REVIEW: Structural Studies on Photosystem II of Cyanobacteria
A. G. Gabdulkhakov* and M. V. Dontsova
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: azat@vega.protres.ru* To whom correspondence should be addressed.
Received May 21, 2013
Photosynthesis is one of the most important chemical processes in the biosphere responsible for the maintenance of life on Earth. Light energy is converted into energy of chemical bonds in photoreaction centers, which, in particular, include photosystem II (PS II). PS II is a multisubunit pigment–protein complex located in the thylakoid membrane of cyanobacteria, algae and plants. PS II realizes the first stage of solar energy conversion that results in decomposition of water to molecular oxygen, protons, and bound electrons via a series of consecutive reactions. During recent years, considerable progress has been achieved in determination of the spatial structures of PS II from various cyanobacteria. In the present review, we outline the current state of crystallographic studies on PS II.
KEY WORDS: photosystem II, water oxidation, manganese cluster, lipids, plastoquinone, channelsDOI: 10.1134/S0006297913130105
Abbreviations: DGDG, digalactosyl-diacylglycerol; MALDI, matrix-activated laser desorption/ionization; MGDG monogalactosyl-diacylglycerol; PG, phosphatidylglycerol; PQ, plastoquinone; PS II, photosystem II; SQDG, sulfoquinovosyl-diacylglycerol.
The Sun is one of the greatest energy sources on our planet. The
conservation of this energy with minimal losses is a vitally important
task in studies concerning energy resources. Biological systems capable
of converting solar energy with maximal efficiency emerged about
two-three billion years ago, and their activities resulted in
generation of oil, carbon and gas stores. The succession of reactions
resulting in light energy conservation as energy-rich compounds is
termed photosynthesis. But the mechanism of this process remains poorly
studied. There is an immense variety of photosynthesizing enzymes, from
the simplest photoreaction centers located in membranes of
photosynthesizing bacteria to multisubunit complexes, so-called
photosystems, which are located in thylakoids of plants, algae and
cyanobacteria.
In the first stage of photosynthesis, light energy is absorbed. The energy is absorbed by light-collecting pigments and passed to trapping pigments in the reaction center. In the reaction center, charges are primarily separated resulting in the light conversion into energy of chemical bonds. During light absorption, the pigments loose electrons, and for re-reduction they need an electron donor. In oxygenic photosynthesis, the water molecule is the major donor of electrons. The enzyme complex responsible for decomposition of water into protons, electrons, and molecular oxygen is termed photosystem II (PS II).
This review considers structural studies on PS II of cyanobacteria. It is the reaction catalyzed by this enzyme complex that resulted in appearance of oxygen stores in Earth’s biosphere.
LOCALIZATION AND COMPOSITION OF PS II
PS II is an ensemble of proteins, lipids, and pigments. It is localized in the depth of the thylakoid membrane. In plants and algae this membrane is a part of specialized organelles – chloroplasts. In cyanobacteria the whole system is localized directly in protrusions of the cytoplasmic membrane.
During some decades, different groups of researchers tried to determine the spatial structure of components of the PS II complex. Finally, in 2001, Zouni and colleagues for the first time succeeded in obtaining by X-ray crystallographic analysis the spatial structure of PS II from the cyanobacterium Synechococcus elongatus with resolution of 3.8 Å [1]. The enzyme was in an active form, i.e. crystals of PS II decomposed water under the influence of light [2]. Then, for some years, the quality of PS II structure and the limit of its resolution were gradually increased [3-6]. As a result, in 2009 the PS II three-dimensional model was determined with resolution of 2.9 Å [7]. In the spatial organization of PS II from Thermosynechococcus elongatus all 20 protein subunits, 35 chlorophyll molecules, 12 carotenoid molecules and 25 molecules of incorporated lipids could be distinguished. For today, this is the most complete model of PS II including its multisubunit composition. Unfortunately, the catalytic center of the photosynthetic oxidation of water, which is a unique complex of metals, was poorly distinguishable at this resolution. Therefore, experiments for improving the model of PS II were continued, and now the structure of PS II from the thermophilic cyanobacterium Thermosynechococcus vulcanus is determined with resolution of 1.9 Å [8]. This resolution allowed researchers to determine positions of individual atoms of the cluster responsible for oxidation of water to oxygen, protons and electrons.
