grein, J., and Janatpour, M. J. (2010)
Breast Cancer Res., 12, R32; doi: 10.1186/bcr2586.
38.Ambolet-Camoit, A., Buim, L. C., Pierre, S.,
Chevallier, A., Marchand, A., Coumoul, X., Garlatti, M., Andreau, K.,
Barouki, R., and Aggerbeck, M. (2010) Toxicol. Sci., 115,
501-512.
39.Hong, X. Y., Wang, J., and Li, Z. (2013) Cell.
Biochem. Biophys., May 28 [Epub ahead of print] PMID: 23712868.
40.Lu, X., and Kang, Y. (2010) Clin. Cancer
Res., 16, 5928-5935.
41.Gardner, L. B., and Corn, P. G. (2008) Cell
Cycle, 7, 1916-1924.
42.Wang, Z., Hao, Y., and Lowe, A. W. (2008)
Cancer Res., 68, 492-497.
43.Pohler, E., Craig, A. L., Cotton, J., Lawrie, L.,
Dillon, J. F., Ross, P., Kernohan, N., and Hupp, T. R. (2004) Mol.
Cell. Proteom., 3, 534-547.
44.Murray, E., McKenna, E. O., Burch, L. R., Dillon,
J., Langridge-Smith, P., Kolch, W., Pitt, A., and Hupp, T. R. (2007)
Biochemistry, 46, 13742-13751.
45.Wu, J., Wang, C., Li, X., Song, Y., Wang, W., Li,
C., Hu, J., Zhu, Z., Li, J., Zhang, W., Lu, Z., and Yang, C. J. (2012)
PLoS One, 7, e46393; doi:
10.1371/journal.pone.0046393.
46.Norris, A. M., Gore, A., Balboni, A., Young, A.,
Longnecker, D. S., and Korc, M. (2012) Oncogene, Sep 3; doi:
10.1038/onc.2012.394.
47.Maslon, M. M., Hrstka, R., Vojtesek, B., and
Hupp, T. R. (2010) J. Mol. Biol., 404, 418-438.
48.Yu, H., Zhao, J., Lin, L., Zhang, Y., Zhong, F.,
Liu, Y., Yu, Y., Shen, H., Han, M., He, F., and Yang, P. (2012) Mol.
Biosyst., 8, 2710-2718.
49.Ryu, J., Park, S. G., Lee, P. Y., Cho, S., Lee,
do H., Kim, G. H., Kim, J. H., and Park, B. C. (2013) Biochem.
Biophys. Res. Commun., 430, 610-615.
50.Kaser, A., Adolph, T. E., and Blumberg, R. S.
(2013) Semin. Immunopathol., 35, 307-319.
51.Patel, P., Clarke, C., Barraclough, D. L.,
Jowitt, T. A., Rudland, P. S., Barraclough, R., and Lian, L. Y. (2013)
J. Mol. Biol., 425, 929-943.
52.Gray, T. A., Murray, E., Nowicki, M. W., Remnant,
L., Scherl, A., Muller, P., Vojtesek, B., and Hupp, T. R. (2013)
Protein Sci., Jun 18; doi: 10.1002/pro.2299 [Epub ahead of
print].
53.Hirano, N., Shibasaki, F., Sakai, R., Tanaka, T.,
Nishida, J., Yazaki, Y., Takenawa, T., and Hirai, H. (1995) Eur. J.
Biochem., 234, 336-342.
54.Bourdi, M., Demady, D., Martin, J. L., Jabbour,
S. K., Martin, B. M., George, J. W., and Pohl, L. R. (1995) Arch.
Biochem. Biophys., 323, 397-403.
55.Alanen, H. I., Salo, K. E., Pekkala, M.,
Siekkinen, H. M., Pirneskoski, A., and Ruddock, L. W. (2003)
Antioxid. Redox. Signal., 5, 367-374.
56.Russell, S. J., Ruddock, L. W., Salo, K. E.,
Oliver, J. D., Roebuck, Q. P., Llewellyn, D. H., Roderick, H. L.,
Koivunen, P., Myllyharju, J., and High, S. (2004) J. Biol.
Chem., 279, 18861-18869.
57.Coe, H., and Michalak, M. (2010) Int. J.
Biochem. Cell. Biol., 42, 796-799.
58.Frickel, E. M., Frei, P., Bouvier, M., Stafford,
W. F., Helenius, A., Glockshuber, R., and Ellgaard, L. (2004) J.
Biol. Chem., 279, 18277-18287.
59.Lindquist, J. A., Jensen, O. N., Mann, M., and
Hammerling, G. J. (1998) EMBO J., 17, 2186-2195.
