REVIEW: Hydroxylamine Derivatives for Regulation of Spermine and Spermidine Metabolism
M. A. Khomutov1, J. Weisell2, M. Hyvönen2, T. A. Keinänen2, J. Vepsäläinen2, L. Alhonen2, A. R. Khomutov1*, and S. N. Kochetkov1
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; E-mail: alexkhom@list.ru2School of Pharmacy, Biocenter Kuopio, University of Eastern Finland, Yliopistonranta 1E, 70210, Kuopio, Finland
* To whom correspondence should be addressed.
Received July 17, 2013
The biogenic polyamines spermine, spermidine, and their precursor putrescine are present in micro-to-millimolar concentrations in all cell types and are vitally important for their normal growth. High intracellular content of spermine and spermidine determines the multiplicity of the cellular functions of the polyamines. Many of these functions are not well characterized at the molecular level, ensuring the ongoing development of this field of biochemistry. Tumor cells have elevated polyamine level if compared with normal cells, and this greatly stimulates the search for new opportunities to deplete the intracellular pool of spermine and spermidine resulting in decrease in cell growth and even cell death. O-Substituted hydroxylamines occupy their own place among chemical regulators of the activity of the enzymes of polyamine metabolism. Varying the structure of the alkyl substituent made it possible to obtain within one class of chemical compounds highly effective inhibitors and regulators of the activity of all the enzymes of putrescine, spermine and spermidine metabolism (with the exception of FAD-dependent spermine oxidase and acetylpolyamine oxidase), effectors of the polyamine transport system, and even actively transported in cells “proinhibitor” of ornithine decarboxylase. Some principles for the design of specific inhibitors of these enzymes as well as the peculiarities of cellular effects of corresponding O-substituted hydroxylamines are discussed.
KEY WORDS: polyamines, spermine, spermidine, O-substituted hydroxylamines, ornithine decarboxylase, S-adenosylmethionine decarboxylase, spermidine/spermine-N1-acetyltransferase, cell cultureDOI: 10.1134/S0006297913130051
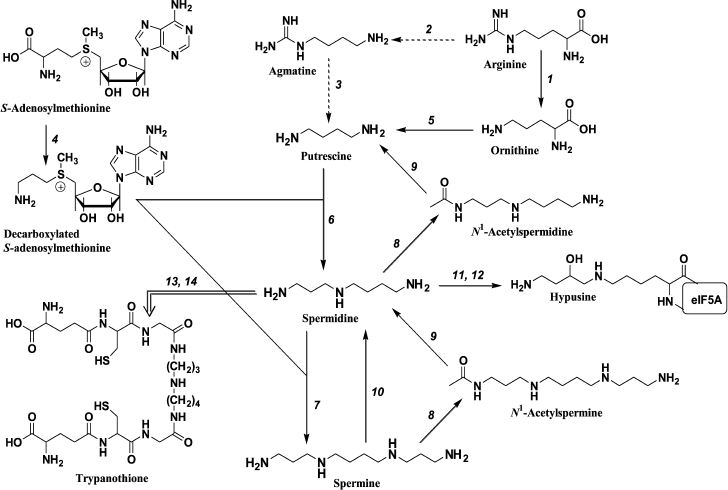
The history of biogenic polyamines spermine (Spm) and spermidine (Spd) rises up to the second half of XVII century, when Antoine van Leeuwenhoek discovered unknown crystals in semen [1]. Two hundred and fifty years later this compound was identified as Spm phosphate [2]. Nevertheless, polyamine biochemistry is a young and fast-developing field – its modern history started in the late 1960s when mammalian ornithine decarboxylase was discovered [3, 4]. This finding was the key to bringing the system of Spm and Spd biosynthesis into its modern form (Fig. 1). In the beginning of the 1970s, it was established that tumor cells contain higher amounts of polyamines as compared with normal cells [5], and this finding greatly stimulated the development of molecular biology and biochemistry of polyamines.
The biogenic polyamines Spm, Spd, and their precursor putrescine (Put) are present in micro-to-millimolar concentrations in all living organisms and are vitally important for normal growth of cells [6, 7]. The pancreas has the highest concentration (7-8 mM) of Spm and Spd in humans [8]. Depletion of the intracellular pool of Spm and Spd slows cell growth and may even result in cell death. High concentration of Spm/Spd, whose amino groups are protonated at physiological pH, a priori determines the multiplicity of cellular functions of polyamine, and these are presumably described in terms of specific electrostatic interactions with DNA, RNA, and other negatively charged macromolecules, nucleoprotein complexes, receptors, and membrane fragments [6, 7, 9].
Fig. 1. Polyamine metabolism: 1) arginase; 2) arginine decarboxylase; 3) agmatinase; 4) S-adenosylmethionine decarboxylase; 5) ornithine decarboxylase; 6) spermidine synthase; 7) spermine synthase; 8) spermidine/spermine N1-acetyltransferase; 9) acetylpolyamine oxidase; 10) spermine oxidase; 11) deoxyhypusine synthase; 12) hypusine synthase; 13) glutathionespermidine synthetase; 14) tripanothione synthetase. Transformations that are typical for mammals (→), bacteria (-->), and pathogenic trypanosomatides (= = >) are shown.
The rate-limiting enzymes of polyamine biosynthesis are pyridoxal-5′-phosphate-dependent ornithine decarboxylase (ODC) and pyruvate-dependent S-adenosylmethionine decarboxylase (AdoMetDC), while the key enzyme of polyamine catabolism is spermidine/spermine-N1-acetyltransferase (SSAT) (Fig. 1). Biosynthesis of these enzymes is precisely controlled at the transcriptional and translational levels, and key regulatory mechanisms are established both for normal and tumor cells [7, 9]. All cells are equipped with a system of polyamine active transport, which has an important impact on cellular polyamine pool supplying up to 30% of total amount of Spm, Spd, and Put. The small (27 kDa) short-living protein antizyme (AZ) is one of the most important regulators of polyamine homeostasis in cells – its biosynthesis is triggered by increased intracellular concentration of Spm and Spd [10]. AZ is known to inhibit uptake of polyamines, and it forms a tight complex with the ODC subunit and delivers it to the 26S proteasome [11]. All these achieve fast and efficient control of intracellular polyamine content [12].
Design and synthesis of specific inhibitors of the enzymes of Spm and Spd biosynthesis, as well as inducers of SSAT, was for many years one of the key vectors of polyamine biochemistry [13-15]. This goal is closely related to practical medicine, since inhibitors of polyamine biosynthesis have expressed anti-parasite activity (polyamines are of crucial importance for the life cycle of such disease-causing trypanosomatides as Plasmodium falciparum, Trypanosoma brucei gambiense, Leishmania donovani, etc.), and because inhibitors of polyamine biosynthesis have a potential for the treatment of some oncological diseases (polyamine level in tumor cells is higher compared with that in normal cells). Many data disclosing the role and functions of Spm and Spd, as well as regulatory mechanisms responsible for maintaining of polyamine homeostasis in a cell, were obtained using inhibitors of polyamine biosynthesis (reviewed in [13, 15]). A set of highly effective inducers of SSAT was synthesized, and their ability to efficiently deplete the intracellular pool of Spm and Spd was studied [16, 17]. The best of these compounds are active in vivo and are at different stages of clinical trials as potential antitumor and anti-parasite drugs (reviewed in [7, 9, 18, 19]).
O-Substituted hydroxylamines occupy their own place among chemical regulators of the activity of the enzymes of polyamine metabolism. Varying the structure of the alkyl substituent made it possible to obtain within one class of chemical compounds highly effective inhibitors and regulators of the activity of all the enzymes of Put, Spm, and Spd metabolism (with the exception of FAD-dependent SMO and APAO), effectors of the polyamine transport system, and even actively transported in cells proinhibitor of ODC. Hydroxylamine-containing inhibitors of ODC and AdoMetDC (the most investigated representatives of this family of compounds) were found to be essential instruments for the investigation of cellular functions and metabolism of Spm and Spd and provided mile-stone results for the field.
HYDROXYLAMINE-CONTAINING ANALOGS OF DECARBOXYLATED
S-ADENOSYLMETHIONINE
S-Adenosylmethionine decarboxylase (AdoMetDC, EC 4.1.1.50) belongs to a small family of enzymes having covalently bonded pyruvate residue as a prosthetic group [20]. AdoMetDC, like other pyruvate-dependent decarboxylases, is synthesized as a proenzyme that after autocatalytic splitting gives rise to the α-subunit containing 266 amino acids (human enzyme) with an N-terminal pyruvate residue and a smaller β-subunit (67 amino acid residues) [20].
