REVIEW: Hypochlorous Acid as a Precursor of Free Radicals in Living Systems
O. M. Panasenko1,2*, I. V. Gorudko3, and A. V. Sokolov1,4
1Research Institute of Physico-Chemical Medicine, Malaya Pirogovskaya str. 1a, 119435 Moscow, Russia; E-mail: o-panas@mail.ru2Pirogov Russian National Research Medical University, Ostrovityanova str. 1, 117997 Moscow, Russia
3Department of Biophysics, Belarusian State University, Nezavisimosti Avenue 4, 220050 Minsk, Belarus
4Institute of Experimental Medicine, North West Branch of the Russian Academy of Medical Sciences, Akademika Pavlova str. 12, 197376 St. Petersburg, Russia
* To whom correspondence should be addressed.
Received December 12, 2012; Revision received June 19, 2013
Hypochlorous acid (HOCl) is produced in the human body by the family of mammalian heme peroxidases, mainly by myeloperoxidase, which is secreted by neutrophils and monocytes at sites of inflammation. This review discusses the reactions that occur between HOCl and the major classes of biologically important molecules (amino acids, proteins, nucleotides, nucleic acids, carbohydrates, lipids, and inorganic substances) to form free radicals. The generation of such free radical intermediates by HOCl and other reactive halogen species is accompanied by the development of halogenative stress, which causes a number of socially important diseases, such as cardiovascular, neurodegenerative, infectious, and other diseases usually associated with inflammatory response and characterized by the appearance of biomarkers of myeloperoxidase and halogenative stress. Investigations aimed at elucidating the mechanisms regulating the activity of enzyme systems that are responsible for the production of reactive halogen species are a crucial step in opening possibilities for control of the development of the body’s inflammatory response.
KEY WORDS: myeloperoxidase, hypochlorous acid, free radicals, reactive halogen species, halogenative stress, biomarker of myeloperoxidase, inflammationDOI: 10.1134/S0006297913130075
Abbreviations: DMPO, 5,5-dimethyl-1-pyrroline-N-oxide; EPO, eosinophil peroxidase; Hal, halogen; LOOH, lipid hydroperoxide; LPO, lipid peroxidation; MPO, myeloperoxidase; RHS, reactive halogen species; ROS, reactive oxygen species; SOD, superoxide dismutase.
Endogenous radicals produced in vivo are divided into the
categories primary, secondary, and tertiary [1].
Primary radicals (semiquinones, •O2‾,
•NO) are formed in enzymatic reactions (mitochondrial
respiratory chain, enzymatic systems of blood cells and vessels) and
are essential components for the normal function of cells and the
organism as a whole. Secondary radicals (•OH and lipid
radicals L•, LO•, LOO•,
etc.) tend to arise in nonenzymatic reactions involving molecular
precursors such as H2O2, lipid hydroperoxides
(LOOH), and other reactants. Tertiary radicals are formed as a result
of the interception, mainly of secondary radicals, by antioxidant traps
(α-tocopherol, thyroxin, etc.). Secondary radicals, unlike
primary radicals, have cytotoxic activity. Indeed, formation of
secondary radicals entering reactions with many biologically important
molecules leads to the development of various socially significant
diseases [1].
One of the most important molecular precursors of free radicals in the body is hypochlorous acid (HOCl), the salt of which is called hypochlorite (OCl–):
H2O2 + Cl– + H+ → HOCl + H2O, HOCl ↔ OCl– + H+. (1)
HOCl is formed by the reaction catalyzed by enzymes related to the mammalian heme peroxidase family. HOCl has pKa 7.5 [2]. This means that at physiological pH HOCl and OCl– are present in the aqueous medium at approximately equal concentrations. On one hand, being a strong oxidant, HOCl is the most important component of the bactericidal system of human and other mammals’ bodies. On the other hand, HOCl, due to high reactivity and ability to perform the role of free radical precursor, reacting with many biologically important molecules, has cytotoxic effect and can provoke the development of a number of diseases one way or another associated with the inflammatory response of the body (cardiovascular disease, cancer, neurodegenerative and other diseases) [3].
In this review, we focus on the mechanisms of HOCl formation in vivo and on its reactions with different biologically important inorganic and organic molecules leading to the formation of highly reactive free radicals. Let us briefly consider the implications of these reactions causing various human diseases.
PATHWAY OF HOCl FORMATION IN THE HUMAN BODY
The family of mammalian heme peroxidases (donor:H2O2-oxidoreductase, EC 1.11.1.7) consists of four main enzymes: myeloperoxidase (MPO), eosinophil peroxidase (EPO), lactoperoxidase, and thyroid peroxidase. MPO is mainly found in azurophilic granules of neutrophils, where it accounts for 2-5% of the total cellular protein or 2-4 mg per 109 cells [4, 5]. The enzyme is also found in the lysosomes of monocytes but in much lower quantities (0.9% by weight), gradually disappearing after their differentiation into macrophages [6, 7]. EPO is contained in specific matrix granules of eosinophils, where it accounts for about 5% of the total protein of these granules or 15 mg per 109 cells [8, 9]. The similarity of the functional and enzymatic properties of MPO and EPO are explained by their amino acid sequence homology, reaching 70% [10-12]. Lactoperoxidase is contained in exocrine gland secretions: milk, saliva, and tears, and it performs mainly bactericidal function [13]. Iodination is required for the synthesis of thyroid hormones catalyzed by thyroid peroxidase [14]. The last two enzymes are not found in blood under physiological conditions and are not capable of oxidizing chloride to HOCl. Considering this circumstance, they can hardly be attributed to the potential sources of HOCl in vivo.
A diagram of the major enzymatic reactions catalyzed by mammalian heme peroxidases is shown in Fig. 1. As a result of the catalytic cycle, heme iron undergoes successive oxidation and reduction. The ferri form of the native enzyme (MPO-Fe3+) is quickly (2·107 M–1·s–1 [15]) oxidized by hydrogen peroxide, turning into Compound I (MPO•+-Fe4+=O) [15-17]. This is a species of oxy-ferryl-porphyrin-cation-radical, where oxygen is attached by a double bond to iron in the ferryl state (reaction 1 in Fig. 1) [16, 18]. Compound I, having two-electron redox potential of 1.16 V [12], has high reactivity and catalyzes the oxidation of halide ions (Cl–, Br–, I–) to the corresponding hypohalous acids (HOCl, hypobromous (HOBr), and hypoiodous (HOI) acids) with the formation of the native ferri form of the enzyme (reaction 2, Fig. 1) [11, 12, 16, 18]. Reactions 1 and 2 (Fig. 1) are called the halogenating cycle, which describes the total Eq. (1).
Fig. 1. Scheme of halogenating and peroxidase cycle of MPO. Hal, halogen.
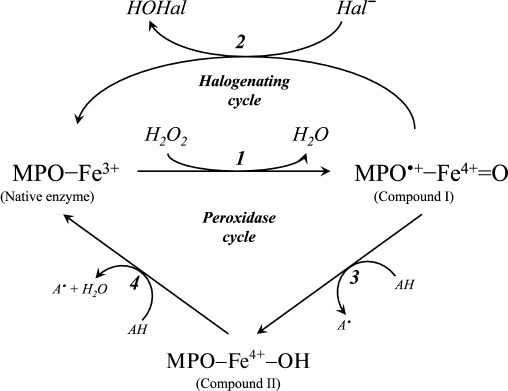
In the presence of electron donors AH (Tyr, ascorbate, urate, nitrite, catecholamines, and many aromatic xenobiotics), Compound I can be turned into Compound II (MPO-Fe4+-OH) and can oxidize AH by one-electron transfer with formation of A•-radicals (reaction 3, Fig. 1) [12, 16, 19]. Compound II is catalytically inactive in hypohalous acid formation, but, like Compound I, it can cause one-electron oxidation of substrate (AH) with regeneration of the native enzyme (reaction 4, Fig. 1) [12, 16, 19]. Thus, reactions 1, 3, and 4 (see Fig. 1) unite in the classic peroxidase cycle.
A number of works have shown that during stimulation of leukocytes, approximately 20 to 70% of the hydrogen peroxide is spent on HOCl formation [20, 21]. EPO effectively oxidizes chloride only at low pH values [22]. Given these circumstances, and also the fact that the content of neutrophils in blood is approximately 20 times greater than the number of eosinophils, and the rate constant for oxidation of chloride by MPO at physiological pH is about an order of magnitude higher than that by EPO (2.5·104 and 3.1·103 M–1·s–1, respectively [11, 12]), we conclude that the main source of HOCl in the body is leukocytic MPO.
Normal leukocytes are quietly found in an inactive state. The first reaction in response to pathogenic microorganisms is accompanied by activation of leukocytes and a change in their morphology, adhesion, directed movement toward the trauma, increased oxygen consumption (“respiratory burst”), and degranulation [23]. Degranulation is characterized by the fusion of cytoplasmic granules with phagosomes, entry of the granule enzymes into the phagosomes, and partial secretion of these enzymes (including MPO) into the extracellular space [23, 24]. Using ELISA, it was found that the content of MPO in the serum of healthy people is 10-250 ng/ml [25], and this can be elevated in inflammatory diseases, in particular, in acute coronary syndrome, reaching 1110 ng/ml [26]. It is not difficult to calculate from the data given in [27] that the local concentration of HOCl/OCl– in the inflammation foci can reach about 25-50 mM.
FREE RADICAL FORMATION IN REACTIONS INVOLVING HOCl
Reactions of HOCl with inorganic compounds. All radical-forming reactions involving HOCl can be divided into two groups: reaction with low molecular weight inorganic compounds and reaction with functional groups of lipids, proteins, carbohydrates, and other biopolymers. Among the first the reactions in which •OH radical is formed must be emphasized, because this radical is the most reactive. The lifetime of •OH is less than 10–9 s [28]. In 1980, it was suggested that •OH is formed in the reaction of HOCl with superoxide anion-radical [29]:
HOCl + •O2‾ → •OH + O2 + Cl–. (2)
This suggestion was later confirmed experimentally [30].
Another important reaction leading to •OH formation involves transition metal ions. So, using the continuous-flow EPR spectroscopy, it was established that HOCl causes one-electron oxidation of titanium ions (Ti3+) with •OH radical formation [31]:
Ti3+ + HOCl → Ti4+ + •OH + Cl–.
Later it was shown that Fe2+ in the Fe(CN)64– complex ion is oxidized by HOCl with a bimolecular reaction rate constant of about 114 ± 7 M–1·s–1 (pH 7.2) [19, 30] according to the equation:
Fe(CN)64– + HOCl → Fe(CN)63– + •OH + Cl–. (3)
With deprotonation of HOCl (pKa 7.5), the rate constant also is decreased, indicating that the reactive form is a molecule of acid (HOCl) and not hypochlorite anion (OCl–) [19, 30]. Data from chemiluminescence and EPR studies revealed the features of the reaction of HOCl with Fe2+, the famous Fenton reaction in which •OH radical is formed:
Fe2+ + H2O2 → Fe3+ + •OH + OH–. (4)
It was found that the kinetics of luminescence and the spectra of chemiluminescence and parameters of the EPR spectra of the spin adduct trap N-tert-butyl-α-phenylnitrone registered in both reactions are similar [32, 33]. This can be easily explained if we suppose that the compared reactions are of the same type. In this case the reaction of HOCl with Fe2+ can be described by the equation:
Fe2+ + HOCl → Fe3+ + •OH + Cl–. (5)
For comparison, the rate constant of the Fenton reaction measured by others varied at about 42-79 M–1·s–1 [34].
Interestingly, reaction (3) is catalyzed by Cu2+, most likely according to the scheme:
Cu2+ + Fe(CN)64– → Cu+Fe3+(CN)62–.
Cu+ then acts as an electron donor:
Cu+Fe3+(CN)62– + HOCl → Cu2+ + Fe(CN)63– + •OH + Cl–.
This leads to increase in reaction rate constant to 1.8·105 M–1 (copper)·s–1 [35].
It should also be noted that the reaction with HOCl itself does not lead to the formation of free radicals but forms molecules breaking free radicals or entering further reactions with the formation of free radicals. First of all, this is a reaction with H2O2:
HOCl + H2O2 → 1O2 + Cl– + H+ + H2O, (6)
in which singlet oxygen (1O2) is formed. The latter interacts with unsaturated bonds of lipids 1500 times faster than triplet oxygen, with formation of lipid hydroperoxides, which leads to the initiation of lipid peroxidation (LPO) [36].
Another important reaction is that of HOCl with nitrite, the overall equation (k = 7.4·103 M–1·s–1 at pH of 7.2, 25ºC [37]) being:
HOCl + NO2– → Cl– + H+ + NO3–. (7)
It was established that in the course of this nitryl chloride (NO2Cl) is formed as an intermediate [37, 38]. The latter causes not only nitration, but also an expressed chlorination effect with respect to phenolic compounds, in particular, with tyrosine [39]. Most likely, these effects are due to the decomposition of NO2Cl to radicals [39]:
NO2Cl → •NO2 + Cl•, (8)
that leads to the initiation of free-radical reactions in proteins and lipids, in particular, tyrosyl radical formation [39] and LPO [37, 40].
The reaction of HOCl with NO also leads to Cl• formation [41]:
HOCl + NO → Cl• + HNO2. (9)
Judging by its pH-dependence, for this reaction the molecular form of HOCl is important. In an exothermic reaction with hypochlorite-anion (OCl–), instead of the radical products nitrogen dioxide (NO2) is formed. Nitrite formed in reaction (9) can react with HOCl according to reaction (7). Thus, taking into account reactions (7), (8), and (9), the HOCl molecule can be regarded as the precursor of free radicals.