All models of PS II were obtained from crystals of homodimeric PS II isolated from thermophilic cyanobacteria, whereas attempts to obtain the structure of this complex from other organisms how not been successful up so far. There are reports about PS II crystals from red algae, but the structure is not yet published [9].
A monomer of PS II includes 20 different protein subunits [7]. Seventeen of the proteins are located inside the membrane, and three are located from the side of the lumen (Fig. 1). Pigments of the monomer are represented by 35 different molecules of chlorophyll a (Chla), two molecules of pheophytin a (Pheoa), two heme molecules (components of cytochromes b559 and c550), 12 molecules of β-carotene, and three molecules of plastoquinone PQ (QA, QB, and QC). The PS II monomer also includes 25 molecules of incorporated lipids, as well as calcium, magnesium, chlorine, iron and bicarbonate ions.
Fig. 1. General appearance of spatial structure of PS II dimer from Thermosynechococcus elongatus with 1.9 Å resolution. The arrow indicates a non-crystallographic C2 axis. To the left the protein part of the PS II monomer is presented (proteins D1, CP47, CP43, D2, PsbE, PsbF, PsbO, PsbU, PsbV, etc.). To the right the non-protein part of PS II is presented (chlorophyll a, pheophytin, heme, carotenoids, plastoquinones, lipids and water).
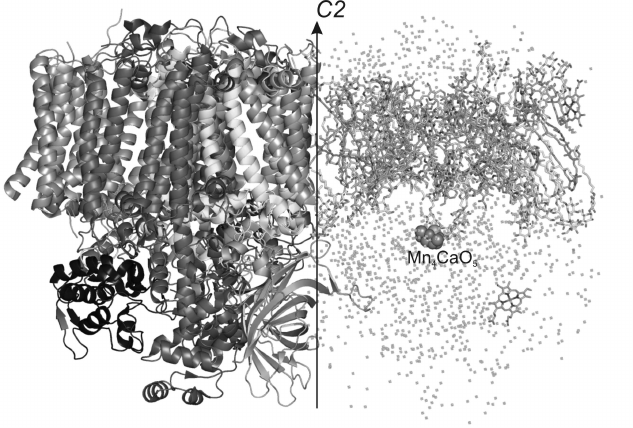
The carcass of the PS II monomer consists of two large subunits PsbA and PsbD traditionally known as proteins D1 and D2, respectively. These subunits are homologous [10] and have similar spatial structures. They are α/β proteins mainly consisting of α-helices. The central five transmembrane α-helices in every subunit are organized in two semicircles and are connected to one another with a “handshake” motif connected by a second-order local axis. Just the D1 and D2 subunits form the main membrane part of PS II and create a photosynthetic reaction center. These proteins interact with all the pigments of the electron transfer chain, which is represented by two symmetric branches of electron carriers bound by second-order pseudosymmetry. One branch of the electron transfer chain consists of a special pair of chlorophylls PD1 consisting of two Chla molecules and also of ChlD1, PheoD1, and QA, whereas the other branch consists of their symmetric analogs PD2, ChlD2, PheoD2, and QB. A nonheme iron atom is situated between molecules of plastoquinones QA and QB. Each of these proteins, as well as D1 and D2, also interacts with one carotenoid molecule. The whole complex of subunits and pigments is surrounded by two other large proteins, PsbB and PsbC, which form the nucleus of the light-absorbing system and are also termed antennal complexes CP47 and CP43, respectively. They are bound with the majority of Chla molecules, except Chla molecules belonging to the reaction center. In particular, the proteins CP47 and CP43 interact, respectively, with 16 and 13 molecules of Chla. The CP47 subunit is located close to the surface of interaction between the PS II monomers and protein D2, whereas CP43 is located on the complex periphery near the D1 subunit. In addition to their functioning as antennas, CP47 and CP43 play an important role in stabilization of the whole PS II ensemble and also of the oxygen-evolving center. The antennal proteins are homologous [10] and have similar spatial structures. They consist of α-helices and β-strands. Each of these proteins has six transmembrane α-helices. From the lumenal side, these proteins are different in the number and size of the β-strands. Both subunits have a large number of non-structured loops from the lumenal side. These loops outgoing beyond the membrane into the lumen are involved in interactions with external proteins. From the cytoplasm side, proteins CP47 and CP43 also have non-structured regions.