Hydroxylamine-containing inhibitors of AdoMetDC were first synthesized in early 1980s and were derived from sulfonium-containing hydroxylamines, analogs of decarboxylated AdoMet (deAdoMet) and methylmethionine [21, 22]. The most active of these compounds was S-(5′-deoxy-5′-adenosyl)[(methylthio)ethyl]hydroxylamine – AMA (Table 1). Substitution of the terminal aminomethylene group of deAdoMet with an aminooxy group is isosteric, which is crucial for structural similarity with the substrate and obeys specific binding of the inhibitor in the enzyme active center. The sulfonium center and adenosine group fulfill anchoring functions and are necessary for the exact recognition of the inhibitor by the enzyme. Therefore, AMA was about five orders of magnitude more effective compared with compounds (I) and (II) (Table 1). The properly positioned aminooxy group of AMA forms an oxime with a pyruvate residue of the active center – this is the reason for irreversible inhibition of AdoMetDC. Inhibition develops in time because in the active center of AdoMetDC the carbonyl group of pyruvate, in contrast to PLP-dependent enzymes, is not activated by forming a protonated internal aldimine with a Lys residue. The substrate is not able to restore the activity of AdoMetDC, but competing with the inhibitor for binding protected the enzyme from the action of AMA. All the above explains why under the standard assay (without preincubation of the enzyme with the inhibitor) AMA was less effective towards the enzyme from L1210 cells (IC50 3·10–8 M [23]) when compared with that for the enzyme from rat liver (IC50 3·10–9 M [22]) – in the latter case 30 min preincubation of the inhibitor with AdoMetDC was used.
Inhibition of AdoMetDC with deAdoMet hydroxylamine-containing analogs
[39]
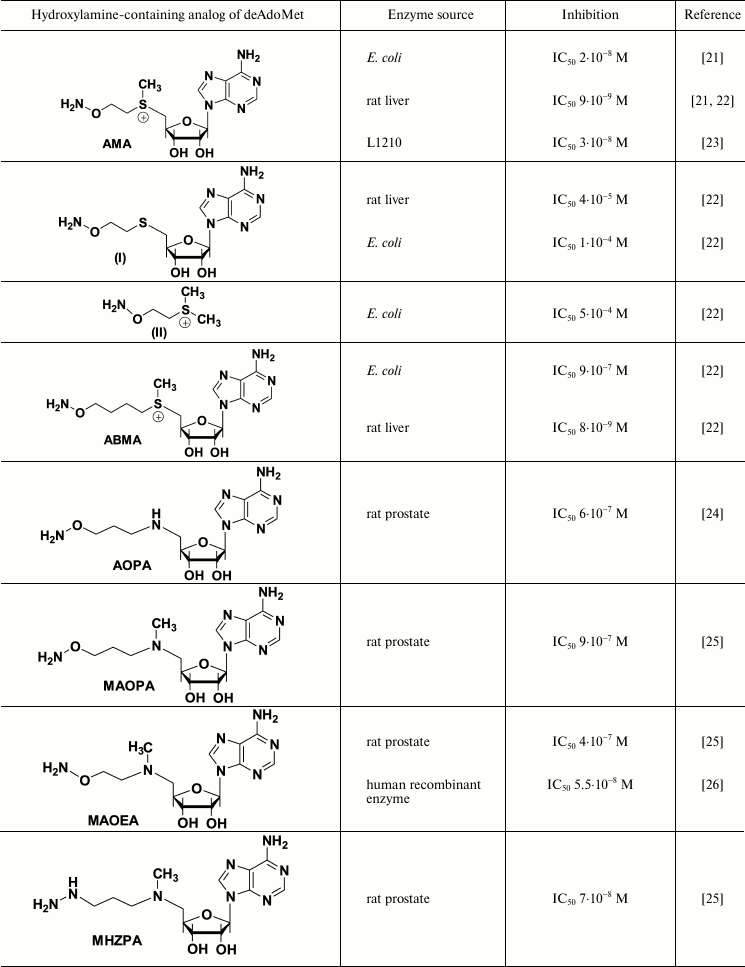
AMA is a monocation at physiological pH value (the pKa of the H2NO-group is ~4.5, which is for five orders of magnitude lower than that of the amino group of deAdoMet), and this is essential for the recognition of the inhibitor by the enzyme. Application of the scheme of Kitz and Wilson [30] to describe the inhibition of bacterial AdoMetDC with AMA clearly showed the process to be two-stage. Moreover, the affinity of the inhibitor towards the enzyme at the reversible step was 100 time better than the Km of the substrate and 10 times better than the affinity of deAdoMet to the enzyme, while kinact of AdoMetDC from E. coli was determined as 0.8·10–2 sec–1 [22]. The specificity of the action of AMA towards AdoMetDC was confirmed with low activity of AMA towards PLP-dependent ODC and aspartate aminotransferase. These two enzymes were inhibited only by micro- and millimolar concentrations of AMA, respectively.
The activity of most eukaryotic AdoMetDC is allosterically regulated by putrescine. The mechanism of this activation apparently includes putrescine-induced conformational change of the protein structure, which results in 10-fold decrease in Km of the substrate (0.06 mM at pH 7.5), changes in the pH profile of enzymatic reaction, and also decrease in kcat to 2.5 sec–1 at 37°C [32]. On the contrary, bacteria including E. coli have Mg2+-dependent AdoMetDC. It turned that Mg2+ is essential for effective inhibition of AdoMet with AMA. In the absence of Mg2+, AMA was inactive against E. coli enzyme even at 6·10–6 M concentration [33], while in the presence of Mg2+ the IC50 was 2·10–8 M (Table 1). A similar decrease in the inhibitory activity in the absence of Mg2+ was observed also for 1-aminooxy-3-aminopropane (APA), which is a nonspecific irreversible inhibitor of AdoMetDC. One can assume that Mg2+ causes conformational changes of the enzyme, and a catalytically important pyruvate residue become available and reactive towards the amino group of the substrate as well as towards the aminooxy residue of AMA [33].
High inhibitory activity and specificity of the action of AMA is most likely determined by the structural similarity between AMA–AdoMetDC oxime and the external aldimine, the first intermediate of the enzymatic decarboxylation of AdoMet, but X-ray investigations of the AMA–AdoMetDC complex have never been performed. However, X-ray studies of the complex of AMA with 1-aminocyclopropanecarboxylate synthase (AdoMet is the substrate of this PLP-dependent enzyme) demonstrated that the oxime of the enzyme mimics the external aldimine [34]. Correspondingly, the same may be true also for the case of the AMA–AdoMetDC complex.
One of the peculiarities of the AdoMetDC reaction is the irreversible inhibition of the enzyme with a substrate taking place once per 6000-7000 decarboxylations [35]. The reason for “substrate inhibition” could be either transamination of the pyruvate residue and/or addition of Cys140 to the acrylate system [36, 37]. It is worth mentioning that the incubation of AdoMetDC with deAdoMet also results in the irreversible inhibition of the enzyme [38]. (Abortive transamination is well-known and typical for PLP-dependent decarboxylases [40].)
Substitution of sulfonium center of deAdoMet with the methylamino group gives rise to a nitrogen-containing analog (aza-deAdoMet), which is a rather poor competitive inhibitor of AdoMetDC having Ki 0.1 mM [38], while the product of the enzymatic reaction has affinity of a few orders of magnitude better (Ki 0.1 µM [31]). Incubation of AdoMetDC with aza-deAdoMet, in contrast to deAdoMet, did not lead to irreversible inhibition of the enzyme. Moreover, in the case of aza-deAdoMet it was impossible to reduce the Schiff base with NaBH3CN, which distinguishes in principal aza-deAdoMet from deAdoMet [38]. One possible explanation for this difference could be improper orientation of the side-chain of the inhibitor in the enzyme active center, making impossible the formation of a Schiff base.
One might assume that non-productive binding of aza-deAdoMet in the active site of AdoMetDC could be determined by abnormally low pKa of the secondary amino group of aza-deAdoMet, like the corresponding amino group of aza-AdoMet having pKa of only 7.09 [41]. Respectively, aza-AdoMet can mimic AdoMet, which is a corepressor of the methionine repressor (MetJ) of E. coli, only at pH 5.5, while at pH 7.4 the analog is not recognized as AdoMet [41].