Reactions of HOCl with amino acids. HOCl reacts both with α-amino group and some functional groups in the side chains of amino acids [42]. From the point of view of possible formation of free radicals, the reaction of HOCl with the amino group deserves special attention. For amino acids without a functional group in the side chain that can react with HOCl (Gly, Ala, Val, Ser), the reaction proceeds only with α-amino group with formation of monochloramine, and dichloramine is formed in the case of excess HOCl:
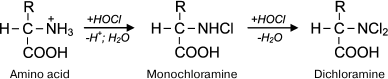
Under physiological conditions, the concentration of HOCl in vivo is not high, and so dichloramine formation is unlikely. The rate of the first reaction is significant: the reaction rate constant depending on the amino acid species ranges from 5.4·104 M–1·s–1 for Ala to 1.7·105 M–1·s–1 for Ser [42]. Chloramines in α-position from the carboxyl group are unstable (half-life ranges from several minutes to several hours [43]) and decompose into aldehydes, passing through a stage of decarboxylation and deamination [44, 45]:
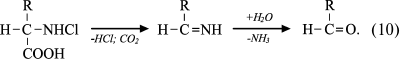 (10)
(10)It has been hypothesized that the side chain of Lys can also be chlorinated and hydrolyzed with aldehyde formation [46, 47]. However, later this mechanism was challenged on the basis that significant generation of carbonyl compounds was detected only in the case when chloramine was in the α-position from the carboxyl group, as in the case of α-amino acid chloramines [48]. There was no evidence that aldehydes are formed from ε-amino-Lys chloramines [49]. Moreover, it was found that the N–Cl bond of ε-amino-Lys chloramines can undergo thermal homolytic bond breakage with the formation of free radicals:
R–CH2–HN–Cl → R–CH2–HN• + •Cl. (11)
Breakage of N–Cl bonds was observed during reduction of Lys chloramine, for example, by transition metal ions:
R–CH2–HN–Cl + Mn+ → R–CH2–HN• + Cl– + M(n+1)+. (12)
The spin trap method using 15N-labeled Lys showed that in both cases the N-centered radical is first formed, which eventually transforms into a C-centered radical, and under aerobic conditions it quickly turns into peroxyl radical [43, 50, 51]. The latter can attach a proton with hydroperoxide formation, which interacts with transition metal ions and turns into alkoxyl radical [52]:
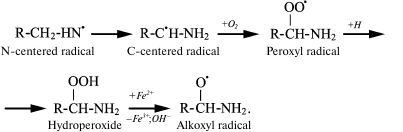 (13)
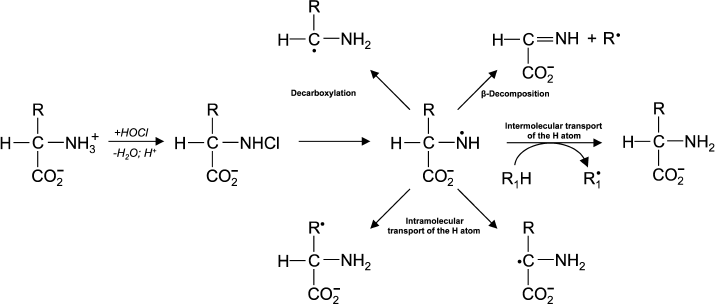
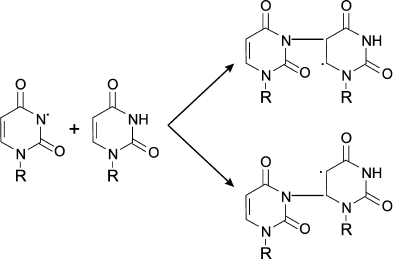
(13)In experiments with Lys and other amino acids, it was found that from α-amino acids chloramine N-centered radicals are formed, which quickly undergo further restructuring, including decarboxylation, β-decomposition, and intra- or intermolecular transfer of the H atom [43]. In all cases C-centered radicals are formed (Fig. 2). Thus, α-amino acid chloramines, in addition to deamination and decarboxylation, can undergo decomposition through stages of free radical formation, thereby increasing cytotoxic effect and contributing to damage of tissues and organs, causing various diseases [47, 53].
Fig. 2. Free radical formation in the reaction of HOCl with the NH2-group of an amino acid.
Using the spin trap method, it was shown that thiyl radicals are formed in the reactions of HOCl with Cys and other low molecular weight thiols (homocysteine, N-acetylcysteine, cysteamine, 3-mercaptopropionic acid, etc.) [54]. Most likely this occurs through formation of a sulfenyl chloride (R–SCl) as an intermediate:
R–SH + HOCl → R–SCl + H2O. (14)
In the presence of transition metal ions, heating, or UV irradiation, the R–SCl decomposes with thiyl radical formation (R–S•) [54].
Reactions of HOCl with proteins. It is clear that all the above reactions of HOCl with functional groups in the side chains of amino acids can also occur in the case of proteins and polypeptides. However, the reactivity of the functional groups in the side chains of amino acids and the same groups in the structure of a polypeptide chain may vary considerably. Treatment of small globular proteins, namely insulin (5.7 kDa) and lysozyme (14.4 kDa), with increasing HOCl concentrations leads to the modification of a number of amino acid residues (His, Lys, Arg, Tyr, etc.). But the nature of the products noticeably differs from the mixture of individual N-acetylated amino acids simulating the amino acid composition of the studied proteins [55]. This indicates that the tertiary structure of the protein globule significantly influences the reactivity of amino acid residue side chains and/or their availability to HOCl.
On addition of HOCl to albumin and other proteins (myoglobin, ribonuclease A, histones, etc.) in the presence of the spin trap 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), spin adducts of N- and C-centered radicals were formed. This is not associated with the presence of SH-groups in proteins, since the enrichment of protein SH-groups by adding 3-mercaptopropionic acid does not affect the intensity and spectral parameters of the recorded spin adduct EPR signal [51]. Preliminary blocking of amino groups of lysine by reductive methylation or reduction of chloramines by methionine to amines before adding the spin trap almost completely prevented the formation of the spin adduct EPR signals, suggesting the involvement of NH2-groups in free radical intermediate formation. Thus, both in a protein as well as in isolated amino acids, the main reaction leading to the formation of free radicals is the reaction of HOCl with NH2-groups, in which an N-centered radical is first formed, which is then transformed into a C-centered radical [51].
The usual result of HOCl–protein interaction is fragmentation, which is mainly due to the cleavage of a peptide bond. When HOCl interacts with a peptide bond, chloramide is formed (Fig. 3). It should be noted that the rate of reaction with the peptide bond amide is much slower than with an amino group. The reaction rate constant greatly depends on the chemical structure of the compound and varies in the range of 3 to 4 orders of magnitude (from 1.2·10–3 M–1·s–1 for N-acetyl-Ala up to 25 M–1·s–1 for cyclo(Gly)2) [42].
Fig. 3. N- and C-centered radical formation with subsequent fragmentation of the polypeptide chain during the reaction of HOCl with the peptide bond.
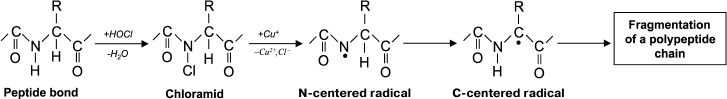
In aqueous media, chloramides are slowly hydrolyzed with peptide bond cleavage and fragmentation of the protein [56]. In addition, the N–Cl bond can undergo homolytic cleavage with N-centered radical formation, for example, in the presence of transition metal ions. Using spin traps, it was shown that a short-lived N-centered radical is the result of intramolecular rearrangement, transforming into a C-centered radical, which leads to fragmentation of the polypeptide chain (Fig. 3) [43].
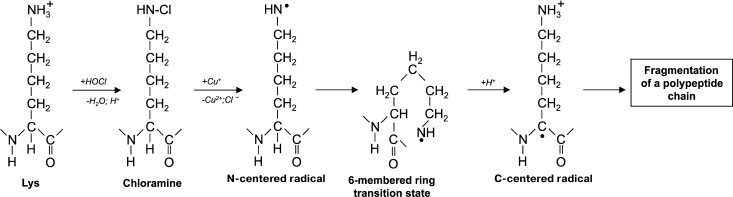
Another reason for peptide bond cleavage can be the formation of chloramide or chloramine in the reaction of HOCl with a side-chain amide group of Gln [50] or an amino group of Lys [51], as shown in Fig. 4 in the case of Lys. First an N-centered radical is formed in the side chain of Lys (or Gln), which is result of 6-membered ring intramolecular rearrangement transforming the radical to the C-atom of the peptide bond. Under aerobic conditions, such a C-centered radical quickly converts into peroxyl radical (see reaction (13)) with subsequent degradation of the polypeptide chain [57].
Fig. 4. Scheme of N-centered Lys radical formation with subsequent transformation to a C-centered radical of the peptide bond and degradation of the polypeptide chain during the reaction of HOCl with the NH2-group of a Lys side chain.
There are many examples of degradation of proteins after treatment with HOCl formed in MPO-dependent reactions. Already in early studies, it was found that three-fold excess of HOCl relative to the number of amino acid residues in ovalbumin caused fragmentation [58]. Low molecular weight peptides were found after the reaction of HOCl with immunoglobulins (IgA, IgG, and IgM) [59]. On reaction of transferrin, ceruloplasmin, and superoxide dismutase (SOD) with HOCl, fragments of the proteins were formed [60]. HOCl formed by the MPO-dependent mechanism during activation of neutrophils caused degradation of apolipoprotein A-I [61].
HOCl also cleaved albumin, ribonuclease A, myoglobin, and some other proteins. Moreover, the effect is strictly dependent on HOCl concentration and incubation time [51]. Despite the fact that one molecule of albumin has many more side-chain functional groups as targets for HOCl (1 Cys, 4 Met, 2 Trp, 59 Lys, totally 66) than ribonuclease A (10 Lys, 4 Met, totally 14), the fragmentation of the latter required more HOCl per peptide bond. Moreover, if Lys in serum albumin was previously blocked by reductive methylation, less HOCl was required for protein fragmentation. This result confirms a hypothesis [56] according to which the fragmentation of the protein is more effective if HOCl reacts directly with the nitrogen of a peptide bond with formation of a chloramide (Fig. 3). However, this can happen only if the majority of side-chain functional groups that are targets for HOCl are blocked or unavailable for HOCl, because the rate constant of the reaction of HOCl with a peptide bond is significantly less than those in the case of side-chain groups of Met, Cys, His, Trp, and Lys [42]. Increasing time of incubation of albumin with a slight excess of HOCl (HOCl/protein < 50 moles/moles) also led to increasing fragmentation of the protein. Moreover, in this case the fragmentation was determined by the reaction of HOCl with side-chain functional groups, mainly of Lys residues with chloramine Lys formation and decomposition of the first to N-centered, and then to C-centered radicals, according to the scheme in Fig. 4. This mechanism is confirmed by the fact that scavengers of radicals (ascorbate, glutathione, and Trolox) and methionine, regenerating formed chloramines, prevent degradation of the protein [51].
Similar results were obtained on addition of HOCl to human blood plasma [62]. Adding HOCl to plasma was accompanied by formation of chloramines, followed by decomposition of latter registered by spin adducts of N- and C-centered radicals, as also occurred in proteins isolated from plasma [51]. The detected paramagnetic centers were associated with a protein fraction. After increase in HOCl concentration or the time of its incubation with plasma, fragmentation of proteins increased, as was indicated by gel electrophoresis, as well as by increase in the mobility of spin adducts of radicals. Fragmentation of proteins was inhibited by addition of methionine and other reagents (ascorbate, urate, glutathione, and Trolox) eliminating chloramines or intercepting free radicals. These results confirm the hypothesis that the chloramines formed in the reaction of HOCl with NH2-groups of proteins are decomposed with formation of N-centered radicals, which are converted into C-centered radicals with subsequent fragmentation of the polypeptide chain [62]. This free radical protein degradation can occur in plasma under conditions of oxidative stress and inflammation.
It should be noted that spin adducts of thiyl radicals were not registered on the reaction of HOCl with plasma. They appeared only after adding glutathione to the plasma [62]. Thus, if thiyl radicals formed during HOCl treatment of plasma (see reaction (14)), they are minor products. Fragmentation of proteins by free radical mechanisms is due to homolytic cleavage of the N–Cl bonds of chloramines and chloramides with the formation of N-centered radicals.
The reaction of HOCl with enzymes is usually accompanied to some extent by their inactivation, which can be due to modification of key amino acid residues that are localized in the active center as well as nonspecific reactions leading to changes in the physical and chemical properties of the polypeptide chain (change in conformation, fragmentation, etc.). In some cases, the involvement of free-radical intermediates in inactivation of enzymes under HOCl treatment or functioning of MPO is confirmed by the fact that scavengers of free radicals partially or fully protect the activity of the enzyme [63].
Table 1 lists enzymes studied by various authors after modification by HOCl, MPO, or activated neutrophils. In most cases some inactivation of the enzymes was found. Exceptions were collagenase and gelatinase from specific granules of neutrophils, which are activated in the presence of HOCl [84-87] and neutrophils induced by the HOCl-dependent pathway [85, 86, 88]. In some cases HOCl-modified proteins became more sensitive to proteolytic enzymes. Thus, the incubation of fibronectin and collagen in the presence of HOCl increased the proteolytic activity of elastase and collagenase, respectively, with respect to the proteins [53, 89]. However, it should be noted that proteolytic enzymes of granulocytes are active only in a narrow range of HOCl concentrations not exceeding HOCl/enzyme molar ratio of 10-40. Higher concentrations of HOCl, as well as longer time of incubation in the presence of activated neutrophils or MPO, resulted in inactivation of collagenase, gelatinase, and elastase [68, 87].
Table 1. List of enzymes inactivated by HOCl, by enzymatically active MPO or by
activated neutrophils

Reactions of HOCl with nucleotides and nucleic acids. It is known that HOCl does not react with ribose [35], and therefore the most likely target for HOCl in nucleotides is the nitrogen in nitrogenous bases. Hypohalous acids do not react with tertiary nitrogen, in particular with 3-methylthymidine [90]. It seems that the target for HOCl is the primary nitrogen in the amino group of cytosine, guanosine, and adenosine (–NH2) or secondary nitrogen of the imino group in the heterocycle of uridine, thymidine, and guanosine (>NH). Table 2 shows rate constants of bimolecular reactions of HOCl with mononucleotides, polymers based on them, and DNA. It is seen that the rate of interaction of HOCl with the primary N-atom outside the cycle is much lower than with the secondary nitrogen in the heterocycle. For guanosine monophosphate containing both types of N-atoms, fast and slow kinetics with HOCl was registered in accordance with chlorination of the N-atom in the heterocycle and nitrogen of NH2-group, respectively (Table 2) [35].
Table 2. Rate constants of bimolecular reactions of HOCl with imino- and
amino-groups of mononucleotides, polymers based on them, and DNA
(20oC, pH 6.9) [35, 90]
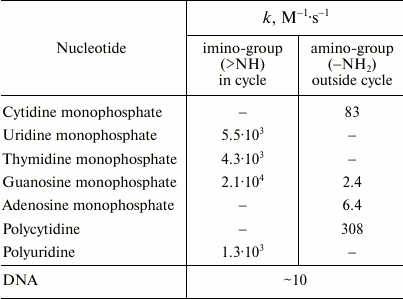
Rate constant of the reaction of HOCl with polynucleotides (polycytidine and polyuridine) is not significantly different from that for the corresponding mononucleotides (cytidine and uridine), suggesting that polymerization does not play a significant role in the reactivity of the N-atoms inside and outside the heterocycle (Table 2). However, the rate of the reaction of HOCl with DNA is significantly lower than with most mononucleotides, although in DNA there are not only slowly reacting N-atoms of NH2-groups but also reactive N-atoms of the heterocycle. This phenomenon can be explained by a steric barrier to penetration of HOCl between the base pairs in DNA structure. This hypothesis is consistent with the fact that HOCl reacts with thermally denatured DNA about 10 times faster than with the native biopolymer [90].