60.Dong, G., Wearsch, P. A., Peaper, D. R.,
Cresswell, P., and Reinisch, K. M. (2009) Immunity, 30,
21-32.
61.Boyle, L. H., Hermann, C., Boname, J. M., Porter,
K. M., Patel, P. A., Burr, M. L., Duncan, L. M., Harbour, M. E.,
Rhodes, D. A., Skjodt, K., Lehner, P. J., and Trowsdale, J. (2013)
Proc. Natl. Acad. Sci. USA, 110, 3465-3470.
62.Aureli, C., Gaucci, E., Arcangeli, V., Grillo,
C., Eufemi, M., and Chichiarelli, S. (2013) Gene, 524,
390-395.
63.Coppari, S., Altieri, F., Ferraro, A.,
Chichiarelli, S., Eufemi, M., and Turano, C. (2002) J. Cell.
Biochem., 85, 325-333.
64.Grillo, C., Coppari, S., Turano, C., and Altieri,
F. (2002) Biochem. Biophys. Res. Commun., 295, 67-73.
65.Turano, C., Coppari, S., Altieri, F., and
Ferraro, A. (2002) J. Cell. Physiol., 193, 154-163.
66.Eufemi, M., Coppari, S., Altieri, F., Grillo, C.,
Ferraro, A., and Turano, C. (2004) Biochem. Biophys. Res.
Commun., 323, 1306-1312.
67.Yang, J., and Stark, G. R. (2008) Cell
Res., 18, 443-451.
68.Ramirez-Rangel, I., Bracho-Valdes, I.,
Vazquez-Macias, A., Carretero-Ortega, J., Reyes-Cruz, G., and
Vazquez-Prado, J. (2011) Mol. Cell Biol., 31,
1657-1671.
69.Topisirovic, I., and Sonenberg, N. (2011) Cold
Spring Harb. Symp. Quant. Biol., 76, 355-367.
70.Laplante, M., and Sabatini, D. M. (2013) J.
Cell. Sci., 126, 1713-1719.
71.Grillo, C., D’Ambrosio, C., Scaloni, A.,
Maceroni, M., Merluzzi, S., Turano, C., and Altieri, F. (2006) Free
Radical Biol. Med., 41, 1113-1123.
72.Bhakat, K. K., Mantha, A. K., and Mitra, S.
(2009) Antioxid. Redox. Signal., 11, 621-638.
73.Cardoso, A. A., Jiang, Y., Luo, M., Reed, A. M.,
Shahda, S., He, Y., Maitra, A., Kelley, M. R., and Fishel, M. L. (2012)
PLoS One, 7, e47462; doi:
10.1371/journal.pone.0047462.
74.Krynetski, E. Y., Krynetskaia, N. F., Bianchi, M.
E., and Evans, W. E. (2003) Cancer Res., 63, 100-106.
75.Chichiarelli, S., Ferraro, A., Altieri, F.,
Eufemi, M., Coppari, S., Grillo, C., Arcangeli, V., and Turano, C.
(2007) J. Cell. Physiol., 210, 343-351.
76.Urade, R., Oda, T., Ito, H., Moriyama, T.,
Utsumi, S., and Kito, M. (1997) J. Biochem., 122,
834-842.
77.Okudo, H., Urade, R., Moriyama, T., and Kito, M.
(2000) FEBS Lett., 465, 145-147.
78.Rutledge, A. C., Zhang, R., Urade, R., and Adeli,
K. (2013) Arch. Biochem. Biophys., Jul 1; pii:
S0003-9861(13)00183-5; doi: 10.1016/j.abb.2013.06.013.
79.Okudo, H., Kito, M., Moriyama, T., Ogawa, T., and
Urade, R. (2002) Biosci. Biotechnol. Biochem., 66,
1423-1426.
80.Cunnea, P. M., Miranda-Vizuete, A., Bertoli, G.,
Simmen, T., Damdimopoulos, A. E., Hermann, S., Leinonen, S., Huikko, M.
P., Gustafsson, J. A., Sitia, R., and Spyrou, G. (2003) J. Biol.
Chem., 278, 1059-1066.
81.Walsh, P., Bursac, D., Law, Y. C., Cyr, D., and
Lithgow, T. (2004) EMBO Rep., 5, 567-571.
82.Sterrenberg, J. N., Blatch, G. L., and Edkins, A.
L. (2011) Cancer Lett., 312, 129-142.
83.Cheong, K., Choi, J., Choi, J., Park, J., Jang,
S., and Lee, Y. H. (2013) Genomics Inform., 11,
52-54.