Nevertheless, a system of hydroxylamine-containing inhibitors of AdoMetDC derived from aza-deAdoMet, i.e. AOPA [24], MAOEA, and MAOPA [25] (Table 1) has been prepared. The hydrazine-containing inhibitor of AdoMetDC, MHZPA [25], also belongs to this family of inhibitors. It was assumed that in all the above compounds the 5′-methylamino group can be protonated at physiological pH value, while the possibility of its abnormal protonation leading to the mimetics of decarboxylated S-adenosylhomocysteine (deAdoHCys), but not of deAdoMet, was not discussed earlier. Investigation of the interaction of prostate AdoMetDC, or the enzyme from L1210 cells with AOPA, MAOPA, and MAOEA (Table 1), demonstrated that these inhibitors are about 100 times less effective compared with the inhibition of rat liver AdoMetDC with AMA (Table 1). These differences can be partially attributed to different methods of determination of the enzymatic activity – 30 min preincubation with AdoMetDC was used in the case of rat liver enzyme [21, 22], while in the case of prostate AdoMetDC and the enzyme from L1210 the inhibitors were added to the substrate mixture just before the reaction was started upon the addition of AdoMetDC [25]. However, in all cases the inhibition was irreversible and developed in time. The substrate was not able to restore the activity of AdoMetDC, but it protected the enzyme. Interestingly, hydrazine-containing MHZPA (IC50 7·10–8 M) was more active compared with the corresponding hydroxylamine analogs of aza-deAdoMet [25].
Incubation of human recombinant AdoMetDC with MHZPA resulted in the appearance of absorbance at 260 nm in the UV spectrum of the α-subunit, which is an evidence of covalent binding with the inhibitor containing the adenine chromophore. Moreover, a fragment of the N-terminal peptide pyruvoyl-Ser-Met-Phe-Val-Ser-Lys containing MHZPA (according to mass-spectrometric data) was isolated after limited proteolysis [42]. However, these data do not confirm that MHZPA binds in the active center similarly to that of the substrate, since as early as in 1970 the phenylhydrazone of AdoMetDC was isolated and identified [43].
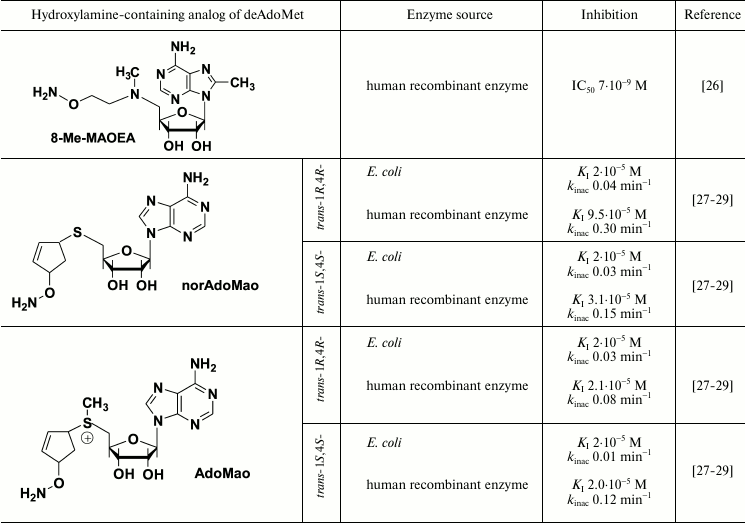
X-Ray investigation of the AdoMetDC complex with AdoMet methyl ester clearly demonstrated Schiff base formation and binding of the adenosine group in the unusual for nucleosides syn-conformation [44]. It is known that the introduction of the substituent in the eighth position of the adenine ring favors this conformation [26]. Respectively, a family of 8-substituted derivatives of MAOEA was synthesized and their inhibitory activity was investigated [26]. 8-Me-MAOEA turned to be the most active compound, and it inhibited human recombinant AdoMetDC 8-fold better compared with that of MAOEA (Table 1).
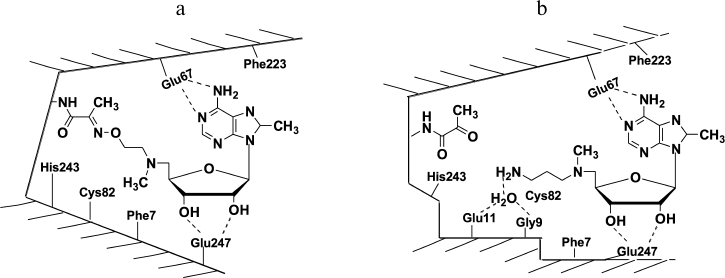
X-Ray investigation of the complex of 8-Me-MAOEA with human recombinant AdoMetDC proved the formation of the oxime of the enzyme (Fig. 2a). However, in the case of 8-Me-aza-deAdoMet (the nitrogen-containing analog of deAdoMet with the N-methylamino group at the sulfonium center of AdoMet), a Schiff base was not detected in the crystal (Fig. 2b). Hence, it was clearly demonstrated at the structural level that deAdoMet and aza-deAdoMet bind differently in the AdoMetDC active center, and a hypothesis of Abdel-Monem [38] about different positions of the aminopropyl group of deAdoMet and aza-deAdoMet in enzyme–inhibitor complexes was confirmed. The formation of AdoMetDC oxime with 8-Me-MAOEA and the hydrazone of the enzyme with MHZPA might take place due to the flexibility of aminooxyethyl and hydrazinopropyl groups in the initial noncovalent complexes of AdoMetDC with these inhibitors and also because the formation of oximes and hydrazones are irreversible processes.
Fig. 2. Schematic structure of AdoMetDC complexes with 8-Me-MAOEA (a) and 8-Me-aza-deAdoMet (b). Adapted from [26].
Two suicide inhibitors of AdoMetDC are known – 5-((Z)-amino-2-butenyl)methylamino-5′-deoxyadenosine (AbeAdo [45]) and S-(5′-deoxy-5′-adenosyl)-1-amino-4-methylthiocyclopentene-2 (AdoMac [27, 46]). Activities of Z-cis-isomer of AbeAdo towards AdoMetDC from E. coli as well as against the rat liver enzyme were practically 1000 higher compared with the corresponding activities of the E-trans-isomer [45, 47]. This may be considered as indirect evidence of “nonproductive” binding of aza-deAdoMet in the active center of AdoMetDC since productive binding requires a stretched conformation of the side-chain of the substrate/inhibitor. It can be assumed that the cis-configuration of the double bond in the AbeAdo structure is enough to compensate structural disturbances caused by the substitution of the sulfonium center with the aminomethyl group. Hence, the proper position of the terminal amino group of the inhibitor in the E–I complex, which is necessary for Schiff base formation, is fixed, and this is one of the driving forces for the irreversible inhibition. In the case of E. coli AdoMetDC, the inhibition was Mg2+-dependent [47], as was observed in the case of AMA and the bacterial enzyme (see above), while in the case of rat liver the inhibitory potency of AdoMetDC was increased in the presence of Put [45]. These data clearly indicate the existence of Mg2+- and Put-determined conformational changes of AdoMetDC, making the carbonyl group available to react with inhibitors.
Third group of hydroxylamine-containing inhibitors of AdoMetDC is derived from AdoMac by the substitution of the amino group in the cyclopentene cycle to the aminooxy group (Table 1). This algorithm might be rather mechanistic, but it gave very powerful inhibitors with interesting spectra of activities that increased our knowledge about the peculiarities of the inhibition of AdoMetDC and existing structure–activity relationships.
Both AdoMao and norAdoMao irreversibly inhibited bacterial and human recombinant AdoMetDC (Table 1). The differences in the activities of (R,R)- and (S,S)-diastereomers in the case of AdoMetDC from E. coli were not large. Moreover, activities of sulfonium-containing AdoMao and norAdoMao with a sulfide bond against the enzyme from E. coli (Table 1) were rather similar, which distinguishes these inhibitors from AMA and the corresponding sulfide (I) – in the latter case AMA was 1000 times more efficient (Table 1). The structure–activity relationships observed in the case of AdoMao/norAdoMao can be attributed to the conformational restrictions induced by the cyclopentene cycle and stereo configuration of the two chiral centers. These fix the spatial orientation of the reactive aminooxy group in the E–I complex. Finally, the differences in the activities of trans-1R,4R- and trans-1S,4S-norAdoMao towards bacterial and human AdoMetDC (Table 1) clearly indicate the existence of differences in the organization of the substrate recognition sites.
Therefore, three types of hydroxylamine-containing inhibitors of AdoMetDC are known (Table 1). The best representatives of this family (AMA, 8-Me-MAOEA, and AdoMao) irreversibly and specifically inhibit the enzyme from different sources at 1-100 nM concentration. High inhibitory activity and specificity of action should allow successful application of these compounds for the regulation of polyamine metabolism in cell culture and yield some mile-stone results for biochemistry of Spm and Spd.