Chlorinated nucleotides have different resistance. Cytidine, adenosine, guanosine chloramines of NH2-group are significantly more resistant than uridine and thymidine chlorimine of heterocycles [91]. Using spin traps, it was established that upon heating, UV irradiation, or in the presence of transition metal ions all nucleotide chloramines and chlorimines decompose with formation of N-centered radicals, as shown for example in the case of uridine [91]:
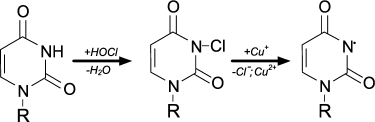
When HOCl is added to a mixture of nucleotides, the ability to form N-centered radicals follows the series: cytidine > adenosine = guanosine > uridine = thymidine. These data are in contrast with rate of interaction of HOCl with nucleotides (Table 2) and the resistance of their chlorinated forms [91]. However, this can be easily explained if we assume that chlorine is rapidly transferred from chlorimine of thymidine, uridine, or guanosine heterocycle to the NH2-group of cytidine and adenosine. This increases the yield of the more stable cytidine and adenosine chloramines, which are then decomposed with N-centered radical formation [91].
The interaction of the resulting N-centered radical with other molecule of nucleotide leads to formation of dimer of C-centered radical [91]:
Similar patterns were observed on treating DNA, polynucleotides, or nucleic acids with HOCl. Chloramines and chlorimines formed first, which then decomposed forming N-centered radicals, which subsequently led to intermolecular crosslinking [92] and denaturation of the DNA [35].
However, after HOCl treatment of nucleosomes the core target for HOCl is the protein, not the DNA. Thus, when nucleosomes were treated with 50-200-fold molar excess of HOCl, around 50-80% of the HOCl reacted with Lys and His forming chloramines in the histones. These chloramines decomposed with the formation of N-centered radicals. The free valence transferred onto pyrimidine nucleotides, resulting in the formation of intermolecular protein–DNA crosslinking and free radical centers of the nucleotides that ultimately lead to DNA fragmentation. Preventing such destruction of DNA by scavengers of free radicals again indicates the participation of free radicals in this process [93].
Recently, the first paper appeared about the possibility of HOCl-induced free radical formation in genomic DNA of living cells [94]. The authors used method based on application of the immunospin-trap DMPO and antibodies against the trap. Addition of H2O2 to HL-60 line cells or H2O2 synthesis in the glucose/glucose oxidase system in the presence of cells led to activation of intracellular MPO and formation of DNA-centered radicals. The latter were fixed by DMPO spin trap and were registered by ELISA using antibodies against DMPO. DNA-centered radicals also were found in RAW 264.7 macrophage line cells preincubated with lipopolysaccharide and then stimulated with phorbol-12-myristate-13-acetate. During incubation of A549 line epithelial cells with MPO, the enzyme penetrated into the cells and localized on the surface of the nuclei. After adding the glucose/glucose oxidase system generating H2O2, thus activating the chlorinating cycle of MPO, DNA-centered adducts of radicals appeared in epithelial cell line A549 cells without noticeable changes in their viability. Coincubation of epithelial cells with activated neutrophils also led to DNA-centered radical formation. In all cases control experiments showed that the DNA radical formation was caused by the functioning of the MPO chlorinating cycle. Published results [94] showed that penetrating into the cell or newly synthesized MPO catalyzed HOCl generation by the chlorinating cycle. This caused radical formation in the genomic DNA.
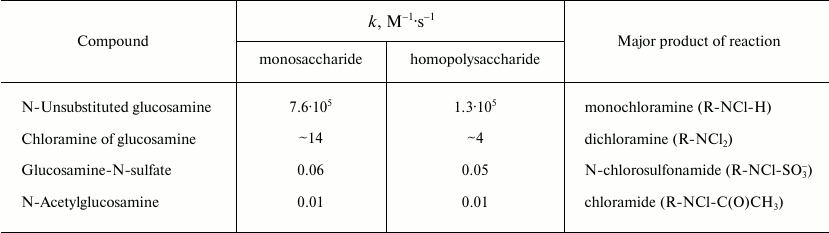
Reactions of HOCl with carbohydrates. It is known that neutrophils cause degradation of proteoglycans, an important component of polysaccharide chains [95], and this process also involves MPO [96]. The main target for HOCl in carbohydrates is the nitrogen in amino sugars (D-glucosamine, D-galactosamine, D-mannosamine, etc.), in which the hydroxyl group at the 2-C atom is replaced by a primary amino group. Rate constants of bimolecular reactions of HOCl with unaltered glucosamine, some of its derivatives, as well as polysaccharides based on them are given in Table 3. The reaction of HOCl with the free NH2-group of unsubstituted glucosamine proceeds quickly, usually even faster than with the NH2-group of amino acids [42]. Substitution by chlorine of the second H atom in monochloramine with dichloramine formation occurs much more slowly and only with excess of HOCl. Finally, substituted glucosamine reacted even more slowly with HOCl. Glucosamine-N-sulfate reacting with HOCl is converted to N-chlorosulfonamide and N-acetylglucosamine – to chloramide. Interestingly, the reaction of HOCl with homopolysaccharides proceeds more slowly than with the corresponding monosaccharides. This can probably be explained by steric interaction and the influence of negatively charged groups in the polysaccharides.
Table 3. Rate constants of second order (bimolecular) reactions of HOCl with
monomer and polymer derivatives of glucosamine (22°C, pH 7.4)
[97, 98]
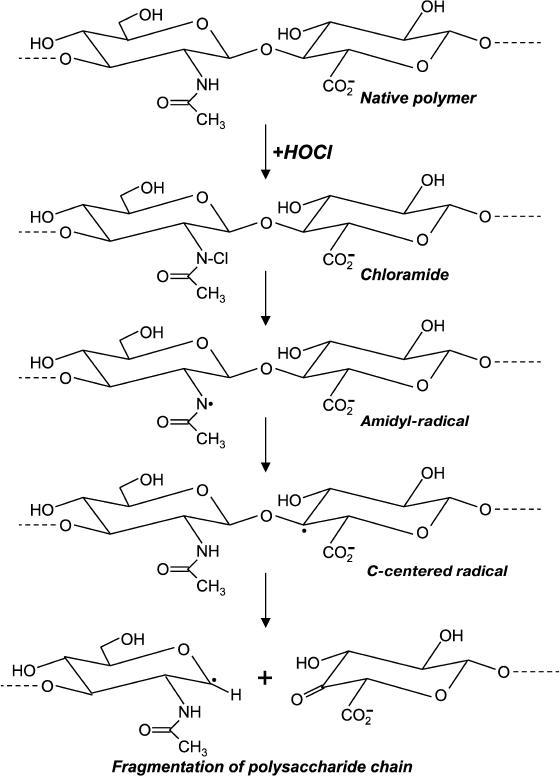
Chloro-derivatives of amino sugars and their substituted analogs are not stable, and the gradually decompose with formation of free radical intermediates. One of the mechanisms of decomposition using the example of hyaluronic acid is shown in Fig. 5. Chloramide, which is formed at the first stage in the reaction with HOCl, transforms into N-centered amidyl-radical as a result of homolytic cleavage of N–Cl bonds in the presence of transition metal ions. The radical undergoes internal rearrangement and after isomerization a C-centered radical is formed. This finally leads to fragmentation of the polysaccharide chain [99, 100]. Probably due to reduction, transition metal ions in the presence of •O2ˉ significantly accelerates homolytic cleavage of N–Cl bonds, thereby increasing HOCl-induced degradation of polysaccharides [101, 102].
Fig. 5. Scheme of N-centered amidyl-radical formation and transformation into C-centered radical with fragmentation of the polysaccharide chain during reaction of HOCl with the acetylamide-group of hyaluronic acid.
A different mechanism of HOCl-induced fragmentation of polysaccharides involving free radicals was recently proposed [103, 104]. This mechanism involves direct interaction of HOCl with a hydroxyl group in the carbohydrate ring (R–CH2–OH) according to the reaction:
R–CH2–OH + HOCl → R–CH2–OCl + H2O.
The intermediate R–CH2–OCl is unstable, and it decomposes to O-centered radical, for example, in the presence of transition metal ions:
R–CH2–OCl + Cu+ → R–CH2–O· + Cu2+ + Cl–.
This leads to the breakage of the carbohydrate ring and fragmentation of the polysaccharide chain [103, 104]. At present the occurrence of this mechanism has not been proven.
The main components of extracellular matrix are proteoglycans, which contain large numbers of negatively charged groups in the carbohydrate part. MPO, a polycationic protein secreted by leukocytes into the extracellular space, can bind to glycosaminoglycans of proteoglycans by electrostatic interactions [96]. The functioning of the enzyme in the vicinity of proteoglycans, which is accompanied by HOCl production, leads to formation in the carbohydrate of chloramines and chloramides. The latter are decomposed by a radical mechanism (Fig. 5) that is accompanied by fragmentation of glycosaminoglycans and extracellular matrix in general [96, 105].
The consequences of this destruction of the extracellular matrix are still little investigated. However, one can expect that degradation of proteoglycans should significantly affect adhesion, migration, proliferation, growth, and phenotype of cells. It is likely that the free radical mechanism of MPO-dependent modifications of glycosaminoglycans of extracellular matrix described in this section may be the cause of cell dysfunction and development of diseases such as atherosclerosis [106], rheumatoid arthritis [107], asthma [108, 109], and others diseases in which intensive MPO-dependent generation of oxidants (including HOCl) is observed.
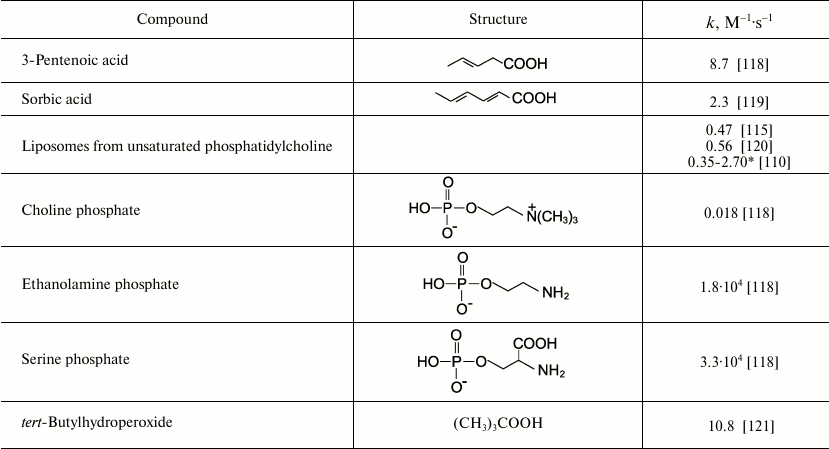
Reactions of HOCl with lipids. It is known that HOCl does not react with saturated fatty acids [110-113] and saturated phosphatidylcholine [110-117]. This means that HOCl does not react with saturated –CH2–CH2– bonds, with carboxyl group, ester, or with the phosphocholine group. The second order reaction rate constants of the reactions of HOCl with compounds modeling functional groups of lipids are given in Table 4. It is obvious that in lipids there are two main targets for HOCl: unsaturated bonds in acyl chains and cholesterol (–CH=CH–) as well as NH2-group of “polar heads” in phosphatidylethanolamine and phosphatidylserine. With regard to unsaturated bonds, there are two main mechanisms: molecular (without involvement of free radicals) and free radical (LPO), i.e. with free radical intermediates being obligatory participants. The interaction of HOCl with unsaturated bonds of fatty acid chains, as well as their models, was studied by luminol-dependent chemiluminescence [19, 110, 113], iodometry [19, 122, 123], 1H-NMR [19, 113], spectrophotometry [120, 124], chromatography [111], and mass spectrometry [111, 112, 125]. It is established that the basic reaction proceeds through the mechanism of electrophilic addition of HOCl to a double bond according to the sum of Eq. (15) with the formation of chlorohydrin isomers [19, 110, 112, 116, 117]:
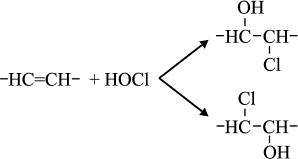 (15)
(15)The rate constant for this reaction with unsaturated bonds of phospholipids in liposomes is noticeably smaller than that measured in the case of solution of 3-pentenoic and sorbic acids (Table 4). This is probably because the availability of double bonds in multilayer liposomes for HOCl is limited, and this is the limiting stage of reaction (15). This hypothesis is confirmed by fact that reduction of the number of layers in liposomes by ultrasonic treatment increased the rate of interaction of HOCl with double bonds by 6-8-fold, bringing it closer to that found in the case of monolamellar liposomes [110, 113].
Table 4. Rate constants of second order reaction of HOCl with compounds modeling
functional groups of lipids (22oC, pH 7.2-7.4)
* Rate constant was increased in the given range with increasing time of
preliminary ultrasonic treatment of liposomes.
It was shown that during incubation of HOCl with phosphatidylcholine liposomes or blood lipoproteins containing unsaturated lipids, in addition to chlorohydrins, typical products were detected that usually are found when initiating LPO in traditional ways: primary molecular products – diene conjugates and hydroperoxides [122, 123, 126-130]; secondary products of carbonyl nature reacting with 2-thiobarbituric acid [115, 122, 123, 126-128, 131-134]; final products with fluorescence in the visible range (Schiff bases) formed during the interaction of oxidized lipids with proteins [132]. HOCl-induced LPO is completely blocked by known scavengers of free radicals (micromolar concentrations of α-tocopherol or butylated hydroxytoluene) [37, 122, 123, 128, 129, 133, 134] that confirms the free radical mechanism of this reaction. HOCl penetrates into the lipid phase of serum lipoproteins [135], destroys lipophilic antioxidants (carotenoids, α-tocopherol) [19, 37, 136], and thereby lowers the resistance of the lipoprotein lipid phase to LPO [136].
Like any free-radical chain reaction, LPO has a stage of initiation. What is the initiation stage in HOCl-induced LPO? It is known that trace amounts of H2O2 (about 10–8 M) are always present in water as result of natural radiolysis [137]. HOCl reacts with H2O2 according to reaction (6) with singlet oxygen formation [138]. The latter can oxidized unsaturated bonds of lipids 1500 times faster than triplet oxygen, forming hydroperoxides and thus initiating LPO [36]. Moreover, HOCl reacts with transition metal ions, in particular with Fe2+ [19, 30, 32, 139], or with superoxide anion-radical (•O2ˉ) [29, 140], with formation of the extremely reactive •OH radical. The latter is an effective initiator of free radical reactions of LPO (see reactions (5) and (2)). Any of the reactions (2), (4), or (5) might be able to perform the role of the initiation stage of HOCl-induced LPO. However, preliminary addition of Fe2+, H2O2, catalase, or SOD to phospholipid liposomes before HOCl did not change the yield of LPO products [122, 123], this suggesting the unlikeliness of the involvement of reactions (2), (5), and (6) in HOCl-induced LPO. Perhaps this is due to fact that the reaction in these experiments occurs in the aqueous phase, and the lifetime of 1O2 and especially •OH in water is so low (10–6 and 10–9 s, respectively [28]) that they do not have time for diffusion into the hydrophobic lipid phase where unsaturated bonds, the substrate of LPO, are localized.