The base of the PS II monomers is composed of four subunits: D1, D2, CP47 and CP43. The other 16 protein subunits surround this complex and stabilize its integrity. Eleven of these subunits are low molecular mass proteins including the only transmembrane α-helix. These are the following subunits: PsbF, PsbH, PsbI, PsbJ, PsbK, PsbL, PsbM, PsbT, PsbX, PsbY and Ycf12. The PsbZ subunit consists of two transmembrane α-helices. The PsbE subunit consists of three α-helices, one of which penetrates across the membrane and two others are situated from the lumenal side of the photosynthesizing complex. The PsbE protein together with the PsbF subunit binds the heme group and present α and β subunits of cyt b559. All these proteins are located within the membrane.
Other subunits are situated on the lumenal side of the complex and are designated as external ones. These proteins include the protein PsbU consisting of six α-helices and two short β-strands. The protein PsbV, bound with the heme group and consisting of five α-helices and four β-strands, forms cyt c550. On the lumenal side of PS II the PsbO protein is also located, the only subunit of the photocomplex, which consists of eight β-strands forming a long β-barrel.
In addition to proteins and pigments, every multisubunit monomer of PS II contains lipid molecules, among which there are 11 molecules of monogalactosyl-diacylglycerol (MGDG), seven molecules of digalactosyl-diacylglycerol (DGDG), five molecules of sulfoquinovosyl-diacylglycerol (SQDG) and two molecules of phosphatidylglycerol (PG). This lipid composition is in good correlation with the statistical mean lipid composition of thylakoid membrane, which usually includes ~45% MGDG, ~25% SQDG, 15-25% DGSQ, and 5-15% PG [11]. The lipid molecules are distributed asymmetrically in the structure of PS II. Negatively charged heads of PG and SQDG are located only on the cytoplasmic side, uncharged DGDG are situated on the lumenal side, and heads of MGDG are located on both sides. Each lipid head forms polar contacts, i.e. hydrogen bonds or salt bridges with at least two different protein subunits. Moreover, the majority of carotenoid molecules interact with lipid molecules, except for two carotenoid molecules bound with the D1 and D2 proteins.
Multisubunit monomers of PS II are connected to each other with a non-crystallographic symmetry axis C2. In the interaction region they have some protein–protein contacts, but the surface between two monomers is mainly occupied by lipids. From the protein subunits only three low molecular weight proteins, PsbL, PsbM and PsbT, form a three-helix knot on the surface of monomer interaction, whereas two symmetric subunits PsbM and PsbM′ form a seven-member motif of aliphatic side chains similarly to a leucine zipper. The lipids are supposed to play an important role in formation of the PS II dimer [12].
During its functioning, the complete PS II complex periodically dissociates and re-associates [13, 14], and this is necessary to provide the replacement of the injured parts of the photosystem damaged by light-induced photooxidation. To by-pass the problem of disassemblage and re-assemblage of the whole photocomplex, there are in PS II complicated repair processes that selectively replace the D1 subunit. This protein is affected more frequently because it is a component of the electron transfer chain [15]. Inside every PS II monomer lipids form a kind of belt that surrounds subunits D1 and D2 and partially separates the reaction center from other intramembrane proteins. These lipids are responsible for a suitable environment that can be important for rapid turnover of the D1 protein during repair of PS II.
OXYGEN-EVOLVING COMPLEX
A manganese cluster also called the oxygen-evolving complex is a catalytic center for the oxidation of water. The cluster is situated in the lumenal part of PS II, and it passes through the so-called Si-state cycles (i = 0-4) [16, 17] (for review see [18]).
As described earlier, the structure of PS II from the two closely related thermophilic cyanobacteria Thermosynechococcus elongatus [1, 4, 6, 7] and T. vulcanus [3, 19] has been determined with resolution from 2.9 to 3.8 Å by different research groups. These structural studies revealed the reaction center position, but the resolution was not sufficiently high for determination of its exact structure. The cluster was supposed to consist of four manganese atoms, one calcium atom, a number of oxygen atoms, and, possibly, also water molecules, which are the substrate of the reaction of the Mn4CaOX cluster. During recording diffraction data, the cluster was destroyed under the influence of X-radiation [20, 21], this resulting in the appearance of some differences in the geometrical positions of atoms and also in the model of the liganding of the cluster [1, 3, 4, 6, 7, 19, 22-24].