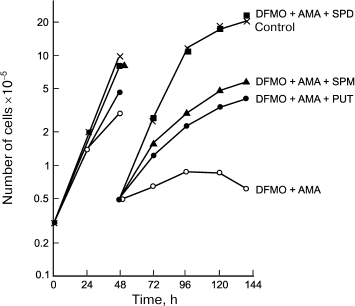
AMA was the first irreversible and very effective inhibitor of AdoMetDC [21], and it inhibited the growth of L1210 at 0.1 mM concentration, depleting the intracellular pool of Spd and Spm, while the amount of Put inside the cells was dramatically increased [23]. These observations confirmed the specificity of action of AMA and are in good agreement with that expected for an inhibitor of AdoMetDC. In the case of L1210 cells, AMA was more effective than DFMO (a suicide inhibitor of ODC) and exhibited lower toxicity [23]. Combined application of AMA and DFMO allowed for the first time to deplete chemically the pool of all three polyamines, i.e. Spm, Spd, and Put [23]. This opened a possibility of investigating the ability of the individual polyamines to support the growth of L1210 cells. Only Spd was able to completely support the growth and viability of L1210 cells with depleted pool of all three polyamines, but not Spm, in spite of its effective catabolism to Spd (Fig. 3). This observation was of crucial importance for polyamine biochemistry since in the case of L1210 cells a weak point in polyamine metabolism (Spd deficiency) was discovered. Later, it was demonstrated that the crucial importance of Spd for growth support is related to the need to supply aminobutyl group for posttranslational modification of initiation translation factor 5A – Spd is the only donor of the aminobutyl group for this vitally important reaction.
Fig. 3. Spd (1 µM), but not Spm (3 µM) or Put (1 µM), reduces L1210 cell growth with completely depleted polyamine pool (simultaneous treatment with 0.1 mM AMA and 1 mM DFMO for 144 h).
Incubation of L1210 with AMA resulted in 50-fold increase in the content of inactive AdoMetDC protein. In addition to the protein with molecular weight ~32 kDa, in this case it was also possible to detect a protein with molecular weight ~37 kDa, which was also revealed with AdoMetDC-specific antibodies [48]. This was the first example of the application of the inhibitor to demonstrate directly that AdoMetDC is synthesized in cells in a form of a proenzyme. Incubation of cells with AMA decreased the intracellular concentration of Spd and Spm, which activated the system of polyamine transport – Vmax increased 4.5-fold, while exogenous Spd and Spm rapidly decreased Vmax to control values [49].
Incubation of L1210 or SV-3T3 cells with MAOEA (0.1 mM) or 0.05 mM MHZPA also resulted in the depletion of the intracellular Spm/Spd pool and increased Put content, while these inhibitors in combination with DFMO depleted the pool of all three polyamines in cells [25]. Incubation of L1210 cells with either MAOEA or MHZPA increased the amount of active ODC, which was due to stimulation of ODC biosynthesis at the stage of translation and also due to decrease in the rate of degradation of ODC [50]. Exogenous Spm and Spd reversed all the effects of the inhibitors. Later, these data were explained in terms of polyamine-dependent regulation of the biosynthesis of antizyme – one of the key regulators of polyamine homeostasis in cells.
It is known that the disease-causing parasite Trypanosoma brucei has no system of purine biosynthesis, but it has a specific purine transporter. AdoMao is able to use this transporter to penetrate inside the parasite, and in vitro it inhibited the growth of T. brucei with IC50 0.9 µM [29].
Therefore, investigation of cellular activity of the inhibitors of AdoMetDC, mainly hydroxylamine-containing compounds of nucleoside nature, provided very important results for polyamine biochemistry.
HYDROXYLAMINE-CONTAINING ANALOGS OF PUTRESCINE
Pyridoxal-5′-phosphate (PLP)-dependent ornithine decarboxylase (ODC, EC 4.1.1.17) is the rate-limiting enzyme of Spm and Spd biosynthesis. The catalytically active α2-dimer has two active centers, each formed with amino acid residues of both subunits. Each subunit has molecular weight of 50-55 kDa depending on the source of the enzyme [51]. Many inhibitors of ODC, including compounds active in vivo, are known (reviewed in [13, 15]). The best known and investigated is α-difluoromethyl ornithine (DFMO) [13, 52, 53]. This compound named Eflornithine® is used for treatment of African sleeping sickness [54, 55] and for facial hirsutism, i.e. excessive masculine-like hair growth in women [56]. DFMO has also advanced to clinical trials with promising results as a chemopreventative agent in the case of elevated risk for specific epithelial cancers [57].
Highly effective and specific inhibitors of ODC were discovered among Put-like O-substituted hydroxylamines (Table 2). The first and simplest was 1-aminooxy-3-aminopropane (APA), which pseudo-irreversibly inhibited the enzymes from rat and mouse liver in nanomolar concentrations [58, 59]. The positively charged amino group of APA serves as an anchor during the interaction with ODCs that display exact binding of the inhibitor in the enzyme active center. The properly positioned and unprotonated at physiological pH aminooxy group (pKa 4.2) [61] reacts with PLP in the active center of ODC, forming the oxime of the enzyme.
Inhibition of ODC with hydroxylamine-containing analogs of putrescine
[60]
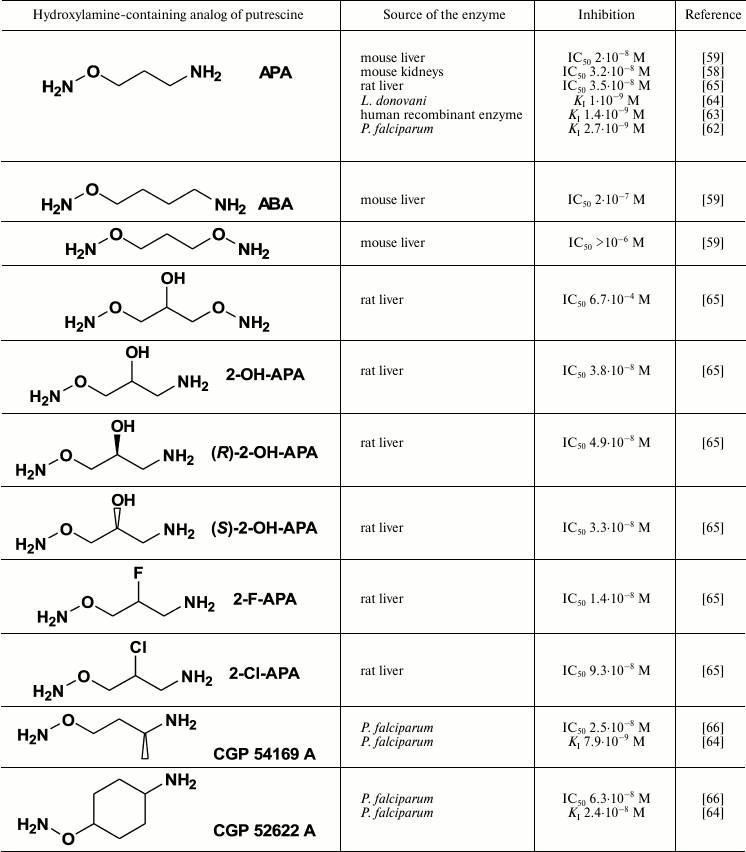
The aminooxy analog of cadaverine (ABA) was 10 times less active than APA, while unprotonated at physiological pH bis-aminooxy derivatives were not effective even at 1 µM concentrations (Table 2). The specificity of action of APA was confirmed by poor activity (IC50 ~ 0.1 mM) of the inhibitor towards PLP-dependent ornithine δ-aminotransferase and AdoMetDC [59].
Introduction of a hydroxyl group in the APA molecule had slight if any effect on the activity of the inhibitor; also, the inhibitory potency of the (R)- and (S)-isomers of 2-OH-APA were about the same (Table 2). 2-F-APA, in contrast with APA, 2-OH-APA, and ABA (all three are pseudo-irreversible inhibitors), irreversibly inhibited ODC [65]. The molecular mechanisms of this irreversible inhibition were not studied, but it may be that first the ODC oxime is formed, followed by the alkylation of the catalytically essential Cys360 residue. An alternative explanation could be the stabilization of the E–I complex with a strong hydrogen bond, which is typical for a fluorine atom.
The high inhibitory activity of APA towards ODC stimulated X-ray investigation of the ODC–APA complex. The inhibitor was properly bound in the enzyme active center (the protonated at physiological pH amino group of the inhibitor served anchoring functions), that like in the case of substrate binding resulted in cleavage of the C=N double bond of the internal aldimine between the carbonyl group of the coenzyme and the amino group of Lys69 [63]. The pyridinium ring of PLP was turned away from amino group of Lys69, and the carbonyl group of the coenzyme was positioned close to the aminooxy group of APA. However, in this case, in contrast to complexes of PLP-dependent GABA transaminase [67], aspartate aminotransferase [68], 1-aminocyclopropane-1-carboxylate synthase [34], and pyruvate-dependent AdoMetDC [26] with aminooxy analogs of substrate/product of the corresponding enzymatic reaction, the formation of the ODC oxime was not observed in the crystal. The carbonyl group of PLP and the aminooxy group APA remained “frozen” at ~3 Å, i.e. at the distance of a hydrogen bond. So, in this particular case ODC–APA complex is crystallized in a conformation not realized in solution, where the ODC oxime is formed.