At the same time, using chemiluminescence and spin traps methods, it was established that HOCl reacts with the hydroperoxide group with forming peroxyl radicals, which are transformed into alkoxyl radicals [121, 129, 141-147]. In the case of fatty acid hydroperoxide (but not tert-butyl or cumene hydroperoxides [147-149]) it is possible that some singlet oxygen production in addition to O-centered radical formation occurs [129, 147]. MPO in the presence of substrates (H2O2 and Cl–), as well as activated neutrophils, destroyed hydroperoxide with the formation of O-centered radicals identified as peroxyl and alkoxyl radicals [145]. Inhibitory analysis using traps of HOCl (taurine, methionine), scavengers of free radicals (butylated hydroxytoluene and mannitol), and inhibitors of MPO (salicylhydroxamic acid and 4-aminobenzoic hydrazide) showed that decomposition of hydroperoxide group in the presence of isolated MPO or activated neutrophils is caused directly by enzymatic activity. However, only some of the radical intermediates occurred as the result of the MPO halogenating cycle – the stage of HOCl formation (Fig. 1). Another part was formed regardless of HOCl, apparently involving the MPO peroxidase cycle [145].
Addition of hydroperoxides to the incubation medium of phospholipid liposomes and HOCl or MPO + H2O2 + Cl– was accompanied by further accumulation of LPO products [115, 129, 134]. This effect was completely prevented by scavengers of free radicals (butylated hydroxytoluene), traps of HOCl (taurine and methionine), and inhibitor of MPO (sodium azide), demonstrating the involvement of the free radical mechanism involving the MPO halogenating cycle [134]. The combination of available experimental results confirms that the MPO-dependent reaction of HOCl with hydroperoxides, which are always present in trace amounts in unsaturated lipids [36, 150], proceeds with free radical formation and can really take place in HOCl-induced LPO.
The presence of protein helps to activate the free radical mechanism of HOCl-induced LPO [151]. In protein–lipid complexes, for example in low density lipoproteins, HOCl primarily reacted with NH2 groups of apolipoprotein B-100 with chloramine formation. The latter, as a result of homolytic cleavage of N–Cl bonds, decays with formation of free radicals (see reactions (11) and (12)), which are involved in initiation of LPO [151]. In all cases, HOCl is a precursor of free radicals.
Taking into account these results, the putative involvement of HOCl in free radical modification of unsaturated lipids can be represented in the scheme shown in Fig. 6. Stimulation of leukocytes is accompanied by activation of a membrane bound enzyme, NADPH-oxidase, which catalyzes •O2‾ formation. The latter dismutates into H2O2 spontaneously or under the influence of another enzyme (SOD). MPO is secreted into the extracellular space upon activation of leukocytes. The latter catalyzes the oxidation of Cl– to HOCl using H2O2 as a substrate. Each of the mentioned compounds (•O2‾, HOCl, and H2O2) can be involved in reactions (2), (5), (6), and the Fenton reaction (4) as a source of •OH radical or singlet oxygen (1O2). The •OH radical, abstracting a hydrogen atom from unsaturated lipid (L), forms an alkyl radical (L•) and thus initiates LPO. Under aerobic conditions, L• attaches an oxygen molecule forming peroxyl radical (LOO•) and subsequent hydroperoxide (LOOH) formation. The latter can also be formed through a molecular mechanism (without the participation of free radicals) after the reaction of unsaturated lipid with singlet oxygen (1O2), which in turn occurs in the reaction of HOCl with H2O2 or fatty acid chain hydroperoxide. Hydroperoxide can be reduced by Fe2+ with formation of alkoxyl radical (LO•), and can be oxidized by HOCl with formation of peroxyl radical. Both radicals give rise to a chain reaction of free radical LPO. Thus, like Fe2+, HOCl turning hydroperoxides in free radicals (in this case peroxyl) plays an important role in intensification of LPO reactions at the stage of branching cycles according to the scheme:
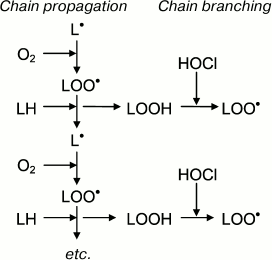
Fig. 6. Scheme of participation of HOCl produced by activated neutrophils in initiation and branching of free radical LPO chains. See notes in text.
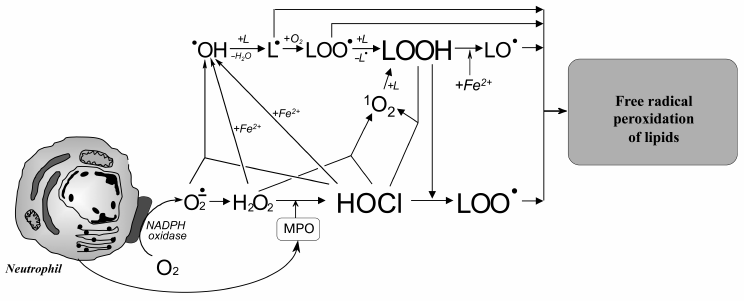
It should be noted that hydroperoxides can be formed in the human body not only as a result of LPO activation of free radical reactions. There are other sources: photoinduced oxidation involving singlet oxygen [152], ozonolysis [153], and catalytic oxidation of unsaturated fatty acid chains by different lipoxygenases [154], for example the oxidation of arachidonic acid to leukotrienes [155].
HOCl reacts with only unsaturated bond of cholesterol [156]. Using thin layer chromatography, NMR, and mass spectrometry, it was established that addition of HOCl to cholesterol-containing phospholipid liposomes, as well as their incubation in the presence of the MPO + H2O2 + Cl– system, led to formation of the α- and β-chlorohydrin isomers [157, 158]. Unlike chlorohydrins of fatty acid chains, cholesterol chlorohydrins were not stable, and as a result of subsequent dehydrochlorination they turned into epoxides. Similar products were formed in low density lipoproteins (LDL) [159], cellular membranes [160] or intact cells (breast cancer, erythrocytes, neutrophils) [160] if they were treated with HOCl or incubated in the presence of the MPO + H2O2 + Cl– system. Hydrolysis and subsequent oxidation of α- and β-epoxides lead to large number of cholesterol hydroxy- and keto-products [156, 161]. However, in the literature there is no experimental evidence for free radical formation during these reactions [162].
Consider the mechanism of interaction of HOCl with polar head groups of phospholipids. There is only one target, namely the nitrogen atom. Table 4 shows rate constants for the second order reactions of HOCl with compounds modeling the polar head group of phospholipids. Using different methods in numerous experiments, it was shown that HOCl did not react with appreciable rate with the phosphocholine group of phosphatidylcholine [19, 110, 113, 115-117, 125, 163, 164] or model compounds [118]. As shown in Table 4, the reaction rate constant measured for choline phosphate is extremely small. This constant was calculated from the decrease in optical density at 290 nm (absorption band of OCl–). This difficult to reproduce reaction was extremely slow. New spectral peaks were not registered [118]. So, diminution of OCl– is not necessarily determined by the reaction of HOCl with choline phosphate. Most likely, hypohalous acids do not interact with the quaternary nitrogen atom in the phosphocholine group.
Free NH2-groups of phosphatidylethanolamine and phosphatidylserine, on the contrary, quickly react with HOCl with formation of chloramine (Table 4). For unsaturated phosphatidylethanolamine, primarily NH2-groups of the polar head groups interact with HOCl, and only when all amines have been converted to dichloramines, chlorohydrins began to be formed [165]. This pathway of events is a result of too great a difference (3-4 orders of magnitude) in the rate constants of reaction of HOCl with NH2-group of the polar head group of phosphatidylethanolamine and unsaturated –HC=CH– bonds of fatty acid chains (Table 4).
Chloramines of polar head groups of phosphatidylethanolamine and phosphatidylserine are quite different in stability. Thus, chloramine of serine phosphate decomposed completely after 60-min incubation at 37ºC, while chloramine ethanolamine phosphate was much more stable and was practically stable for at least 2 h. The chloramine of phosphatidylserine in liposomes under the same conditions decomposed within 15 min [118]. This difference in stability of chloramines of phosphatidylethanolamine and phosphatidylserine is due to the fact that the amino group of the latter is adjacent to a carboxyl group, as in case of amino acids. Such chloramines are unstable and quickly decomposed with aldehyde formation (see reaction (10)).
On reaction with HOCl, the amino group of phosphatidylethanolamine first is converted to a monochloramine, and then a dichloramine [130, 166]. The latter can be transformed into a nitrile via dehydrochlorination through a molecular mechanism [166] and can undergo homolytic cleavage of the N–Cl bond forming an N-centered radical [130]. Due to this difference in the mechanisms of HOCl-induced decomposition of phosphatidylethanolamine and phosphatidylserine, the chloramines can regulate MPO-dependent free radical LPO. It was found that the presence of phosphatidylethanolamine in liposomes of unsaturated phosphatidylcholine contributed to the further accumulation of LPO products in the course of HOCl-induced LPO due the formation of an N-centered radical, which acts as an initiator of LPO. Addition of phosphatidylserine to phosphatidylcholine liposomes, on the contrary, inhibited HOCl-induced LPO, since the HOCl concentration is decreased as result of reaction with amino groups. Thus, only molecular products, which do not influence the LPO reaction, were formed [130].
ROLE OF REACTIVE HALOGEN SPECIES AND HALOGENATIVE STRESS IN
DEVELOPMENT OF HUMAN DISEASES
Because hypohalous acids contain oxygen, they are sometimes referred to reactive oxygen species (ROS) [7 (p. 27), 167, 168]. This is hardly appropriate, since the oxidizer in hypohalous acid (HOHal) is not oxygen, as in the case of ROS, but a halogen atom, taking two electrons and forming halogenide (Hal–):
HOHal + 2e + H+ → Hal– + H2O.
Hypohalous acids are strong two-electron oxidants, and their oxidative ability decreases in the series HOCl > HOBr > HOI. The redox potentials for the HOHal/Hal–,H2O couples are 1.08, 0.93, and 0.57 V, respectively [12].
Moreover, the reactivities of hypohalous acids are manifested not only in redox reactions, but also in reactions of replacement, exchange, and others with modification of various functional groups in important biomolecules [19, 97, 169]. It is now known that MPO- and EPO-dependent reactions involving halogen-containing compounds in living organisms can damage tissues and organs and thereby promote the development of numerous diseases [170-172]. Then reactive halogenated compounds along with ROS and reactive nitrogen species would be correctly considered as a separate group of reactive halogen species (RHS). The response of organisms to RHS is called halogenative stress.
RHS are halogen-containing compounds exhibiting high reactivity, and they are formed in living organisms or enter the organism from the environment. In this case, halogenative stress (taking into account the term “stress” introduced by H. Selye [173]) is a nonspecific reaction of an organism to any influence of RHS when RHS appearance or formation in the organism exceeds the organism’s ability to remove or neutralize them.
The impact of environmental factors often results in poisoning of an organism as accumulation of critical RHS concentrations on contact of a human or animal with air, soil, water, flora, or fauna as well as breathing, eating, etc. They can be pesticides, components of medicines, wastewater, exhaust gases, industrial poisons, and other reactive halogenated compounds. In the literature, investigations of mechanisms of the toxic action of RHS from environment on humans have been repeatedly discussed [174, 175]. So we will leave this topic beyond this review and will focus on endogenous halogenative stress caused by formation and transformation of RHS directly in the human body, as well as the possible role of the above-described reactions of RHS in the pathogenesis of some human diseases.
All endogenous RHS found in mammals can be divided into two groups: primary, formed directly as a result of enzymatic reactions, and secondary, formed in subsequent reactions of primary RHS, including functional groups of biologically important compounds having increased reactivity. HOCl is the primary RHS because it is the result of the MPO halogenating cycle (see reaction (1) and Fig. 1). Secondary RHS include molecular chlorine (Cl2), nitryl chloride (NO2Cl), chloramines, and chloramides of biologically important molecules, and they are usually the precursors of free radicals (see section “Free Radical Formation in Reactions Involving HOCl”).
To reveal the involvement of MPO/EPO in the emergence and/or development of various diseases, the concentration/activity of the enzyme or the presence of so-called biomarkers (usually in blood or the foci of a disease) should be determined. A biomarker is molecule or functional group whose appearance clearly indicates a stage of a studied phenomenon or process in a biological system. A good biomarker should comply with at least the following requirements: 1) being specific in relation to the studied phenomenon; 2) having high rate of formation; 3) having relatively long life (low rate of decomposition); 4) accumulating in the body at sufficient concentration for detection; 5) being detectable by readily available and sensitive methods.
Table 5 lists examples of diseases accompanied by halogenative stress, which are characterized by an increased concentration/activity of MPO or the discovery of biomarkers indicating increased MPO activity in blood, damaged organs, and tissues. First of all, attention is drawn to atherosclerosis and other symptoms of cardiovascular insufficiency. In these diseases, increasing concentration and activity of MPO in blood plasma [187-192, 230], atherosclerotic plaque [176], leukocytes [187], and complexes with LDL and VLDL are found [231].