The recently obtained PS II model with 1.9 Å resolution [8] allowed for the first time to describe the manganese cluster structure (Fig. 2a). In the previously obtained maps of the electron density, five metal ions from the Mn4CaOX cluster were not resolved and appeared as a sphere-shaped electron density [1]. In the electron density map of the PS II model with 1.9 Å resolution, the positions of individual atoms are clearly resolved and every atom of the cluster can be unambiguously identified [8]. Recognizing the positions of manganese and calcium atoms at such resolution was easier because a calcium atom has fewer electrons than a manganese atom. Based on the positions of the five metal ions, the distances between each pair of manganese atoms and between manganese and calcium atoms were established (Fig. 2a and Table 1) [8].
Fig. 2. a) Structure of the Mn4CaO5 cluster; b) structure of the Mn4CaO5 cluster ligands. Distances between atoms are indicated in angstroms.
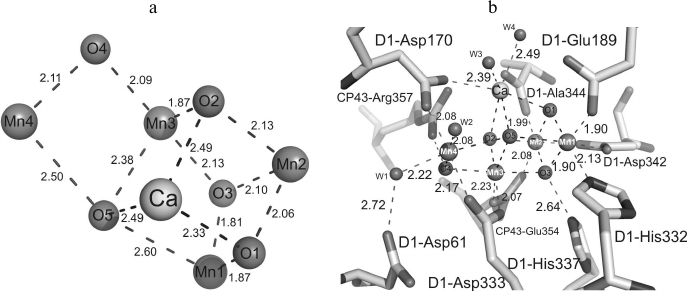
Table 1. Internal and external contacts of
the Mn4CaO5 cluster
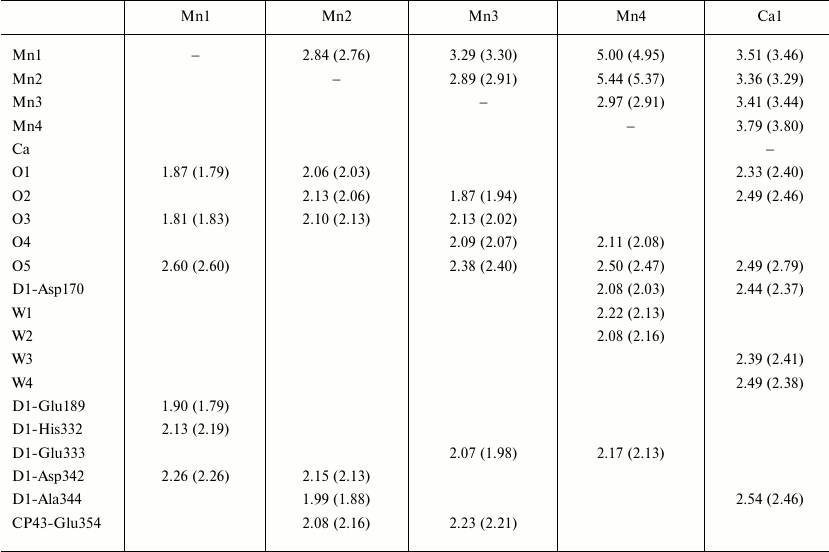
Note: Distances in the second monomer of the PS II dimer are shown in
parentheses. The distances are indicated in angstroms.
It was supposed earlier that single and double µ-oxo-bridges, which bind metal ions, should exist in the Mn4CaOX cluster of the oxygen-evolving complex. In the PS II structure with resolution of 3.5 Å, four oxygen atoms were preliminarily inscribed into the Mn4CaOX cluster [4] mainly based on the assumption that metal ions should bind oxygen. The reliability of positions of these atoms seemed doubtful because it was virtually impossible to identify the oxygen atoms in the electron density map at this resolution. In the PS II structure with the resolution of 2.9 Å, the oxygen atoms are even beyond the metal cluster [7]. Most distinctly, five oxygen atoms binding five metal ions were first determined in the PS II structure with resolution of 1.9 Å.
The general model of the Mn4CaO5 cluster is shaped like a distorted chair with a seat, the so-called cubane, formed by three manganese atoms, four oxygen atoms, and one calcium atom, whereas the chair back is formed by remote manganese and oxygen atoms. Distortions in the shape of the chair are caused by differences in lengths of the bonds between the manganese and oxygen atoms and of those between the calcium and oxygen atoms. While the majority of distances between Mn-O atoms are in the range of 1.9-2.1 Å, the distances between three manganese atoms (Mn1, Mn3, Mn4) and one oxygen atom (O5) are in the range of 2.4-2.6 Å (Table 1).