84.Cunnea, P., Fernandes, A. P., Capitanio, A.,
Eken, S., Spyrou, G., and Bjornstedt, M. (2007) Int. J.
Immunopathol. Pharmacol., 20, 17-24.
85.Corazzari, M., Lovat, P. E., Armstrong, J. L.,
Fimia, G. M., Hill, D. S., Birch-Machin, M., Redfern, C. P., and
Piacentini, M. (2007) Br. J. Cancer, 96, 1062-1071.
86.Dong, M., Bridges, J. P., Apsley, K., Xu, Y., and
Weaver, T. E. (2008) Mol. Biol. Cell, 19, 2620-2630.
87.Ushioda, R., Hoseki, J., Araki, K., Jansen, G.,
Thomas, D. Y., and Nagata, K. (2008) Science, 321,
569-572.
88.Riemer, J., Appenzeller-Herzog, C., Johansson,
L., Bodenmiller, B., Hartmann-Petersen, R., and Ellgaard, L. (2009)
Proc. Natl. Acad. Sci. USA, 106, 14831-14836.
89.Thomas, C. G., and Spyrou, G. (2009) J. Biol.
Chem., 284, 6282-6290.
90.Hosoda, A., Tokuda, M., Akai, R., Kohno, K., and
Iwawaki, T. (2010) Biochem. J., 425, 117-125.
91.Maattanen, P., Gehring, K., Bergeron, J. J., and
Thomas, D. Y. (2010) Semin. Cell. Dev. Biol., 21,
500-511.
92.Hosokawa, N., Wada, I., Hasegawa, K., Yorihuzi,
T., Tremblay, L. O., Herscovics, A., and Nagata, K. (2001) EMBO
Rep., 2, 415-422.
93.Tamura, T., Cormier, J. H., and Hebert, D. N.
(2011) J. Biol. Chem., 286, 24906-24915.
94.Hagiwara, M., Maegawa, K., Suzuki, M., Ushioda,
R., Araki, K., Matsumoto, Y., Hoseki, J., Nagata, K., and Inaba, K.
(2011) Mol. Cell, 41, 432-444.
95.Diamanti, E., Mathieu, S., Jeanneau, C., Kitraki,
E., Panopoulos, P., Spyrou, G., and About, I. (2013) Int. Endod.
J., 46, 160-168.
96.Williams, J. M., Inoue, T., Banks, L., and Tsai,
B. (2013) Mol. Biol. Cell, 24, 785-795.
97.Munoz-Lobato, F., Rodriguez-Palero, M. J.,
Naranjo-Galindo, F. J., Shephard, F., Gaffney, C. J., Szewczyk, N. J.,
Hamamichi, S., Caldwell, K. A., Caldwell, G. A., Link, C. D., and
Miranda-Vizuete, A. (2013) Antioxid. Redox. Signal., Jul 3,
[Epub ahead of print] PMID: 23641861.
98.Oka, O. B., Pringle, M. A., Schopp, I. M.,
Braakman, I., and Bulleid, N. J. (2013) Mol. Cell, 50,
793-804.
99.Jordan, P. A., Stevens, J. M., Hubbard, G. P.,
Barrett, N. E., Sage, T., Authi, K. S., and Gibbins, J. M. (2005)
Blood, 105, 1500-1507.
100.Kaiser, B. K., Yim, D., Chow, I. T., Gonzalez,
S., Dai, Z., Mann, H. H., Strong, R. K., Groh, V., and Spies, T. (2007)
Nature, 447, 482-486.
101.Huergo-Zapico, L., Gonzalez-Rodriguez, A. P.,
Contesti, J., Gonzalez, E., Lopez-Soto, A., Fernandez-Guizan, A.,
Acebes-Huerta, A., de Los Toyos, J. R., Lopez-Larrea, C., Groh, V.,
Spies, T., and Gonzalez, S. (2012) Cancer Immunol. Immunother.,
61, 1201-1210.
102.Matsuo, Y., Akiyama, N., Nakamura, H., Yodoi,
J., Noda, M., and Kizaka-Kondoh, S. (2001) J. Biol. Chem.,
276, 10032-10038.
103.Matsuo, Y., Nishinaka, Y., Suzuki, S., Kojima,
M., Kizaka-Kondoh, S., Kondo, N., Son, A., Sakakura-Nishiyama, J.,
Yamaguchi, Y., Masutani, H., Ishii, Y., and Yodoi, J. (2004) Arch.
Biochem. Biophys., 423, 81-87.
104.Matsuo, Y., Masutani, H., Son, A.,
Kizaka-Kondoh, S., and Yodoi, J. (2009) Mol. Biol. Cell,
20, 4552-4562.