Table 5. Biomarkers of MPO/HOCl in human diseases
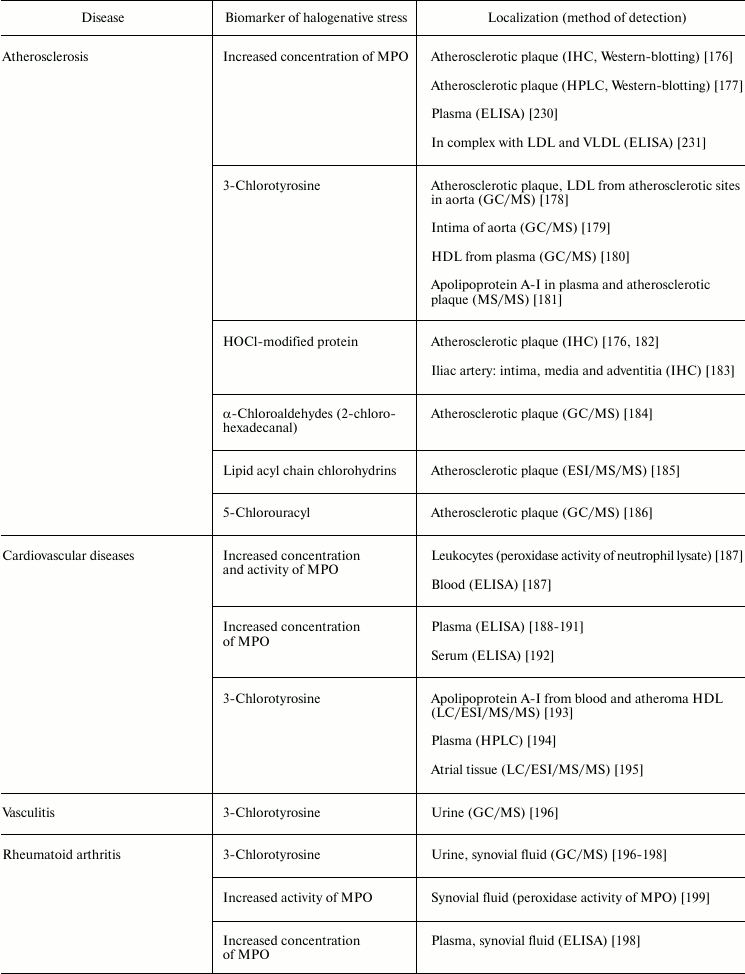
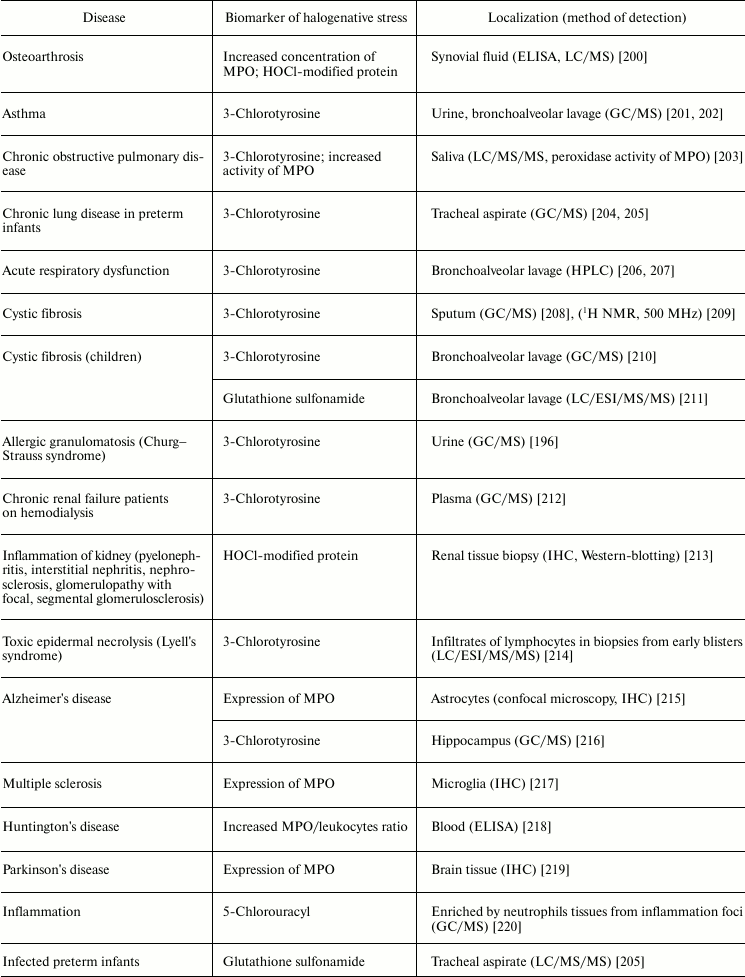
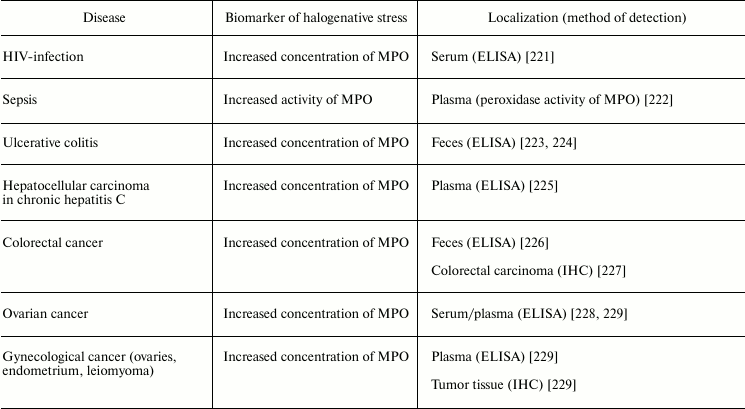
Note: HPLC, high-pressure liquid chromatography; IHC,
immunohistochemistry; ELISA, enzyme-linked immunosorbent assay; HLD,
high density lipoproteins; LDL, low density lipoproteins; VLDL, very
low density lipoproteins; GC/MS, gas chromatography–mass
spectrometry; LC/MS, liquid chromatography–mass spectrometry;
MS/MS, tandem mass spectrometry; LC/MS/MS, liquid chromatography-tandem
mass spectrometry; ESI/MS/MS, electrospray-tandem mass spectrometry;
LC/ESI/MS/MS, liquid chromatography-electrospray tandem mass
spectrometry.
MPO plays an important role in development of atherosclerosis by modifying major components of the surface of LDL (protein and phospholipids), mainly under the influence of RHS, including HOCl generated during the MPO halogenating cycle [132, 169]. Being a polycationic protein, MPO associates due to electrostatic interaction with the LDL surface [231]. One possible binding site for MPO is the region 445EQIQDDCTGDED456 of apolipoprotein B-100, which is rich in negatively charged amino acid residues [232]. This binding of MPO to LDL brings the enzyme to the target and exacerbates modification of LDL surface components. HOCl formed in the halogenating cycle of MPO reacts with functional groups of amino acid residues, including NH2 groups of Lys, Arg, and Gln, resulting in free radical formation (see section “Free Radical Formation in Reactions Involving HOCl”, Fig. 4). After penetration into the lipid phase of the phospholipid monolayer surface [135], HOCl initiates free radical LPO. Also, HOCl reacts by the molecular mechanism with unsaturated bonds of fatty acid chains, resulting in formation of chlorohydrins (see section “Free Radical Formation in Reactions Involving HOCl”, reaction (15)). As a result of such modification, LDL are transformed into their proatherogenic form and are intensely taken up by subendothelial cells. This is accompanied by accumulation of intracellular cholesterol with formation of foam cells – one of the most important and pathognomonic characteristics of early stages of atherosclerosis [37, 169, 233]. Confirmation of this is the fact that biomarkers of halogenative stress have been repeatedly found in the blood plasma of patients with cardiovascular diseases, in atherosclerotic sites of the aorta, in blood lipoproteins, and in cardiac tissue. Such biomarkers include proteins modified by HOCl, 3-chlorotyrosine, 5-chlorouracyl, α-chloroaldehydes, and acyl chains of lipid chlorohydrins, which are formed in living organism only as result of the halogenating cycle of mammalian heme peroxidases, mainly MPO (Table 5).
MPO is also involved in the development of joint diseases: arthritis and arthrosis (Table 5). Increase in MPO concentration in plasma from patients with rheumatoid arthritis was shown [198]. In urine and synovial fluid of such patients, 3-chlorotyrosine has been found [196-198]. The content of 3-chlorotyrosine in synovial fluid increases with increasing MPO concentration [198]. In addition, increased MPO activity in synovial fluid [198, 199] correlates with the level of damage to articular cartilage tissue under the influence of HOCl [199]. In the synovial fluid, MPO concentration was increased and HOCl-modified proteins were detected in the case of osteoarthritis [200]. It is expected that during development of inflammatory reaction in the synovial fluid, MPO is released by activated leukocytes. HOCl formed in MPO-dependent reactions provokes oxidative/halogenative stress. The latter damages carbohydrate components of cartilage tissue in articular surfaces, contributing to softening and reduction of thickness of the articular cartilage.
Respiratory diseases are also associated with increased activity of MPO. The HOCl biomarker 3-chlorotyrosine was detected in urine and bronchoalveolar lavage from patients with asthma [201, 202], saliva from patients with chronic obstructive pulmonary disease [203], tracheal aspirate from preterm infants with respiratory dysfunction [204, 205], bronchoalveolar lavage from patients with acute respiratory failure [206, 207], sputum [208], and bronchoalveolar lavage from patients with cystic fibrosis [209, 210]. Increased activity of MPO was also found in saliva from patients with chronic obstructive pulmonary disease [203]. The product of oxidation of glutathione by HOCl, glutathione sulfonamide, was found in bronchoalveolar lavage from children with cystic fibrosis [211]. If lung disease was accompanied by allergic symptoms, in addition to 3-chlorotyrosine, a biomarker of HOBr, 3-bromotyrosine, was repeatedly found [196, 201, 202, 209, 210]. This is due to the fact that allergies significantly increase the number of eosinophils in blood, and secreted EPO mainly catalyzes Br– oxidation to HOBr [11, 12]. The latter catalyzes bromination of Tyr with formation of 3-bromotyrosine [234].
Kidney failure is accompanied by inflammation connected with development of immune response, increase in antibody and immune complex concentration in the kidney filtering system, migration of neutrophils, and their activation and secretion of active MPO. Enrichment of basal kidney membrane by acidic sialoproteoglycans and proteoglycans (heparin sulfate) demonstrating high affinity to polycationic MPO is a factor amplifying the damaging action of MPO. The involvement of MPO in progressive renal insufficiency is confirmed by development of autoimmune reaction with increased titer of antibody against MPO (MPO-ANCA) in serum of patients. Kidney disease is also characterized by the presence of 3-chlorotyrosine in plasma [212] or HOCl-modified protein in renal tissue biopsy [213], indicating the involvement of MPO in inflammation of the kidneys.
Much attention is given to the role of MPO and RHS in development of neurodegenerative diseases: Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, multiple sclerosis, and others [235]. It was shown that in Alzheimer’s disease [215] and multiple sclerosis [217] MPO is overexpressed in astrocytes and microglia, respectively. The participation of MPO in development of these neurodegenerative diseases is confirmed by the presence of early symptoms of multiple sclerosis and Alzheimer’s disease in women with the G allele in the case of –463G/A polymorphism, which is associated with increased MPO expression in women. Expression of MPO is registered also in the ventral part of the midbrain from patients with Parkinson’s disease [219]. Tyrosyl radicals and reactive nitrogen species were found in neurons affected by the neurotoxin MPTP, which used for experimental modeling of Parkinson’s disease. In blood of patients with Huntington’s disease, increase in MPO/leukocyte ratio was found [218]. 3-Chlorotyrosine was found in the hippocampus from patients with Alzheimer’s disease [216]. Neuroprotective effects of desferoxamine and uric acid can be explained also by their ability to interact with HOCl [236-238]. This suggests the involvement of HOCl in neurodegenerative disorders. These and other results indicate that MPO secretion by microglia and other macrophagal cells in brain can lead to HOCl-induced death of neurons, thus contributing to the development of neurodegenerative diseases [235].
Numerous infectious diseases also show signs of halogenative stress. In tracheal aspirate from infected preterm infants, the HOCl-biomarker glutathione sulfonamide was detected [205]. Increased concentration of MPO was detected in the serum of HIV-infected patients [221] and in the feces of patients with ulcerative colitis [223, 224]. MPO activity was increased in blood plasma during sepsis [222]. Tissues enriched in neutrophils from inflammation foci contained 5-chlorouracil, a biomarker of HOCl-induced damage of nucleic acids [220].
It is now estimated that about 20% of all cancer deaths are associated with chronic infection and inflammation [239]. MPO is an important participant of the inflammatory process. As shown in the section “Free Radical Formation in Reactions Involving HOCl”, HOCl formed in the MPO-dependent reactions can directly react with nucleotides, forming free radicals that induce mutagenesis [240]. That is why the issue about the participation of MPO and RHS in the pathogenesis of cancer has been debated lately. As shown in studies conducted using ELISA or immunohistochemical methods, MPO concentration was increased in serum/plasma from patients with ovarian cancer [228], gynecologic cancer [229], and hepatocellular carcinoma in patients with chronic hepatitis C [225]. In the case of colorectal cancer, MPO concentration was increased in feces [226] and tumor [227]. Increased MPO concentration in tumor tissue was established also in different cases of gynecological cancer (ovaries, endometrium, leiomyoma) [239]. These and other results suggest the involvement of MPO/HOCl in the pathogenesis of cancer.
These examples are only a small part of the results indicating the participation of RHS, including HOCl formed in the living organism, in development of human diseases that are usually accompanied by inflammatory reactions. However, it should be borne in mind that HOCl reacts with almost all biologically important molecules not only through radical, but also through molecular mechanisms, without formation of free radical intermediates, as set out in detail in number of reviews [19, 57, 97, 169, 170, 241, 242]. The question arises: how great is the contribution of free radical reactions involving HOCl compared to the molecular reactions in damage of biomolecules, protein–lipid structures of cells, and development of oxidative/halogenative stress?
Let us try to answer this question using as an example of the reactions of HOCl with unsaturated lipids (see section “Free Radical Formation in Reactions Involving HOCl”). Judging by the number of resulting products and functional groups involved, it can be assumed that this is a molecular mechanism. The concentration of chlorohydrins formed by reaction (15) (especially in vitro experiments [112, 148, 242]) significantly exceeds the concentration of registered products of LPO [122, 127, 131, 132, 150]. Indeed, in LPO reactions with any type of initiation (HOCl [131, 133], MPO + H2O2 + Cl– [132, 134], activated neutrophils/monocytes [243], Fe2+-ascorbate [127], etc.), only a small proportion of unsaturated lipids are involved, and the level of registered products is several orders of magnitude lower than the concentration of double bonds – the main substrate of LPO [123, 150]. However, LPO reactions lead to significant changes in the lipid phase of biological membranes and other protein–lipid structures, causing the development of many cases of so-called “free radical pathology” [7, 244]. It seems that in the case of MPO/RHS-induced oxidative/halogenative stress, the free-radical intermediates forming in the reactions of RHS with biologically important molecules contributes significantly to the development of the above-mentioned socially significant diseases.