APA exists at physiological pH in mono-cationic form and can be considered as the simplest insufficiently protonated analog of Put. Since deprotonation of the terminal amino group of Put must take place in the active site of spermidine synthase (SpdSy) before the transfer of the aminopropyl group from deAdoMet, one can consider APA as a unique analog of the intermediate mono-protonated form of Put. This suggests efficient competitive inhibition of SpdSy by APA, but only if the protonation of an amino acid residue in the SpdSy active center is not essential for catalysis. In fact, APA reversibly inhibited SpdSy from mouse kidney [58] and P. falciparum [69] with KI values of 3.2 and 35 µM, respectively.
Cellular effects of APA were studied in more detail compared with the effects of hydroxylamine-containing analogs of deAdoMet (see previous section). APA is not stable in cell culture, and it is decomposed by serum aminooxidases [70, 71]. On the contrary, its oximes with pyridoxal (PL) and PLP are much more stable, even in spite of the slow dephosphorylation of APA-PLP oxime occurring in cell culture medium [70].
APA, 2-OH-APA, and 2-F-APA inhibited the growth of various tumor cells at 0.01-0.1 mM concentrations [64, 65, 72-76]. APA penetrates inside baby hamster kidney cells (BHK) by passive diffusion, which was clearly shown using 3H-labeled APA [77]. The Put transporter of tumor cells more likely also cannot recognize APA as Put. In all cells treated with APA, decrease in the Put/Spd pool and accumulation of deAdoMet was observed, which is quite typical for ODC inhibitors. Growth of cells and their viability can be restored with exogenous Put or Spd. Interestingly, APA-PL or APA-PLP at 0.01 mM concentration inhibited DNA biosynthesis in BHK cells, i.e. these oximes were as effective as APA itself [70]. Treatment of Ehrlich ascites tumor cells with APA greatly increased activity of AdoMetDC, as well as biosynthesis of both AdoMetDC and ODC – latter enzyme being accumulated in the inhibited form [75]. Treatment of human colon cancer cells (Caco-2 and HT-29) with APA and AMA in combination (0.1 mM of each) resulted in total inhibition of ODC/AdoMetDC activities within 24 h and also decreased the intracellular polyamine pool to 8-23% of the control. Combined application of APA, AMA, and 5-fluorouracil reduced colon cancer cell survival more potently than treatment with either 5-fluorouracil alone or only with APA-AMA [78].
Polyamines are essential for the growth of protozoan parasites (Plasmodium falciparum, Leishmania donovani, Trypanosoma brucei gambiense) causing malaria, leishmaniasis, and African sleeping sickness, respectively; these diseases are widespread in the tropics and subtropics. Therefore, new metabolic pathways that can be targeted to inhibit the growth and development of these parasites are urgently needed. The enzymes of polyamine biosynthesis are promising targets since Put and Spd are required for the growth of the parasite and also because Spd is a constituent of trypanothione (Try) (Fig. 1), fulfilling multiple functions protecting the parasite from unfavorable influences [54]. However, Spd is vitally important also for host (human) cells, and this seriously complicates the application of inhibitors of polyamine biosynthesis for treatment of parasitic diseases. Nevertheless, DFMO (a suicide inhibitor of ODC) is successfully used to treat late stages of African sleeping sickness caused by Trypanosoma brucei gambiense [55].
APA is among the most active inhibitors of ODC (Table 2) slowing the growth of normal and tumor cells. APA is also very active against P. falciparum (IC50 1 µM [64]), as well as promastigotes (IC50 42 µM [62]) and amastigotes (IC50 5 µM [62]) of L. donovani, depleting Put, Spd, and Try pools of these parasites. The growth of L. donovani and P. falciparum can be completely restored upon addition of Put/Spd to the medium, which shows the specificity of APA action. At the same time, some forms of L. donovani hyperexpress ODC, and these forms are resistant to APA, this indicating that ODC is the target of the inhibitor [62].
APA penetrates into parasites by passive diffusion, and in the case of L. donovani there was an attempt to transform APA into an actively penetrating prodrug, i.e. 1-guanidinooxy-3-aminopropane (GAPA). This inhibitor (Fig. 4) is an agmatine isoster, but its terminal guanidinooxy group has pKa of only 6.71 [79], while the pKa of the guanidine group of agmatine is about 12.5-13.0.
Comparison of the activities of APA and GAPA towards ODC (0.001 and 60 µM, respectively) with inhibition of the growth of L. donovani amastigotes (5 and 9 µM, respectively) can be considered as evidence of active penetration of GAPA into the parasite [79], and it suggests GAPA to be first actively transported inhibitor of ODC. The possibility of intracellular transformation of GAPA into APA (Fig. 4) depends on the presence of ureidohydrolases with low substrate specificity. It is known that many arginases can convert canavanine into canaline, while some are even known to cleave Agm to Put [80]. Besides, macrophages where L. donovani is localized in humans have agmatinase activity [81]. Having penetrated inside L. donovani, GAPA depleted the pool of Put and Spd, while exogenous Put and/or Spd completely reversed the effects of the inhibitor. These observations clearly confirmed that the biochemical target of GAPA is related to polyamine metabolism.
Fig. 4. Possible explanation of GAPA activity.
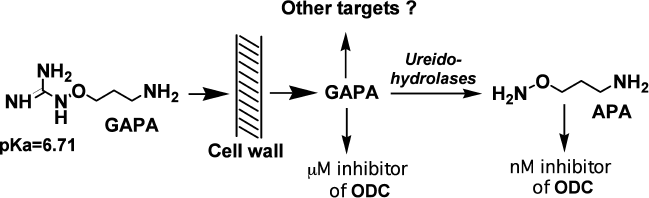
However, GAPA, in contrast to APA, only slightly decreased the level of Try in L. donovani [79]. So, the mechanism of inhibition can hardly be interpreted only in terms of the inhibition of ODC by GAPA itself, or with de novo synthesized APA (Fig. 4). Besides, the relationship between Spd and Try levels in L. donovani might be rather complicated.
Another fundamental difference between GAPA and APA and DFMO is its high activity against ODC-overexpressing forms of L. donovani [82]; they are poorly inhibited by APA and DFMO. Therefore, it seems that GAPA might have other metabolic target(s) different from ODC.
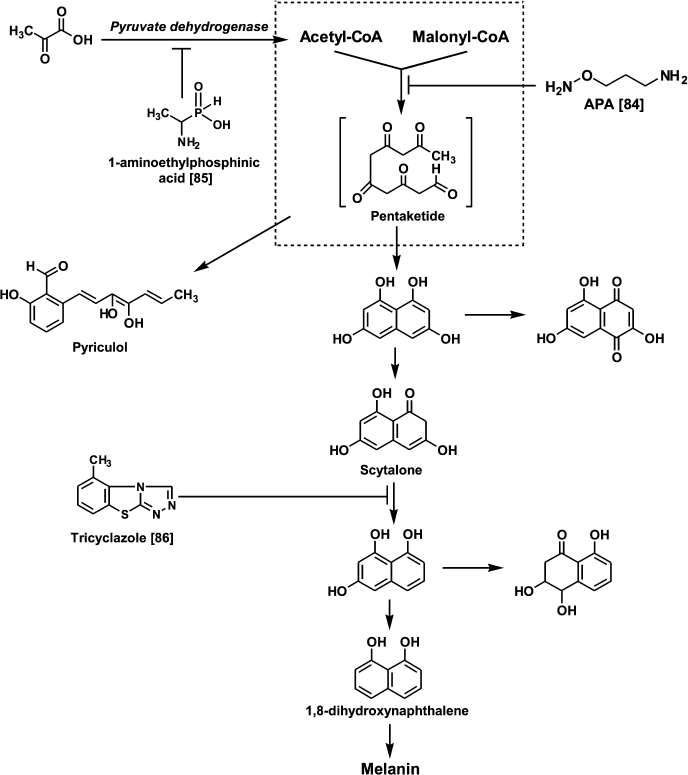
Since the early 1980s, it has been known that DFMO exhibits fungicidal activity against Uromyces phaseoli even in field trials [83]. Investigation of the effects of APA and DFMO on the growth of the phytopathogenic fungus Pyricularia oryzae Cav. causing pyriculariose (the most damaging rice blast disease in all rice-growing regions) demonstrated both that compounds inhibit the growth of P. oryzae at 0.1-1.0 mM concentrations [84]. However, APA, in contrast to DFMO, bleached the mycelium and normally black or deep-brown colonies became small and colorless [84]. Interestingly, the growth and mycelium pigmentation were restored upon the addition of Put [84]. Melanin biosynthesis in P. oryzae starts from the polycondensation of acetyl- and malonyl-CoA. An intermediate pentaketide is a precursor of phytotoxic heptaketide pyriculol and scytalone – the latter undergoing further transformations to dihydroxynaphthalene (Fig. 5). It is known that the inhibition of scytalone conversion to dihydroxynaphthalene with tricyclazole gives rise to pink colonies [86]. Treatment of P. oryzae with APA inhibited the formation of pyriculol and scytalone, this indicating the inhibition of early stages of melanin biosynthesis, presumably the multienzyme complex catalyzing polycondensation of acetyl- and malonyl-CoA, since APA, in contrast to 1-aminoethyl phosphinic acid (treatment with this inhibitor also gives colorless mycelium [85]), did not inhibit pyruvate dehydrogenase complex [84]. These data are the first indication of the involvement of Put in the regulation of the pentaketide biosynthetic pathways in fungi.