105.Lynes, E. M., Bui, M., Yap, M. C., Benson, M.
D., Schneider, B., Ellgaard, L., Berthiaume, L. G., and Simmen, T.
(2012) EMBO J., 31, 457-470.
106.Matsuo, Y., Irie, K., Kiyonari, H., Okuyama,
H., Nakamura, H., Son, A., Lopez-Ramos, D. A., Tian, H., Oka, S.,
Okawa, K., Kizaka-Kondoh, S., Masutani, H., and Yodoi, J. (2013)
Antioxid. Redox. Signal., 18, 1263-1272.
107.Kozlov, G., Maattanen, P., Thomas, D. Y., and
Gehring, K. (2010) FEBS J., 277, 3924-3936.
108.Roth, D., Lynes, E., Riemer, J., Hansen, H. G.,
Althaus, N., Simmen, T., and Ellgaard, L. (2009) Biochem. J.,
425, 195-205.
109.Huber, M., Bahr, I., Kratzschmar, J. R.,
Becker, A., Muller, E. C., Donner, P., Pohlenz, H. D., Schneider, M.
R., and Sommer, A. (2004) Mol. Cell Proteom., 3,
43-55.
110.Kristiansen, G., Pilarsky, C., Wissmann, C.,
Kaiser, S., Bruemmendorf, T., Roepcke, S., Dahl, E., Hinzmann, B.,
Specht, T., Pervan, J., Stephan, C., Loening, S., Dietel, M., and
Rosenthal, A. (2005) J. Pathol., 205, 359-376.
111.Smirnov, D. A., Zweitzig, D. R., Foulk, B. W.,
Miller, M. C., Doyle, G. V., Pienta, K. J., Meropol, N. J., Weiner, L.
M., Cohen, S. J., Moreno, J. G., Connelly, M. C., Terstappen, L. W.,
and O’Hara, S. M. (2005) Cancer Res., 65,
4993-4997.
112.Kovalev, L. I., Shishkin, S. S., Khasigov, P.
Z., Dzeranov, N. K., Kazachenko, A. V., Kovaleva, M. A., Toropygin, I.
Y., and Mamykina, S. V. (2006) Prikl. Biokhim. Mikrobiol.,
42, 480-484.
113.Zhang, Y., Forootan, S. S., Liu, D.,
Barraclough, R., Foster, C. S., Rudland, P. S., and Ke, Y. (2007)
Prostate Cancer Prostatic Dis., 10, 293-300.
114.Maresh, E. L., Mah, V., Alavi, M., Horvath, S.,
Bagryanova, L., Liebeskind, E. S., Knutzen, L. A., Zhou, Y., Chia, D.,
Liu, A. Y., and Goodglick, L. (2010) BMC Cancer, 10, Dec
13; 10:680.doi: 10.1186/1471-2407-10-680.
115.Bu, H., Bormann, S., Schafer, G., Horninger,
W., Massoner, P., Neeb, A., Lakshmanan, V. K., Maddalo, D., Nestl, A.,
Sultmann, H., Cato, A. C., and Klocker, H. (2011) Prostate,
71, 575-587.
116.Kani, K., Malihi, P. D., Jiang, Y., Wang, H.,
Wang, Y., Ruderman, D. L., Agus, D. B., Mallick, P., and Gross, M. E.
(2013) Prostate, 73, 306-315.
117.Ho, M. E., Quek, S. I., True, L. D., Morrissey,
C., Corey, E., Vessella, R. L., Dumpit, R., Nelson, P. S., Maresh, E.
L., Mah, V., Alavi, M., Kim, S. R., Bagryanova, L., Horvath, S., Chia,
D., Goodglick, L., and Liu, A. Y. (2013) Mod. Pathol.,
26, 849-859.
118.Wu, Z. S., Wu, Q., Ding, X. D., Wang, H. Q.,
Shen, Y. X., and Fang, S. Y. (2008) Zhonghua Bing Li Xue Za Zhi,
37, 109-113 (PMID:18681322).
119.Hengel, S. M., Murray, E., Langdon, S.,
Hayward, L., O’Donoghue, J., Panchaud, A., Hupp, T., and
Goodlett, D. R. (2011) J. Proteome Res., 10,
4567-4578.
120.Park, K., Chung, Y. J., So, H., Kim, K., Park,
J., Oh, M., Jo, M., Choi, K., Lee, E. J., Choi, Y. L., Song, S. Y.,
Bae, D. S., Kim, B. G., and Lee, J. H. (2011) Exp. Mol. Med.,
43, 91-100.
121.DiMaio, M. A., Kwok, S., Montgomery, K. D.,
Lowe, A. W., and Pai, R. K. (2012) Hum. Pathol., 43,
1799-1807.