CONCLUSION
HOCl is the most important reactive form of chlorine that is formed in living organisms in the halogenating cycle of MPO. This molecule has high reactivity, and it reacts with all major biologically important molecules: proteins, lipids, nucleic acids, carbohydrates, etc. Many of these reactions proceed with formation of free radical intermediates. Perhaps the most important reaction leading to free radical formation that can be considered is the reaction of HOCl with amino groups, because they are found in all the above-mentioned classes of biologically important compounds. Chlorination of the NH2-group and subsequent free radical formation on decomposition of the chloramine leads to protein degradation, inactivation of enzymes, destruction of carbohydrate-containing biopolymers, denaturation of nucleic acids, and promotion of LPO. HOCl-dependent reactions play a dual role in vivo. On one hand, it is their job in neutrophils in inflammation foci to carry out the bactericidal function. On the other hand, these reactions can provoke damage to supramolecular structures and cells of the host organism, which often promotes oxidative/halogenative stress and development of various diseases associated with inflammation. These diseases are usually characterized by increased MPO concentration and/or activity in blood or inflammation foci and the presence of MPO-biomarkers, indicators of halogenative stress. It becomes obvious that the most important task in the fight against diseases associated with halogenative stress is the regulation of MPO activity. How to insure that MPO functioning in inflammation foci is predominantly aimed at destruction of a pathogen, “collapse” of inflammatory reaction, and not on its transformation into a chronic form? At present, we can name some of candidates for the role of natural regulators of MPO. For example, there is an acidic protein of acute phase inflammation, ceruloplasmin (pI 4.7), that forms a complex with MPO (pI > 10) inhibiting its chlorinating and peroxidase activity [245, 246]. Some substrates of the MPO peroxidase cycle, for example Trp, are able to transform MPO in Compound II (Fig. 1), which is inactive in the halogenating cycle, resulting in a decrease in MPO halogenating activity [247]. The other aromatic amino acid, Tyr, in contrast quickly transforms Compound II of MPO into the native form of the enzyme with regeneration of its chlorinating activity [248]. A similar effect was demonstrated for NO. The functional relationship of this signaling molecule with MPO is underscored by their colocalization with inducible NO-synthase in neutrophils [249]. An important modulator of MPO activity is •O2‾ [250]. It is possible to regulate MPO activity by changing the pH of the medium [251]. In this regard, it is important to elucidate the mechanisms regulating the enzyme systems responsible for RHS formation in the body (MPO and EPO), to develop sensitive methods for determination of low concentrations of MPO and EPO themselves as well as their biomarkers. Such studies will allow in future the development of new approaches and methods for prevention, diagnostics, and timely intervention in halogenative stress and the associated development of inflammatory human diseases.
This study was financially supported by the Russian Foundation for Basic Research grant No. 11-04-01262-a and 12-04-90003-Bel_a.
REFERENCES
1.Vladimirov, Iu. A. (1998) Vestnik Ros. Akad.
Med. Nauk, No. 7, 43-51.
2.Morris, J. C. (1966) J. Phys. Chem.,
70, 3798-3805.
3.Panasenko, O. M., and Sergienko, V. I. (2010)
Vestnik Ros. Akad. Med. Nauk, No. 1, 27-39.
4.Edwards, S. W. (1994) Biochemistry and
Physiology of the Neutrophil, Cambridge University Press,
Cambridge–New York–Melbourn.
5.Schultz, J., and Kaminker, K. (1962) Arch.
Biochem. Biophys., 96, 465-467.
6.Deby-Dupont, G., Deby, C., and Lamy, M. (1999)
Intensive Med., 36, 500-513.
7.Halliwell, B., and Gutteridge, J. M. C. (1999)
Free Radical in Biology and Medicine, Oxford University
Press.
8.Carlson, M. G., Peterson, C. G., and Venge, P.
(1985) J. Immunol., 134, 1875-1879.
9.Giembycz, M. A., and Lindsay, M. A. (1999)
Pharmacol. Rev., 51, 213-340.
10.Sakamaki, K., Tomonaga, M., Tsukui, K., and
Nagata, S. (1989) J. Biol. Chem., 264, 16828-16836.
11.Furtmuller, P. G., Burner, U., and Obinger, C.
(1998) Biochemistry, 37, 17923-17930.
12.Furtmuller, P. G., Jantschko, W., Zederbauer, M.,
Jakopitsch, C., Arnhold, J., and Obinger, C. (2004) Jpn. J. Infect.
Dis., 57, S30-S31.
13.Kussendrager, K. D., van Hooijdonk, A. C. (2000)
Br. J. Nutr., 84, Suppl. 1, S19-S25.
14.Ruf, J., and Carayon, P. (2006) Arch. Biochem.
Biophys., 445, 269-277.
15.Marquez, L. A., Huang, J. T., and Dunford, H. B.
(1994) Biochemistry, 33, 1447-1454.
16.Kettle, A. J., and Winterbourn, C. C. (1997)
Redox Report, 3, 3-15.
17.Kettle, A. J., Sangster, D. F., Gebicki, J. M.,
and Winterbourn, C. C. (1988) Biochim. Biophys. Acta,
956, 58-62.
18.Arnhold, J. (2004) Biochemistry (Moscow),
69, 4-9.
19.Panasenko, O. M., Arnhold, J., and Sergienko, V.
I. (2002) Biol. Membr. (Moscow), 19, 403-434.
20.Weiss, S. J., Klein, R., Slivka, A., and Wei, M.
(1982) J. Clin. Invest., 70, 598-607.
21.Foote, C. S., Goyne, T. E., and Lehrer, R. I.
(1983) Nature, 301, 715-716.
22.Furtmuller, P. G., Burner, U., Regelsberger, G.,
and Obinger, C. (2000) Biochemistry, 39, 15578-15584.
23.Mayanski, A. N., and Mayanski, D. N. (1983)
Essays about Neutrophils and Macrophages [in Russian], Nauka,
Novosibirsk.
24.Zemskov, V. M. (1984) Uspekhi Sovrem.
Biol., 98, 219-233.
25.Gorudko, I. V., Cherkalina, O. S., Sokolov,
A. V., Pulina, M. O., Zakharova, E. T., Vasil’ev, V. B.,
Cherenkevich, S. N., and Panasenko, O. M. (2009) Russ. J. Bioorg.
Chem., 35, 566-575.
26.Baldus, S., Heeschen, C., Meinertz, T., Zeiher,
A. M., Eiserich, J. P., Munzel, T., Simoons, M. L., and Hamm, C. W.
(2003) Circulation, 108, 1440-1445.
27.Weiss, S. J. (1989) N. Engl. J. Med.,
320, 365-376.
28.Pryor, W. A. (1986) Annu. Rev. Physiol.,
48, 657-667.
29.Long, C. A., and Bielski, B. H. J. (1980) J.
Phys. Chem., 84, 555-557.
30.Candeias, L. P., Stratford, M. R., and Wardman,
P. (1994) Free Radic. Res., 20, 241-249.
31.Kozlov, Yu. A., Moravskii, A. P., Purmal’,
A. P., and Shuvalov, V. F. (1981) Zh. Fiz. Khim., 55,
764-766.
32.Yakutova, E. Sh., Dremina, Ye. S., Yevgina, S.
A., Osipov, A. N., Sharov, V. S., Panasenko, O. M., and Vladimirov, Yu.
A. (1994) Biophysics, 39, 241-245.
33.Osipov, A. N., Yakutova, E. Sh., and Vladimirov,
Yu. A. (1993) Biofizika, 38, 390-396.
34.Sychev, A. Ya., and Isak, V. G. (1995) Russ.
Chem. Rev., 64, 1105-1129.
35.Prutz, W. A. (1996) Arch. Biochem.
Biophys., 332, 110-120.
36.Kagan, V. E., Orlov, O. N., and Prilipko, L. L.
(1986) Advances in Science and Technology, Ser. Biophysics [in
Russian], Vol. 18, VINITI, Moscow.
37.Panasenko, O. M., Briviba, K., Klotz, L.-O., and
Sies, H. (1997) Arch. Biochem. Biophys., 343,
254-259.
38.Johnson, D. W., and Margerum, D. W. (1991)
Inorg. Chem., 30, 4845-4851.
39.Eiserich, J. P., Cross, C. E., Jones, A. D.,
Halliwell, B., and van der Vliet, A. (1996) J. Biol. Chem.,
271, 19199-19208.
40.Byun, J., Mueller, M., Fabjan, J. S., and
Heinecke, J. W. (1999) FEBS Lett., 455, 243-246.
41.Ghibaudi, E., Barker, J. R., and Benson, S. W.
(1979) Int. J. Chem. Kinet., 11, 843-851.
42.Pattison, D. I., and Davies, M. J. (2001)
Chem. Res. Toxicol., 14, 1453-1464.
43.Hawkins, C. L., and Davies, M. J. (1998) J.
Chem. Soc. Perkin Trans., 2, 1937-1945.
44.Zgliczynski, J. M., Stelmaszynska, T., Domanski,
J., and Ostrowski, W. (1971) Biochim. Biophys. Acta, 235,
419-424.
45.Zgliczynski, J. M., Stelmaszynska, T.,
Ostrowiski, W., Naskalski, J., and Sznajd, J. (1968) Eur. J.
Biochem., 4, 540-547.
46.Clark, R. A., Szot, S., Williams, M. A., and
Kagan, H. M. (1986) Biochem. Biophys. Res. Commun., 135,
451-457.
47.Hazell, L. J., van den Berg, J. J., and Stocker,
R. (1994) Biochem. J., 302, 297-304.
48.Hazen, S. L., d’Avignon, A., Anderson, M.
A., Hsu, F. F., and Heinecke, J. W. (1998) J. Biol. Chem.,
273, 4997-5005.
49.Hazen, S. L., Hsu, F. F., d’Avignon, A.,
and Heinecke, J. W. (1998) Biochemistry, 37,
6864-6873.
50.Hawkins, C. L., and Davies, M. J. (2005) Free
Radic. Biol. Med., 39, 900-912.
51.Hawkins, C. L., and Davies, M. J. (1998)
Biochem. J., 332, 617-625.
52.Hawkins, C. L., and Davies, M. J. (2001)
Biochim. Biophys. Acta, 1504, 196-219.
53.Davies, J. M., Horwitz, D. A., and Davies, K. J.
(1993) Free Radic. Biol. Med., 15, 637-643.
54.Davies, M. J., and Hawkins, C. L. (2000) Free
Rad. Res., 33, 719-729.
55.Pattison, D. I., Hawkins, C. L., and Davies, M.
J. (2007) Biochemistry, 46, 9853-9864.
56.Thomas, E. L. (1979) Infect. Immun.,
23, 522-531.
57.Hawkins, C. L., Pattison, D. I., and Davies, M.
J. (2003) Amino Acids, 25, 259-274.
58.Baker, R. W. R. (1947) Biochem. J.,
41, 337-342.
59.Drozdz, R., Guevara, I., and Naskalski, J. W.
(1995) Clin. Chem. Acta, 236, 155-160.
60.Sharonov, B. P., Govorova, N. I., and Lyzlova, S.
N. (1989) Biochem. Int., 19, 27-35.
61.Bergt, C., Marsche, G., Panzenboeck, U.,
Heinecke, J. W., Malle, E., and Sattler, W. (2001) Eur. J.
Biochem., 268, 3523-3531.
62.Hawkins, C. L., and Davies, M. J. (1999)
Biochem. J., 340, 539-548.
63.Matheson, N. R., and Travis, J. (1985)
Biochemistry, 24, 1941-1945.
64.Aruoma, O. I., and Halliwell, B. (1987)
Biochem. J., 248, 973-976.
65.Sharonov, B. P., and Churilova, I. V. (1990)
Dokl. Akad. Nauk SSSR, 314, 1500-1502.
66.Sharonov, B. P., and Churilova, I. V. (1992)
Biochem. Biophys. Res. Commun., 189, 1129-1135.
67.Mashino, T., and Fridovich, I. (1988) Biochim.
Biophys. Acta, 953, 63-69.
68.Vissers, M. C., and Winterbourn, C. C. (1987)
Biochem. J., 245, 277-280.
69.Floris, R., and Wever, R. (1992) Eur. J.
Biochem., 207, 697-702.
70.Stark, J. A., and Henderson, A. R. (1993)
Clin. Chem., 39, 986-992.
71.Terada, L. S., Beehler, C. J., Banerjee, A.,
Brown, J. M., Grosso, M. A., Harken, A. H., McCord, J. M., and Repine,
J. E. (1988) J. Appl. Physiol., 65, 2349-2353.
72.Albrich, J. M., McCarthy, C. A., and Hurst, J. K.
(1981) Proc. Natl. Acad. Sci. USA, 78, 210-214.
73.Hawkins, C. L., and Davies, M. J. (2005) Chem.
Res. Toxicol., 18, 1669-1677.
74.Hawkins, C. L., and Davies, M. J. (2005) Chem.
Res. Toxicol., 18, 1600-1610.
75.Dai, J., Meij, J. T. A., Padua, R., and Panagia,
V. (1992) Circ. Res., 71, 970-977.
76.Kukreja, R. C., Weaver, A. B., and Hess, M. L.
(1990) Am. J. Physiol., 259, H1330-H1336.
77.Matsuoka, T., Kato, M., and Kako, K. J. (1990)
Basic. Res. Cardiol., 85, 330-341.
78.Kukreja, R. C., Weaver, A. B., and Hess, M. L.
(1989) Biochim. Biophys. Acta, 990, 198-205.
79.Van Rensburg, C. E., and van Staden, A. M. (1991)
Free Radic. Biol. Med., 11, 285-291.
80.Gorbatenkova, E. A., Naumenko, K. V., and
Sergienko, V. I. (1991) in Electrochemical Methods in Medicine
(Lopukhin, Yu. M., ed.) Research Institute of Physico-Chemical
Medicine, Moscow, pp. 5-6.
81.Wasil, M., Halliwell, B., Hutchison, D. C., and
Baum, H. (1987) Biochem. J., 243, 219-223.
82.Winterbourn, C. C. (1985) Biochim. Biophys.
Acta, 840, 204-210.
83.Bouriche, H., Salavei, P., Lessig, J., and
Arnhold, J. (2007) Arch. Biochem. Biophys., 459,
137-142.
84.Chatham, W. W., Blackburn, W. D., Jr., and Heck,
L. W. (1992) Biochem. Biophys. Res. Commun., 184,
560-567.
85.Saari, H., Sorsa, T., Lindy, O., Suomalainen, K.,
Halinen, S., and Konttinen, Y. T. (1992) Int. J. Tissue React.,
14, 113-120.
86.Peppin, G. J., and Weiss, S. J. (1986) Proc.
Natl. Acad. Sci. USA, 83, 4322-4326.
87.Michaelis, J., Vissers, M. C., and Winterbourn,
C. C. (1992) Arch. Biochem. Biophys., 292, 555-562.
88.Weiss, S. J., Peppin, G., Ortiz, X., Ragsdale,
C., and Test, S. T. (1985) Science, 227, 747-749.
89.Vissers, M. C., and Winterbourn, C. C. (1991)
Arch. Biochem. Biophys., 285, 53-59.
90.Prutz, W. A. (1998) Arch. Biochem.
Biophys., 349, 183-191.
91.Hawkins, C. L., and Davies, M. J. (2001) Chem.
Res. Toxicol., 14, 1071-1081.
92.Hawkins, C. L., and Davies, M. J. (2002) Chem.
Res. Toxicol., 15, 83-92.
93.Hawkins, C. L., Pattison, D. I., and Davies, M.