Fig. 5. Chemical regulators of melanin biosynthesis in the phytopathogenic fungus P. oryzae.
There is another example confirming differences in the cellular effects of APA and DFMO. In the case of BHK cells treated with APA, but not with DFMO, disturbances of the Golgi apparatus were found [87].
Only a few experiments describing antitumor activity of APA and related hydroxylamines in vivo have been reported. Antitumor activity of 2-F-APA (at dose 1/20 of the maximal tolerated dose) is about the same as DFMO (at dose 1/5 of the maximal tolerated dose) in the case of female CD-1 nude mice bearing human bladder carcinoma T24 [88, 89]. These results suggest that compounds like 2-F-APA might have therapeutic value as cytostatic drugs.
HYDROXYLAMINE-CONTAINING ANALOGS OF SPERMINE AND
SPERMIDINE
The biochemical properties of Spm, Spd, and Put are determined by the superposition of the structure and the charge of the molecules, i.e. proper spatial localization of amino groups that are protonated at physiological pH. However, the impact of the charged constituent is not so well investigated. A rational approach to study the influence of the charge on biological properties of a polyamine is to design isosteric but incompletely protonated analogs with pKa of one or two amino groups decreased by a few orders of magnitude. A possible approach to altering the pKa of the amino groups of the polyamine is substitution of the terminal NH2CH2-group with an NH2O-group, causing minimal steric disturbance. The pKa value of the terminal aminooxy group depends on the structure of the analog (Fig. 6) and is ~3.2-4.5, i.e. about five orders of magnitude lower than that of the amino group. The pKa values of -CH2-NH-O-CH2-groups are already in the interval 2.5-2.7. Hence, hydroxylamine-containing analogs of polyamines are structurally similar to the parent polyamines, but they are incompletely protonated, which allows investigation of the effect of the amino group protonation on the biochemical properties of Spm and Spd.
Fig. 6. Structure of some aminooxy- and oxa-analogs of polyamines.
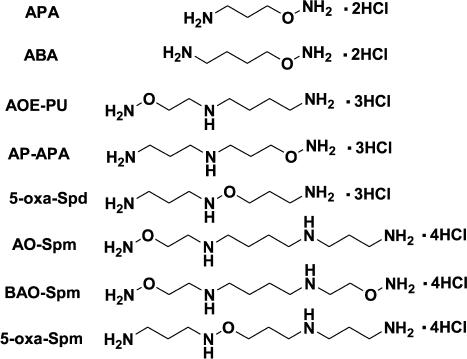
The inhibitory activity of hydroxylamine-containing analogs of Spd – AOE-PU and AP-APA towards ODC and AdoMetDC was not high, and it was comparable with that of simple O-substituted esters of hydroxylamine. Thus, AOE-PU and AP-APA at 1 mM concentration inhibited AdoMetDC by 54 and 70%, respectively, while at 0.01 mM they inhibited ODC by 12 and 20%, respectively [90].
The interactions of AOE-PU and AP-APA with SpmSy were more complicated and provided interesting data. AP-APA is an isoster of doubly protonated (at N1- and N4-nitrogens) Spd, i.e. it can be considered as a mimetic of the doubly protonated state of Spd, which occurs in the active site of SpmSy. On the contrary, AOE-PU is an isoster of Spd being doubly protonated at the N4- and N8-nitrogen atoms, and it cannot be considered as an isoster of the first intermediate of the SpmSy reaction. Hence, AP-APA was 100 times more active then AOE-PU, and its affinity to SpmSy was ~10-fold better than that of Spd [90]. These data confirm that AP-APA is a good mimetic of the doubly protonated form of Spd, which in accordance with transition state theory must have higher affinity to SpmSy compared with the triply protonated Spd. Moreover, AP-APA was a substrate of SpmSy, but the rate of 5-oxa-Spm formation was low, and the kinetic parameters were not determined. Low rate of AP-APA alkylation with deAdoMet (SpmSy catalyzes direct transfer of the aminopropyl group of deAdoMet to the N8-amino group of Spd) was more likely associated with much higher nucleophilicity of the unprotonated amino group compared with the H2NO-group – nucleophilicity of the acceptor of the aminopropyl group is one of the rate-determining factors of the SpmSy reaction [90]. Besides, deprotonation of Spd is obviously associated with the protonation of the enzyme active site amino acid residue, and we do not know anything about the impact of the side chain of this amino acid on catalysis.
Both AOE-PU and AP-APA are substrates of the crude acetylase activity of BHK cell extracts [90], but the experiments with BHK cells demonstrated that only AOE-PU metabolized to the corresponding N1-acetylated derivative in spite of the fact that both AOE-PU and AP-APA penetrated inside the cells [91].
Enzymatic acetylation of Spm and Spd is catalyzed by Spd/Spm-N1-acetyltransferase and takes place via direct transfer of the acetyl group of Ac-CoA to the N1-amino group of Spm/Spd. Hence, for the inhibition of SSAT conjugates of HS-CoA with Spm, or nor-Spd, where HS-CoA and polyamine were connected with acetate linker (Fig. 7) were used. Conjugates of symmetrical polyamines inhibited SSAT with IC50 0.5-5.0 µM depending on the structure of the polyamine residue [92]. Later, to conjugate SH-CoA or D-pantetheine selectively, either to N1- or N8-amino groups of the Spd molecule, a complicated and multistep synthetic procedure was developed [93].
Fig. 7. Mimetics of the transition bisubstrate complex of the SSAT reaction with acetyl and acetone linkers.
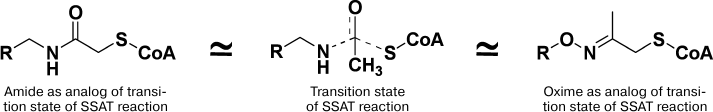
Aminooxy analogs of Spm and Spd (Fig. 6) are not only insufficiently protonated polyamine isosters, but they are also classic carbonyl reagents. The latter provided the possibility of developing simple two step synthesis of the required conjugates using different reactivity of H2NO- and H2N-groups and using acetone linker to combine polyamine and CoA constituents of the conjugate [94, 95]. The most active was the conjugate mimicking the adduct of the N8-position of the Spd molecule (IC50 1 µM), while the conjugation of the N1-position of Spd was not so effective (IC50 6 µM) [94, 95]. The conjugate of AP-APA and HS-CoA was 22-fold more active compared with the starting ketone, which confirmed the impact of the polyamine residue in the inhibition of SSAT. The impact of the adenosine group of CoA in the inhibition of SSAT was crucial, since corresponding derivatives of D-pantetheine were poor inhibitors (IC50 > 100 μM). This structure–activity relationship was not self-evident because it is known that, at least in the case of succinyl-CoA-acetoacetate transferase, the pantetheine group makes a significant contribution in the efficiency of the substrate binding [96].
Among other data on the interaction of aminooxy analogs of Spm/Sd with the enzymes of polyamine metabolism, it is necessary to point out the substrate properties of N1-Ac-AOE-PU in APAO reaction (kcat 0.54 sec–1) [97] and the substrate properties of AO-Spm (Fig. 6) towards SSAT from colon cancer cells (Caco-2): the Km was 2.2 mM [73].
AP-APA and AOE-PU were not stable in cell culture medium, and like APA they were the substrates of serum aminooxidases [71]. These Spd aminooxy analogs efficiently inhibited the growth of L1210 cells but not of BHK cells [74, 91], while AO-Spm did not inhibit the growth of either Caco-2 [73] or BHK cells [74], but it was effective against L1210 cells [74]. Surprisingly, AO-Spm was able to restore the growth of APA-treated BHK cells [98], i.e. one O-substituted hydroxylamine reversed the effect of another O-substituted hydroxylamine. One can assume that AO-Spm is acetylated in BHK cells, as it is in Caco-2 cells [73], and subsequent APAO-catalyzed splitting of N1-Ac-AO-Spm results in formation of Spd.