122.Lee, D. H., Lee, Y., Ryu, J., Park, S. G., Cho,
S., Lee, J. J., Choi, C., and Park, B. C. (2011) Mol. Cells,
31, 563-571.
123.Chung, K., Nishiyama, N., Wanibuchi, H.,
Yamano, S., Hanada, S., Wei, M., Suehiro, S., and Kakehashi, A. (2012)
Osaka City Med. J., 58, 13-24.
124.Barraclough, D. L., Platt-Higgins, A., de Silva
Rudland, S., Barraclough, R., Winstanley, J., West, C. R., and Rudland,
P. S. (2009) Am. J. Pathol., 175, 1848-1857.
125.Hapangama, D. K., Raju, R. S., Valentijn, A.
J., Barraclough, D., Hart, A., Turner, M. A., Platt-Higgins, A.,
Barraclough, R., and Rudland, P. S. (2012) Hum. Reprod.,
27, 394-407.
126.Shishkin, S. S., Dzeranov, N. K., Totrov, K.
I., Kazachenko, A. V., Kovalev, L. I., Eremina, L. S., Kovaleva, M. A.,
and Toropygin, I. Y. (2009) Urologiya, No. 1, 56-58.
127.Pizzi, M., Fassan, M., Realdon, S., Balistreri,
M., Battaglia, G., Giacometti, C., Zaninotto, G., Zagonel, V., De Boni,
M., and Rugge, M. (2012) Hum. Pathol., 43, 1839-1844.
128.Chen, R., Pan, S., Duan, X., Nelson, B. H.,
Sahota, R. A., de Rham, S., Kozarek, R. A., McIntosh, M., and
Brentnall, T. A. (2010) Mol. Cancer, 9, 149; doi:
10.1186/1476-4598-9-149.
129.Gupta, A., Wodziak, D., Tun, M., Bouley, D. M.,
and Lowe, A. W. (2013) J. Biol. Chem., 288,
4321-4333.
130.Izquierdo, L., Mengual, L., Gazquez, C.,
Ingelmo-Torres, M., and Alcaraz, A. (2010) BJU Int., 106,
868-872.
131.Ko, W. C., Sugahara, K., Sakuma, T., Yen, C.
Y., Liu, S. Y., Liaw, G. A., and Shibahara, T. (2012) Oncol.
Lett., 3, 75-81.
132.Kozaki, K., Miyaishi, O., Asai, N., Iida, K.,
Sakata, K., Hayashi, M., Nishida, T., Matsuyama, M., Shimizu, S.,
Kaneda, T., et al. (1994) Exp. Cell. Res., 213,
348-358.
133.Guo, G. G., Patel, K., Kumar, V., Shah, M.,
Fried, V. A., Etlinger, J. D., and Sehgal, P. B. (2002) J.
Interferon Cytokine Res., 22, 555-563.
134.Frank, D. A. (1999) Mol. Med., 5,
432-456.
135.Celli, C. M., and Jaiswal, A. K. (2003)
Cancer Res., 63, 6016-6025.
136.Adikesavan, A. K., and Jaiswal, A. K. (2007)
Mol. Cancer Ther., 6, 2719-2727.
137.Grindel, B. J., Rohe, B., Safford, S. E.,
Bennett, J. J., and Farach-Carson, M. C. (2011) J. Cell.
Biochem., 112, 2606-2615.
138.Liao, C. J., Wu, T. I., Huang, Y. H., Chang, T.
C., Wang, C. S., Tsai, M. M., Lai, C. H., Liang, Y., Jung, S. M., and
Lin, K. H. (2011) Cancer Sci., 102, 2255-2263.
139.Garg, A. D., Krysko, D. V., Vandenabeele, P.,
and Agostinis, P. (2012) Cancer Immunol. Immunother., 61,
215-221.
140.Elisa, G., Fabio, A., Turano, C., and Silvia,
C. (2013) J. Cell. Biochem., 2013 May 20; doi: 10.1002/jcb.24590
[Epub ahead of print] PMID: 23696074.
141.Laurie, S. A., and Goss, G. D. (2013) J.
Clin. Oncol., 31, 1061-1069.
142.Santana-Codina, N., Carretero, R.,
Sanz-Pamplona, R., Cabrera, T., Guney, E., Oliva, B., Clezardin, P.,
Olarte, O. E., Loza-Alvarez, P., Mendez-Lucas, A., Perales, J. C., and
Sierra, A. (2013) Mol. Cell. Proteomics, 2013 Apr 26 [Epub ahead
of print] PMID: 23625662.