J. (2002) Biochem. J., 365, 605-615.
94.Gomez-Mejiba, S. E., Zhai, Z., Gimenez, M. S.,
Ashby, M. T., Chilakapati, J., Kitchin, K., Mason, R. P., and Ramirez,
D. C. (2010) J. Biol. Chem., 285, 20062-20071.
95.McGowan, S. E., and Thompson, R. J. (1989) J.
Appl. Physiol., 66, 400-409.
96.McGowan, S. E. (1990) Am. J. Respir. Cell Mol.
Biol., 2, 271-279.
97.Pattison, D. I., and Davies, M. J. (2006)
Curr. Med. Chem., 13, 3271-3290.
98.Rees, M. D., Pattison, D. I., and Davies, M. J.
(2005) Biochem. J., 391, 125-134.
99.Hawkins, C. L., and Davies, M. J. (1998) Free
Radic. Biol. Med., 24, 1396-1410.
100.Rees, M. D., Hawkins, C. L., and Davies, M. J.
(2003) J. Am. Chem. Soc., 125, 13719-13733.
101.Rees, M. D., Hawkins, C. L., and Davies, M. J.
(2004) Biochem. J., 381, 175-184.
102.Rees, M. D., and Davies, M. J. (2006) J. Am.
Chem. Soc., 128, 3085-3097.
103.Rychly, J., Syoltes, L., Stankovska, M.,
Janigova, I., Csomorova, K., Sasinkova, V., Kogan, G., and Gemeiner, P.
(2006) Polym. Degrad. Stab., 91, 3174-3184.
104.Syoltes, L., Kogan, G., Stankovska, M.,
Mendichi, R., Rychly, J., Schiller, J., and Gemeiner, P. (2007)
Biomacromolecules, 8, 2697-2705.
105.Woods, A. A., and Davies, M. J. (2003)
Biochem. J., 376, 219-227.
106.Heinecke, J. W. (1999) J. Lab. Clin.
Med., 133, 321-325.
107.Baskol, G., Demir, H., Baskol, M., Kilic, E.,
Ates, F., Karakukcu, C., and Ustdal, M. (2006) Cell Biochem.
Funct., 24, 307-311.
108.Wu, W., Samoszuk, M. K., Comhair, S. A. A.,
Thomassen, M. J., Farver, C. F., Dweik, R. A., Kavuru, M. S., Erzurum,
S. C., and Hazen, S. L. (2000) J. Clin. Invest., 105,
1455-1463.
109.Andreadis, A. A., Hazen, S. L., Comhair, S. A.
A., and Erzurum, S. C. (2003) Free Radic. Biol. Med., 35,
213-225.
110.Panasenko, O. M., Arnhold, J., Sergienko, V.
I., Arnold, K., and Vladimirov, Yu. A. (1996) Membr. Cell Biol.,
10, 291-302.
111.Carr, A. C., Winterbourn, C. C., and van den
Berg, J. J. (1996) Arch. Biochem. Biophys., 327,
227-233.
112.Winterbourn, C. C., van den Berg, J. J.,
Roitman, E., and Kuypers, F. A. (1992) Arch. Biochem. Biophys.,
296, 547-555.
113.Arnhold, J., Panasenko, O. M., Schiller, J.,
Vladimirov, Yu. A., and Arnold, K. (1995) Chem. Phys. Lipids,
78, 55-64.
114.Panasenko, O. M., Arnhold, J., Vladimirov, Yu.
A., Arnold, K., and Sergienko, V. I. (1995) Biophysics,
40, 1249-1257.
115.Panasenko, O. M., Arnhold, J., Vladimirov, Yu.
A., Arnold, K., and Sergienko, V. I. (1997) Free Rad. Res.,
27, 1-12.
116.Arnhold, J., Osipov, A. N., Spalteholz, H.,
Panasenko, O. M., and Schiller, J. (2001) Free Radic. Biol.
Med., 31, 1111-1119.
117.Panasenko, O. M., Osipov, A. N., Schiller, J.,
and Arnhold, J. (2002) Biochemistry (Moscow), 67,
889-900.
118.Pattison, D. I., Hawkins, C. L., and Davies, M.
J. (2003) Chem. Res. Toxicol., 16, 439-449.
119.Prutz, W. A. (1998) Arch. Biochem.
Biophys., 357, 265-273.
120.Spalteholz, H., Wenske, K., and Arnhold, J.
(2005) BioFactors, 24, 67-76.
121.Chekanov, A. V., Osipov, A. N., Vladimirov, Yu.
A., Sergienko, V. I., and Panasenko, O. M. (2007) Biophysics,
52, 1-7.
122.Panasenko, O. M., Arnhold, J., Schiller, J.,
Arnold, K., and Sergienko, V. I. (1994) Biochim. Biophys. Acta,
1215, 259-266.
123.Panasenko, O. M., and Arnhold, J. (1996)
Membr. Cell Biol., 10, 93-104.
124.Skaff, O., Pattison, D. I., and Davies, M. J.
(2007) Chem. Res. Toxicol., 20, 1980-1988.
125.Panasenko, O. M., Spalteholz, H., Schiller, J.,
and Arnhold, J. (2006) Biochemistry (Moscow), 71,
571-580.
126.Stelmaszynska, T., Kukovetz, E., Egger, G., and
Schaur, R. J. (1992) Int. J. Biochem., 24, 121-128.
127.Panasenko, O. M., Arnhold, J., Arnold, K.,
Vladimirov, Yu. A., and Sergienko, V. I. (1996) Biophysics,
41, 321-328.
128.Panasenko, O. M. (1997) BioFactors,
6, 181-190.
129.Noguchi, N., Nakada, A., Itoh, Y., Watanabe,
A., and Niki, E. (2002) Arch. Biochem. Biophys., 397,
440-447.
130.Kawai, Y., Kiyokawa, H., Kimura, Y., Kato, Y.,
Tsuchiya, K., and Terao, J. (2006) Biochemistry, 45,
14201-14211.
131.Evgina, S. A., Panasenko, O. M., Sergienko, V.
I., and Vladimirov, Yu. A. (1992) Biol. Membr., 6,
1247-1254.
132.Panasenko, O. M., Evgina, S. A., Aidyraliev, R.
K., Sergienko, V. I., and Vladimirov, Yu. A. (1994) Free Radic.
Biol. Med., 16, 143-148.
133.Panasenko, O. M., Evgina, S. A., Driomina, E.
S., Sharov, V. S., Sergienko, V. I., and Vladimirov, Yu. A. (1995)
Free Radic. Biol. Med., 19, 133-140.
134.Panasenko, O. M., and Arnhold, J. (1999)
Free Rad. Res., 30, 479-487.
135.Panasenko, O. M., Evgina, S. A., and Sergienko,
V. I. (1993) Bull. Exper. Biol. Med., 115, 373-375.
136.Panasenko, O. M., Panasenko, O. O., Briviba,
K., and Sies, H. (1997) Biochemistry (Moscow), 62,
1140-1145.
137.Arnhold, J., Muller, S., Arnold, K., and Grimm,
E. (1991) J. Biolumin. Chemilumin., 6, 189-192.
138.Held, A. M., Halko, D. J., and Hurst, J. K.
(1978) J. Am. Chem. Soc., 100, 5732-5740.
139.Folkes, L. K., Candeias, L. P., and Wardman, P.
(1995) Arch. Biochem. Biophys., 323, 120-126.
140.Candeias, L. P., Patel, K. B., Stratford, M.
R., and Wardman, P. (1993) FEBS Lett., 333, 151-153.
141.Osipov, A. N., Panasenko, O. M., Chekanov, A.
V., and Arnhold, J. (2002) Free Rad. Res., 36,
749-754.
142.Panasenko, O. M., Osipov, A. N., Chekanov, A.
V., Arnhold, J., and Sergienko, V. I. (2002) Biochemistry
(Moscow), 67, 880-888.
143.Chekanov, A. V., Panasenko, O. M., Osipov, A.
N., Arnhold, J., Kazarinov, K. D., Vladimirov, Yu. A., and Sergienko,
V. I. (2002) Biophysics, 47, 731-737.
144.Chekanov, A. V., Panasenko, O. M., Osipov, A.
N., Matveeva, N. S., Kazarinov, K. D., Vladimirov, Yu. A., and
Sergienko, V. I. (2005) Biophysics, 50, 8-14.
145.Panasenko, O. M., Chekanov, A. V., Arnhold, J.,
Sergienko, V. I., Osipov, A. N., and Vladimirov, Yu. A. (2005)
Biochemistry (Moscow), 70, 998-1004.
146.Chekanov, A. V., Osipov, A. N., Vladimirov, Yu.
A., Sergienko, V. I., and Panasenko, O. M. (2006) Biol. Membr.,
23, 426-432.
147.Miyamoto, S., Martinez, G. R., Rettori, D.,
Augusto, O., Medeiros, M. H. G., and Di Mascio, P. (2006) Proc.
Natl. Acad. Sci. USA, 103, 293-298.
148.Arnhold, J., Panasenko, O. M., Schiller, J.,
Arnold, K., Vladimirov, Yu. A., and Sergienko, V. I. (1996) Z.
Naturforsch., 51c, 386-394.
149.Panasenko, O. M., Arnhold, J., and Schiller, J.
(1997) Biochemistry (Moscow), 62, 951-959.
150.Rice-Evans, C., Leake, D., Bruckdorfer, K. R.,
and Diplock, A. T. (1996) Free Rad. Res., 25,
285-311.
151.Hazell, L. J., Davies, M. J., and Stocker, R.
(1999) Biochem. J., 339, 489-495.
152.Girotti, A. W. (1998) J. Lipid Res.,
39, 1529-1542.
153.Freeman, B. A., Sharman, M. C., and Mudd, L.
(1979) Arch. Biochem. Biophys., 197, 264-272.
154.Nelson, M. J., and Seitz, S. P. (1994) Curr.
Opin. Struct. Biol., 4, 878-884.
155.Kuhn, H., Chaitidis, P., Roffeis, J., and
Walther, M. (2007) J. Cardiovasc. Pharmacol., 50,
609-620.
156.Sergienko, V. I., Panasenko, O. M., and Murina,
M. A. (1999) Efferent. Terap., 5, 8-17.
157.Heinecke, J. W., Li, W., Mueller, D. M.,
Bohrer, A., and Turk, J. (1994) Biochemistry, 33,
10127-10136.
158.Carr, A. C., Winterbourn, C. C., Blunt, J. W.,
Philipps, A. J., and Abell, A. D. (1997) Lipids, 32,
363-367.
159.Hazen, S. L., Hsu, F. F., Duffin, K., and
Heinecke, J. W. (1996) J. Biol. Chem., 271,
23080-23088.
160.Carr, A. C., van den Berg, J. J., and
Winterbourn, C. C. (1996) Arch. Biochem. Biophys., 332,
63-69.
161.Van den Berg, J. J., Winterbourn, C. C., and
Kuypers, F. A. (1993) J. Lipid Res., 34, 2005-2012.
162.Iuliano, L. (2011) Chem. Phys. Lipids,
164, 457-468.
163.Panasenko, O. M., Vakhrusheva, T., Tretyakov,
V., Spalteholz, H., and Arnhold, J. (2007) Chem. Phys. Lipids,
149, 40-51.
164.Arnhold, J., Osipov, A. N., Spalteholz, H.,
Panasenko, O. M., and Schiller, J. (2002) Biochim. Biophys.
Acta, 1572, 91-100.
165.Carr, A. C., van den Berg, J. J., and
Winterbourn, C. C. (1998) Biochim. Biophys. Acta, 1392,
254-264.
166.Richter, G., Schober, C., Suβ, R., Fuchs,
B., Birkemeyer, C., and Schiller, J. (2008) Anal. Biochem.,
376, 157-159.
167.Kulinsky, V. I. (1999) Soros. Obrazovat.
Zh., No. 1, 2-7.
169.Vladimirov, Yu. A. (2002) Biol. Membr.,
19, 356-377.
169.Malle, E., Marsche, G., Arnhold, J., and
Davies, M. J. (2006) Biochim. Biophys. Acta, 1761,
392-415.
170.Klebanoff, S. J. (2005) J. Leukoc.
Biol., 77, 598-625.
171.Winterbourn, C. C., Vissers, M. C. M., and
Kettle, A. J. (2000) Curr. Opin. Hematol., 7, 53-58.
172.Hoy, A., Leininger-Muller, B., Kutter, D.,
Siest, G., and Visvikis, S. (2002) Clin. Chem. Lab. Med.,
40, 2-8.
173.Selye, H. (1957) The Stress of Life,
McGraw-Hill Book Company Inc., New York.
174.Kolomiets, A. F. (1991) Russ. Chem.
Rev., 60, 256-260.
175.Fedorov, L. A. (1993) Dioxins as an
Environmental Hazard: Retrospect and Prospect [in Russian], Nauka,
Moscow.
176.Sugiyama, S., Okada, Y., Sukhova, G. K.,
Virmani, R., Heinecke, J. W., and Libby, P. (2001) Am. J.
Pathol., 158, 879-891.
177.Daugherty, A., Dunn, J. L., Rateri, D. L., and
Heinecke, J. W. (1994) J. Clin. Invest., 94, 437-444.
178.Hazen, S. L., and Heinecke, J. W. (1997) J.
Clin. Invest., 99, 2075-2081.
179.Hazen, S. L., Crowley, J. R., Mueller, D. M.,
and Heinecke, J. W. (1997) Free Radic. Biol. Med., 23,
909-916.
180.Pennathur, S., Bergt, C., Shao, B., Byun, J.,
Kassim, S. Y., Singh, P., Green, P. S., McDonald, T. O., Brunzell, J.,
Chait, A., Oram, J. F., O’Brien, K., Geary, R. L., and Heinecke,
J. W. (2004) J. Biol. Chem., 279, 42977-42983.
181.Shao, B., Pennathur, S., and Heinecke, J. W.
(2012) J. Biol. Chem., 287, 6375-6386.
182.Hazell, L. J., Arnold, L., Flowers, D., Waeg,
G., Malle, E., and Stocker, R. (1996) J. Clin. Invest.,
97, 1535-1544.
183.Hazell, L. J., Baernthaler, G., and Stocker, R.
(2001) Free Radic. Biol. Med., 31, 1254-1262.
184.Thukkani, A. K., McHowat, J., Hsu, F. F.,
Brennan, M. L., Hazen, S. L., and Ford, D. A. (2003)
Circulation, 108, 3128-3133.
185.Messner, M. C., Albert, C. J., McHowat, J., and
Ford, D. A. (2008) Lipids, 43, 243-249.
186.Takeshita, J., Byun, J., Nhan, T. Q.,
Pritchard, D. K., Pennathur, S., Schwartz, S. M., Chait, A., and
Heinecke, J. W. (2006) J. Biol. Chem., 281,
3096-3104.