Isosteric Spd analogs – AOE-PU and AP-APA – exhibited different ability to modulate the NMDA receptor; AP-APA was an antagonist, while AOE-PU was an agonist [99]. These data were the first direct indications on different recognition of terminal amino groups of Spd in the polyamine–receptor complex.
Therefore, aminooxy analogs of Spd turned out to be useful tools to study the enzymes of polyamine metabolism, and their use confirmed the functional non-equivalence of the terminal amino groups of Spd.
Hydroxylamine derivatives occupy an important place among inhibitors and chemical regulators of the enzymes of polyamine metabolism. Within this class of chemical compounds, it turned possible to design irreversible inhibitors of carbonyl-dependent AdoMetDC and ODC, those being active at nM concentrations; effective inhibitors of SSAT, SpdSy, and SpmSy; effectors of polyamine transport; and an original proinhibitor of ODC. Using these hydroxylamine derivatives, data being of fundamental value for polyamine biochemistry and dealing with cellular functions of Spm/Spd and also mechanisms controlling biosynthesis and degradation of short-living ODC, AdoMetDC, and SSAT were obtained. The coordinated activities of these enzymes are responsible for maintaining cellular polyamine homeostasis.
This work was supported by the Russian Foundation for Basic Research grant 12-04-31682 (MAK), the Russian Academy of Sciences Presidium Program “Molecular and Cellular Biology” (ARK and SNK), strategic funding from the University of Eastern Finland (JW, MH, TAK, JV), and a grant from the Academy of Finland (LA).
REFERENCES
1.Van Leeuwenhoek, A. (1678) Philos. Trans. R.
Soc. London, 12, 1040-1043.
2.Dudley, H. W., Rosenheim, M. C., and Rosenheim, O.
(1924) Biochem. J., 18, 1263-1272.
3.Russell, D., and Snyder, S. H. (1968) Proc.
Natl. Acad. Sci. USA, 60, 1420-1427.
4.Janne, J., and Raina, A. (1968) Acta Chem.
Scand., 22, 1349-1351.
5.Russell, D. H. (1973) Polyamines in Normal and
Neoplastic Growth, Raven Press, New York.
6.Cohen, S. S. (1998) A Guide to the
Polyamines, Oxford University Press, New York.
7.Pegg, A. E., and Casero, R. A. (2011)
Polyamines: Methods and Protocols. Methods in Molecular Biology,
720, 3-35.
8.Rosenthal, S. M., and Tabor, C. W. (1956) J.
Pharm. Exp. Ther., 116, 131-138.
9.Casero, R. A., and Marton, L. J. (2007) Nat.
Rev. Drug Discov., 6, 373-390.
10.Kahana, C. (2009) Cell. Mol. Life Sci.,
66, 2479-2488.
11.Coffino, Ph. (2001) Nature Rev. Mol. Cell
Biol., 2, 188-194.
12.Kurian, L., Palanimurugan, R., Godderz, D., and
Dohmen, R. J. (2011) Nature, 477, 490-494.
13.Seiler, N. (2003) Curr. Drug Targets,
4, 537-564.
14.Wallace, H. M., Fraser, A. V., and Hughes A.
(2003) Biochem. J., 376, 1-14.
15.Wallace, H. M., and Fraser, A. V. (2004) Amino
Acids, 26, 353-365.
16.Casero, R. A., and Woster, P. M. (2001) J.
Med. Chem., 44, 1-26.
17.Casero, R. A., and Woster, P. M. (2009) J.
Med. Chem., 52, 4551-4573.
18.Wallace, H. M. (2007) Expert. Opin.
Pharmacother., 8, 2109-2116.
19.Birkholz, L.-M., Williams, M., Niemand, J., Louw,
A. I., Persson, L., and Heby, O. (2011) Biochem. J., 438,
229-244.
20.Pegg, A. E., and McCann, P. P. (1992)
Pharmacol. Ther., 56, 359-377.
21.Khomutov, R. M., Zavalova, L. L., Syrku, V. I.,
Artamonova, E. Yu., and Khomutov, A. R. (1983) Bioorg. Khim.,
9, 130-131.
22.Artamonova, E. Yu., Khomutov, A. R., Zavalova, L.
L., and Khomutov, R. M. (1986) Bioorg. Khim., 12,
206-212.
23.Kramer, D. L., Khomutov, R. M., Bukin, Yu. V.,
Khomutov, A. R., and Porter, C. W. (1989) Biochem. J.,
259, 325-331.
24.Kolb, M., and Barth, J. (1985) Liebigs Ann.
Chem., 1035-1040.
25.Pegg, A. E., Jones, D. B., and Secrist III, J. A.
(1988) Biochemistry, 27, 1408-1415.
26.McCloskey, D. E., Bale, S., Secrist 3rd, J. A.,
Tiwari, A., Moss 3rd, T. H., Valiyaveettil, J., Brooks, W. H., Guida,
W. C., Pegg, A. E., and Ealick, S. E. (2009) J. Med. Chem.,
52, 1388-1407.
27.Wu, Y. Q., and Woster, P. M. (1993) Bioorg.
Med. Chem., 1, 349-360.
28.Wu, Y. Q., and Woster, P. M. (1995) Biochem.
Pharmacol., 49, 1125-1133.
29.Guo, J. Q., Wu, Y. Q., Rattendi, D., Bacchi, C.
J., and Woster, P. M. (1995) J. Med. Chem., 38,
1770-1777.
30.Kitz, R., and Wilson, I. B. (1962) J. Biol.
Chem., 237, 3245-3249.
31.Pegg, A. E. (1983) Methods Enzymol.,
94, 239-247.
32.Pegg, A. E. (1984) Cell Bochem. Funct.,
2, 11-15.
33.Wetkamp, E. L. C., Dixon, H. B. F., Khomutov, A.
R., and Khomutov, R. M. (1991) Biochem. J., 277,
643-645.
34.Capitani, G., Eliot, A. C., Gut, H., Khomutov, R.
M., Kirsch, J. F., and Grutter, M. G. (2003) Biochim. Biophys.
Acta, 1647, 55-60.
35.Anton, D. L., and Kutney, R. (1987)
Biochemistry, 26, 6444-6447.
36.Diaz, E., and Anton, D. L. (1991)
Biochemistry, 30, 4078-4081.
37.Li, Y-F., Hess, S., Pannell, L. K., Tabor, C. W.,
and Tabor, H. (2001) Proc. Natl. Acad. Sci. USA, 98,
10578-10583.
38.Pankaskie, M., and Abdel-Monem, M. M. (1980)
J. Med. Chem., 23, 121-127.
39.Khomutov, M. A. (2012) Novel
Biologically-Active Esters of Hydroxylamine and C-Methylated Polyamine
Analogues: Ph. D. Thesis [in Russian], Engelhardt Institute of
Molecular Biology, Moscow.
40.O’Leary, M. H., and Banghin, R. L. (1977)
J. Biol. Chem., 252, 7168-7173.
41.Parsons, I. D., Persson, B., Mekhalfia, A.,
Blackburn, G. M., and Stockley, P. G. (1995) Nucleic Acids Res.,
23, 211-216.
42.Shantz, L. M., Stanley, B. A., Secrist III, J.
A., and Pegg, A. E. (1992) Biochemistry, 31,
6848-6855.
43.Wickner, R. B., Tabor, C. W., and Tabor, H.
(1970) J. Biol. Chem., 245, 2132-2139.
44.Tolbert, D. W., Ekstrom, J. L., Mathews, I. I.,
Secrist III, J. A., Kapoor, P., Pegg, A. E., and Ealick, S. E. (2001)
Biochemistry, 40, 9484-9494.
45.Danzin, C., Marchal, P., and Casara, P. (1990)
Biochem. Pharmacol., 40, 1499-1503.
46.Wu, Y., and Woster, P. M. (1992) J. Med.
Chem., 35, 3196-3201.
47.Casara, P., Marchal, P., Wagner, J., and Danzin,
C. (1989) J. Am. Chem. Soc., 111, 9111-9113.
48.Antelli, R., Stjernborg, L., Khomutov, A. R.,
Khomutov, R. M., and Persson, L. (1991) Eur. J. Biochem.,
196, 551-556.
49.Kramer, D. L., Miller, J. M., Bergeron, R. J.,
Khomutov, R. M., Khomutov, A. R., and Porter, C. W. (1993) J. Cell.
Physiol., 155, 399-407.
50.Madhubala, R., Secrist III, J. A., and Pegg, A.
E. (1988) Biochem. J., 254, 45-50.
51.McCann, P. P., and Pegg, A. E. (1992)
Pharmacol. Ther., 54, 195-215.