187.Zhang, R., Brennan, M. L., Fu, X., Aviles, R.
J., Pearce, G. L., Penn, M. S., Topol, E. J., Sprecher, D. L., and
Hazen, S. L. (2001) JAMA, 286, 2136-2142.
188.Roman, R. M., Camargo, P. V., Borges, F. K.,
Rossini, A. P., and Polanczyk, C. A. (2010) Coron. Artery Dis.,
21, 129-136.
189.Wainstein, R. V., Wainstein, M. V., Ribeiro, J.
P., Dornelles, L. V., Tozzati, P., Ashton-Prolla, P., Ewald, I. P.,
Vietta, G., and Polanczyk, C. A. (2010) Clin. Biochem.,
43, 57-62.
190.Cojocaru, I. M., Cojocaru, M., Iliescu, I.,
Botnaru, L., Gurban, C. V., Sfrijan, F., and Tanasescu, R. (2010)
Rom. J. Int. Med., 48, 101-104.
191.Tang, W. H., Wu, Y., Nicholls, S. J., and
Hazen, S. L. (2011) Clin. Chem., 57, 33-39.
192.Karakas, M., Koenig, W., Zierer, A., Herder,
C., Rottbauer, W., Baumert, J., Meisinger, C., and Thorand, B. (2012)
J. Int. Med., 271, 43-50.
193.Zheng, L., Nukuna, B., Brennan, M. L., Sun, M.,
Goormastic, M., Settle, M., Schmitt, D., Fu, X., Thomson, L., Fox, P.
L., Ischiropoulos, H., Smith, J. D., Kinter, M., and Hazen, S. L.
(2004) J. Clin. Invest., 114, 529-541.
194.Cheng, M. L., Chen, C. M., Gu, P. W., Ho, H.
Y., and Chiu, D. T. (2008) Clin. Biochem., 41,
554-560.
195.Rudolph, V., Andrie, R. P., Rudolph, T. K.,
Friedrichs, K., Klinke, A., Hirsch-Hoffmann, B., Schwoerer, A. P., Lau,
D., Fu, X., Klingel, K., Sydow, K., Didie, M., Seniuk, A., von Leitner,
E. C., Szoecs, K., Schrickel, J. W., Treede, H., Wenzel, U., Lewalter,
T., Nickenig, G., Zimmermann, W. H., Meinertz, T., Boger, R. H.,
Reichenspurner, H., Freeman, B. A., Eschenhagen, T., Ehmke, H., Hazen,
S. L., Willems, S., and Baldus, S. (2010) Nat. Med., 16,
470-474.
196.Higashi, N., Mita, H., Taniguchi, M.,
Turikisawa, N., Higashi, A., Ozawa, Y., Tohma, S., Arimura, K., and
Akiyama, K. (2004) J. Allergy Clin. Immunol., 114,
1353-1358.
197.Wu, S. M., and Pizzo, S. V. (2001) Arch.
Biochem. Biophys., 391, 119-126.
198.Stamp, L. K., Khalilova, I., Tarr, J. M.,
Senthilmohan, R., Turner, R., Haigh, R. C., Winyard, P. G., and Kettle,
A. J. (2012) Rheumatology (Oxford), 51, 1796-1803.
199.Schiller, J., Arnhold, J., Sonntag, K., and
Arnold, K. (1996) Magn. Reson. Med., 35, 848-853.
200.Steinbeck, M. J., Nesti, L. J., Sharkey, P. F.,
and Parvizi, J. (2007) J. Orthop. Res., 25,
1128-1135.
201.Mita, H., Higashi, N., Taniguchi, M., Higashi,
A., Kawagishi, Y., and Akiyama, K. (2004) Clin. Exp. Allergy,
34, 931-938.
202.MacPherson, J. C., Comhair, S. A., Erzurum, S.
C., Klein, D. F., Lipscomb, M. F., Kavuru, M. S., Samoszuk, M. K., and
Hazen, S. L. (2001) J. Immunol., 166, 5763-5772.
203.O’Donnell, C., Newbold, P., White, P.,
Thong, B., Stone, H., and Stockley, R. A. (2010) COPD, 7,
411-417.
204.Buss, I. H., Senthilmohan, R., Darlow, B. A.,
Mogridge, N., Kettle, A. J., and Winterbourn, C. C. (2003) Pediatr.
Res., 53, 455-462.
205.Harwood, D. T., Darlow, B. A., Cheah, F. C.,
McNeill, N., Graham, P., and Winterbourn, C. C. (2011) Pediatr.
Res., 69, 28-33.
206.Lamb, N. J., Gutteridge, J. M., Baker, C.,
Evans, T. W., and Quinlan, G. J. (1999) Crit. Care Med.,
27, 1738-1744.
207.Lamb, N. J., Quinlan, G. J., Westerman, S. T.,
Gutteridge, J. M., and Evans, T. W. (1999) Am. J. Respir. Crit. Care
Med., 160, 1031-1034.
208.Van Der Vliet, A., Nguyen, M. N., Shigenaga, M.
K., Eiserich, J. P., Marelich, G. P., and Cross, C. E. (2000) Am. J.
Physiol. Lung Cell Mol. Physiol., 279, L537-L546.
209.Saude, E. J., Lacy, P., Musat-Marcu, S., Mayes,
D. C., Bagu, J., Man, S. F., Sykes, B. D., and Moqbel, R. (2004)
Magn. Reson. Med., 52, 807-814.
210.Thomson, E., Brennan, S., Senthilmohan, R.,
Gangell, C. L., Chapman, A. L., Sly, P. D., Kettle, A. J., Balding, E.,
Berry, L. J., Carlin, J. B., Carzino, R., de Klerk, N., Douglas, T.,
Foo, C., Garratt, L. W., Hall, G. L., Harrison, J., Kicic, A., Laing,
I. A., Logie, K. M., Massie, J., Mott, L. S., Murray, C., Parsons, F.,
Pillarisetti, N., Poreddy, S. R., Ranganathan, S. C., Robertson, C. F.,
Robins-Browne, R., Robinson, P. J., Skoric, B., Stick, S. M., Sutanto,
E. N., and Williamson, E. (2010) Free Radic. Biol. Med.,
49, 1354-1360.
211.Harwood, D. T., Kettle, A. J., Brennan, S., and
Winterbourn, C. C. (2009) J. Chromatogr. B Analyt. Technol. Biomed.
Life Sci., 877, 3393-3399.
212.Himmelfarb, J., McMenamin, M. E., Loseto, G.,
and Heinecke, J. W. (2001) Free Radic. Biol. Med., 31,
1163-1169.
213.Malle, E., Woenckhaus, C., Waeg, G.,
Esterbauer, H., Grone, E. F., and Grone, H.-J. (1997) Am. J.
Pathol., 150, 603-615.
214.Paquet, P., De Groote, D., and Pierard, G. E.
(2010) Dermatology, 220, 201-207.
215.Maki, R. A., Tyurin, V. A., Lyon, R. C.,
Hamilton, R. L., DeKosky, S. T., Kagan, V. E., and Reynolds, W. F.
(2009) J. Biol. Chem., 284, 3158-3169.
216.Green, P. S., Mendez, A. J., Jacob, J. S.,
Crowley, J. R., Growdon, W., Hyman, B. T., and Heinecke, J. W. (2004)
J. Neurochem., 90, 724-733.
217.Nagra, R. M., Becher, B., Tourtellotte, W. W.,
Antel, J. P., Gold, D., Paladino, T., Smith, R. A., Nelson, J. R., and
Reynolds, W. F. (1997) J. Neuroimmunol., 78, 97-107.
218.Sanchez-Lopez, F., Tasset, I., Aguera, E.,
Feijoo, M., Fernandez-Bolanos, R., Sanchez, F. M., Ruiz, M. C., Cruz,
A. H., Gascon, F., and Tunez, I. (2012) Neurol. Res., 34,
721-724.
219.Choi, D. K., Pennathur, S., Perier, C., Tieu,
K., Teismann, P., Wu, D. C., Jackson-Lewis, V., Vila, M., Vonsattel, J.
P., Heinecke, J. W., and Przedborski, S. (2005) J. Neurosci.,
25, 6594-6600.
220.Henderson, J. P., Byun, J., Takeshita, J., and
Heinecke, J. W. (2003) J. Biol. Chem., 278,
23522-23528.
221.Borato, D. C., Parabocz, G. C., Ribas, S. R.,
Kalva-Filho, C. A., Borba, L. M., Ito, C. A., Bail, L., Dos Santos, F.
A., and Vellosa, J. C. (2012) Metabolism, 61,
1353-1360.
222.Kothari, N., Keshari, R. S., Bogra, J., Kohli,
M., Abbas, H., Malik, A., Dikshit, M., and Barthwal, M. K. (2011) J.
Crit. Care, 26, 435.e1-435.e7.
223.Masoodi, I., Kochhar, R., Dutta, U., Vaishnavi,
C., Prasad, K. K., Vaiphei, K., Kaur, S., and Singh, K. (2009) J.
Gastroenterol. Hepatol., 24, 1768-1774.
224.Masoodi, I., Kochhar, R., Dutta, U., Vaishnavi,
C., Prasad, K. K., Vaiphei, K., Hussain, S., and Singh, K. (2012)
Dig. Dis. Sci., 57, 1336-1340.
225.Do Carmo, R. F., de Almeida, D. B., Aroucha, D.
C., Vasconcelos, L. R., de Moraes, A. C., de Mendonça Cavalcanti
Mdo, S., de Morais, C. N., Pereira, L. M., and Moura, P. (2012) Hum.
Immunol., 73, 1127-1131.
226.Chalkias, A., Nikotian, G., Koutsovasilis, A.,
Bramis, J., Manouras, A., Mystrioti, D., and Katergiannakis, V. (2011)
Am. J. Clin. Oncol., 34, 561-566.
227.Roncucci, L., Mora, E., Mariani, F., Bursi, S.,
Pezzi, A., Rossi, G., Pedroni, M., Luppi, D., Santoro, L., Monni, S.,
Manenti, A., Bertani, A., Merighi, A., Benatti, P., Di Gregorio, C.,
and de Leon, P. M. (2008) Cancer Epidemiol. Biomarkers Prev.,
17, 2291-2297.
228.Fletcher, N. M., Jiang, Z., Ali-Fehmi, R.,
Levin, N. K., Belotte, J., Tainsky, M. A., Diamond, M. P., Abu-Soud, H.
M., and Saed, G. M. (2011-2012) Cancer Biomark., 10,
267-275.
229.Song, M., and Santanam, N. (2001) Antioxid.
Redox Signal., 3, 1139-1146.
230.Chen, L. Q., Rohatgi, A., Ayers, C. R., Das, S.
R., Khera, A., Berry, J. D., McGuire, D. K., and de Lemos, J. A. (2011)
Atherosclerosis, 219, 833-838.
231.Sokolov, A. V., Ageeva, K. V., Cherkalina, O.
S., Pulina, M. O., Zakharova, E. T., Prozorovskii, V. N., Aksenov, D.
V., Vasilyev, V. B., and Panasenko, O. M. (2010) Chem. Phys.
Lipids, 163, 347-355.
232.Sokolov, A. V., Chekanov, A. V., Kostevich, V.
A., Aksenov, D. V., Vasilyev, V. B., and Panasenko, O. M. (2011)
Chem. Phys. Lipids, 164, 49-53.
233.Panasenko, O. M., Melnichenko, A. A., Aksenov,
D. V., Vakhrusheva, T. V., Suprun, I. V., Yanushevskaya, E. V., Vlasik,
T. N., Sobenin, I. A., and Orekhov, A. N. (2004) Biol. Membr.,
21, 498-505.
234.Pattison, D. I., and Davies, M. J. (2004)
Biochemistry, 43, 4799-4809.
235.Yap, Y. W., Whiteman, M., and Cheung, N. S.
(2007) Cell Signal., 19, 219-228.
236.Kaur, H., and Halliwell, B. (1990) Chem.
Biol. Interact., 73, 235-247.
237.Becker, B. F. (1993) Free Radic. Biol.
Med., 14, 615-631.
238.Panasenko, O. M., Arnhold, J., Arnold, K.,
Sergienko, V. I., and Vladimirov, Yu. A. (1995) Membr. Cell
Biol., 9, 299-308.
239.Porta, C., Larghi, P., Rimoldi, M., Totaro, M.
G., Allavena, P., Mantovani, A., and Sica, A. (2009)
Immunobiology, 214, 761-777.
240.Panasenko, O. M., Gorudko, I. V., Kovaleva, A.
M., Gusev, S. A., Sergienko, V. I., and Matishev, D. G. (2010)
Vestnik Yuzhnogo Nauchnogo Tsentra RAN, 6, No. 3,
73-90.
241.Davies, M. J., Hawkins, C. L., Pattison, D. I.,
and Rees, M. D. (2008) Antioxid. Redox Signal., 10,
1199-1234.
242.Spickett, C. M., Jerlich, A., Panasenko, O. M.,
Arnhold, J., Pitt, A. R., Stelmaszynska, T., and Schaur, R. J. (2000)
Acta Biochim. Pol., 47, 889-899.
243.Panasenko, O. M., Vol’nova, T. V.,
Osipov, A. N., Azizova, O. A., and Vladimirov, Yu. A. (1991) Biomed.
Sci., 2, 581-589.
244.Vladimirov, Yu. A., and Archakov, A. I. (1972)
Lipid Peroxidation in Biological Membranes [in Russian], Nauka,
Moscow.
245.Sokolov, A. V., Ageeva, K. V., Pulina, M. O.,
Cherkalina, O. S., Samygina, V. R., Vlasova, I. I., Panasenko, O. M.,
Zakharova, E. T., and Vasilyev, V. B. (2008) Free Rad. Res.,
42, 989-998.
246.Panasenko, O. M., Chekanov, A. V., Vlasova, I.
I., Sokolov, A. V., Ageeva, K. V., Pulina, M. O., Cherkalina, O. S.,
and Vasil’ev, V. B. (2008) Biophysics, 53,
268-272.
247.Galijasevic, S., Abdulhamid, I., and Abu-Soud,
H. M. (2008) Free Radic. Biol. Med., 44, 1570-1577.
248.Vlasova, I. I., Sokolov, A. V., and Arnhold, J.
(2012) J. Inorg. Biochem., 106, 76-83.
249.Galijasevic, S., Saed, G. M., Hazen, S. L., and
Abu-Soud, H. M. (2006) Biochemistry, 45, 1255-1262.
250.Kettle, A. J., Anderson, R. F., Hampton, M. B.,
and Winterbourn, C. C. (2007) Biochemistry, 46,
4888-4897.
251.Vlasova, I. I., Arnhold, J., Osipov, A. N., and
Panasenko, O. M. (2006) Biochemistry (Moscow), 71,
667-677.