52.Metcalf, B. W., Bey, P., Danzin, C., Jung, M. J.,
Casara, J., and Vevert, J. P. (1978) J. Am. Chem. Soc.,
100, 2551-2553.
53.Raul, F. (2007) Biochem. Soc. Trans.,
35, 353-355.
54.Heby, O., Persson, L., and Rentala, M. (2007)
Amino Acids, 33, 359-366.
55.Burri, C. C., and Brun, R. (2003) Parasitol.
Res., 90, 49-52.
56.Shapiro, J., and Lui, H. (2001) Skin. Ther.
Lett., 6, 1-3.
57.Babbar, N., and Gerner, E. W. (2011) Recent
Results Cancer Res., 188, 49-64.
58.Khomutov, R. M., Hyvonen, T., Karvonen, E.,
Kauppinen, L., Paalanen, T., Paulin, L., Eloranta, T., Pajula, R. L.,
Andersson, L. C., and Poso, H. (1985) Biochem. Biophys. Res.
Commun., 130, 596-602.
59.Khomutov, R. M., Denisova, G. F., Khomutov, A.
R., Belostotskaia, K. M., Shlosman, R. B., and Artamonova, E. Yu.
(1985) Bioorg. Khim., 11, 1574-1576.
60.Weisell, J. M. (2012) Structure and Function
of Charge Deficient Polyamines: Ph. D. Thesis, Kuopio, University
of Eastern Finland.
61.Weisell, J., Hyvonen, M. T., Alhonen, L.,
Vepsalainen, J., Keinanen, T. A., and Khomutov, A. R. (2013) Curr.
Pharm. Des., May 16 [Epub ahead of print].
62.Singh, S., Mukherjee, A., Khomutov, A. R.,
Persson, L., Heby, O., Chatterjee, M., and Madhubala, R. (2007)
Antimicrob. Agents Chemother., 51, 528-534.
63.Dufe, V. T., Ingner, D., Heby, O., Khomutov, A.
R., Persson, L., and Al-Karadaghi, S. (2007) Biochem. J.,
405, 261-268.
64.DasGupta, R., Krause-Ihle, T., Bergmann, B.,
Muller, I. B., Khomutov, A. R., Muller, S., Walter, R. D., and Luersen,
K. (2005) Antimicrob. Agents Chemother., 49,
2857-2864.
65.Stanek, J., Frei, J., Mett, H., Schneider, P.,
and Regenass, U. (1992) J. Med. Chem., 35, 1339-1344.
66.Birkholtz, L.-M., Joubert, F., Neitz, A. W. H.,
and Louw, A. I. (2003) Protein Struct. Funct. Genet., 50,
464-473.
67.Liu, W., Peterson, P. E., Carter, R. J., Zhou,
X., Langston, J. A., Fisher, A. J., and Toney, M. D. (2004)
Biochemistry, 43, 10896-10905.
68.Markovic-Housley, Z., Schirmer, T., Hohenester,
E., Khomutov, A. R., Khomutov, R. M., Karpeisky, M. Ya., Sandmeier, E.,
Christen, Ph., and Jansonius, J. N. (1996) Eur. J. Biochem.,
236, 1025-1032.
69.Haider, N., Eschbach, M. L., Dias, Sde S.,
Gilberger, T. W., Walter, R. D., and Luersen, K. (2005) Mol.
Biochem. Parasitol., 142, 224-236.
70.Keinanen, T. A., Hyvonen, T., Pankaskie, M. C.,
Vepsalainen, J. J., and Eloranta, T. O. (1994) J. Biochem.
(Tokyo), 116, 1056-1062.
71.Hyvonen, T., Keinanen, T. A., Khomutov, A. R.,
Khomutov, R. M., and Eloranta, T. O. (1992) J. Chromatogr.,
574, 17-21.
72.Poulin, R., Secrist III, J. A., and Pegg, A. E.
(1989) Biochem. J., 263, 215-221.
73.Turchanowa, L., Shvetsov, A. S., Demin, A. V.,
Khomutov, A. R., Wallace, H. M., Stein, J., and Milovic, V. (2002)
Biochem. Pharmacol., 64, 649-655.
74.Khomutov, A. R., Shvetsov, A. S., Vepsalainen,
J., Kramer, D. L., Hyvonen, T., Keinanen, T. A., Eloranta, T. O.,
Porter, C. W., and Khomutov, R. M. (1996) Bioorg. Khim.,
22, 557-559.
75.Persson, L., Khomutov, A. R., and Khomutov, R. M.
(1989) Biochem. J., 257, 929-931.
76.Hyvonen, T., Alakuijala, L., Andersson, L.,
Khomutov, A. R., Khomutov, R. M., and Eloranta, T. (1988) J. Biol.
Chem., 263, 11138-11144.
77.Hyvonen, T., Khomutov, A. R., Khomutov, R. M.,
Lapinjoki, S., and Eloranta, T. O. (1990) J. Biochem. (Tokyo),
107, 817-820.
78.Milovic, V., Turchanowa, L., Khomutov, A. R.,
Khomutov, R. M., Caspary, W. F., and Stein, J. (2001) Biochem.
Pharmacol., 61, 199-206.
79.Singh, S., Jhingran, A., Sharma, A., Simonian, A.
R., Soininen, P., Vepsalainen, J., Khomutov, A. R., and Madhubala, R.
(2008) Biochem. Biophys. Res. Commun., 375, 168-172.
80.Dabir, S., Dabir, S., and Somranshi, B. (2005)
Int. J. Biol. Sci., 1, 114-122.
81.Sastre, M., Galea, E., Feinstein, D., Reis, D.
J., and Regunathan, S. (1998) Biochem. J., 330,
1405-1409.
82.Khomutov, M. A., Mandal, S., Weisell, J., Saxena,
N., Simonian, A. R., Vepsalainen, J., Madhubala, R., and Kochetkov, S.
N. (2010) Amino Acids, 38, 509-517.
83.Rajam, M. V., Weinstein, L. H., and Galston, A.
W. (1985) Proc. Natl. Acad. Sci. USA, 82, 6874-6878.
84.Khomutov, A. R., Dzavakhia, V. G., Voinova, T.
M., Ermolinsky, B. S., and Khomutov, R. M. (1989) Bioorg. Khim.,
15, 707-709.
85.Khomutov, R. M., Khurs, E. N., Dzavakhia, V. G.,
Voinova, T. M., and Ermolinsky, B. S. (1989) Bioorg. Khim.,
13, 1422-1424.
86.Yamaguchi, I. (1982) J. Pest. Sci.,
7, 307-316.
87.Parkkinen, J. J., Lammi, M. J., Agren, U., Tammi,
M., Keinanen, T. A., Hyvonen, T., and Eloranta, T. O. (1997) J.
Cell. Biochem., 66, 165-174.
88.Mett, H., Stanek, J., Lopez-Ballester, J. A.,
Janne, J., Alhonen, L., Sinervirta, R., Frei, J., and Regenass, U.
(1993) Cancer Chemother. Pharmacol., 32, 39-45.
89.Frei, J., and Stanek, J. (1997) United States
Patent US005610195A.
90.Eloranta, T. O., Khomutov, A. R., Khomutov, R.
M., and Hyvonen, T. (1990) J. Biochem. (Tokyo), 108,
593-598.
91.Hyvonen, T., Keinanen, T. A., Khomutov, A. R.,
Khomutov, R. M., and Eloranta, T. O. (1995) Life Sci.,
56, 349-360.
92.Ervin, B. G., Persson, L., and Pegg, A. E. (1984)
Biochemistry, 23, 4250-4255.
93.Roblot, G., Wylde, R., Martin, A., and Parello,
J. (1993) Tetrahedron, 29, 6381-6398.
94.Simonian, A., Khomutov, A., Hyvonen, T.,
Grigorenko, N., Keinanen, T., Vepsalainen, J., Alhonen, L., and Janne,
J. (2007) Nucleosides, Nucleotides & Nucleic
Acids, 26, 1245-1248.
95.Keinanen, T., Hyvonen, T., Vepsalainen, J.,
Alhonen, L., Khomutov, A., and Janne, J. (2014) Russ. J. Bioorg.
Chem., 40, in press.
96.Fierke, C. A., and Jencks, W. P. (1986) J.
Biol. Chem., 261, 7603-7606.
97.Jarvinen, A. J., Cerrada-Gimenez, M., Grigorenko,
N. A., Khomutov, A. R., Vepsalainen, J. J., Sinervirta, R. M.,
Keinanen, T. A., Alhonen, L. I., and Janne, J. E. (2006) J. Med.
Chem., 49, 399-406.
98.Keinanen, T. A., unpublished data.
99.Berger, M. L., Khomutov, A. R., and Rebernik, P.
(1996) Neuro. Sci. Lett., 203, 25-28.