In addition to the five oxygen atoms, four water molecules (W) bound with the Mn4CaO5 cluster were identified [8]. Two of these water molecules (W1, W2) are bound with the Mn4 atom, whereas two other water molecules (W3, W4) are bound with the calcium atom (Fig. 2b). Distances between the two water molecules and the Mn4 atom are 2.1-2.2 Å, and distances between the water molecules and calcium atom are 2.4-2.5 Å. No other water molecules bound with three other manganese atoms were found. Therefore, at least one of these four water molecules is supposed to serve as the substrate for oxidation. Among the four water molecules bound with the Mn4CaO5 cluster, the W4 molecule is bound with a hydrogen bond directly with tyrosine YZ (D1-Tyr161), the amino acid residue mediating electron transfer between the PS II reaction center and the Mn4CaO5 cluster [8]. The water molecules W1-W3 form hydrogen bonds with YZ indirectly, through the other three water molecules W5-W7. It should be noted that the distance between W7 and YZ is 2.6 Å, and it seems to be a strong hydrogen bond. Moreover, YZ interacts with D1-His190 through a short hydrogen bond of 2.5 Å. Then the net of hydrogen bonds is extended onto D1-Asn298 and further into the lumenal aqueous fraction through several water molecules and some hydrophilic or charged amino acid residues [8]. Hence, proton-bound electron transfer through YZ is suggested to exist, in accordance with numerous previous reports about the possible existence of such a pathway [25-29].
It is important feature of Mn4CaO5 structure that the distances between the O5 atom and metal ions are noticeably greater as compared to other oxygen atoms situated within the oxygen-evolving center. This suggests a weak bond with the O5 atom within the cluster, which suggests a higher reactivity of this oxygen atom. It is interesting that two water molecules, W2 and W3, bound with the Mn4 and Ca atoms, respectively, are in the limits of the hydrogen bonds with the O5 atom (Fig. 2b). These results indicate convincingly that two of the three species — W2, W3, and O5 – are substrates for generation of the O–O bond during the oxidation of water [30].
Liganding of the Mn4CaO5 cluster. The majority of amino acid residues that are ligands interacting with the Mn4CaO5 cluster have been described in some works [3, 4, 6, 7]. However, because of limited resolution of the PS II spatial structures and possible X-ray-caused damage to the molecules, the reliability of locations and composition of the metal cluster ligands was insufficiently reliable. The length of bonds between the metal ions and atoms interacting with them were not accurately determined. Significant differences were also found in the regularity of liganding within the PS II structures with resolution of 3.5, 3.0 and 2.9 Å [4, 6, 7]. Whereas the majority of carboxylate ligands acted as monodentate ligands in the PS II structures with resolution of 3.5 and 3.0 Å, many of them were determined as bidentate ligands in the PS II structure with resolution of 2.9 Å. As a result, all amino acid residues acting as ligands of the Mn4CaO5 cluster were unambiguously determined in the PS II structure with the resolution of 1.9 Å [8]. Table 1 indicates that amino acids related to the D1 and CP43 subunits form together six carboxyl ligands and one histidine ligand. These are the following residues: D1-Asp170, D1-Glu189, D1-Glu333, D1-Asp342, D1-Ala344, CP43-Glu354 and D1-His332. They form the so-called first coordination sphere around the cluster. Among them D1-Asp170, D1-Glu333, D1-Asp342, D1-Ala344 and CP43-Glu354 are bidentate ligands, whereas D1-Glu189 and D1-His332 are monodentate ligands. These amino acid residues combined with oxo-bridges and water molecules form a saturated surrounding of the cluster. According to Table 1, there are six ligands for each of four Mn atoms and seven ligands for the Ca atom [8].
Structure and possible role of residues of the second coordination sphere of the manganese cluster. In addition to the described close ligands of the Mn4CaO5 cluster, three amino acid residues have been found in the second coordination sphere of the cluster that are capable of influencing the cluster, namely the following ligands: D1-Asp61, D1-His337 and CP43-Arg357. The imidazole ε-nitrogen of D1-His337 is bound directly by a hydrogen bond with oxygen atom O3. One of the guanidine η-nitrogens of CP43-Arg357 forms hydrogen bonds with oxygen atoms O2 and O4. Thus, these two amino acid residues can provide partial positive charges and somewhat compensate negative charges caused by oxo-bridges and carboxylate ligands and thus stabilize the cluster structure. In other words, in the absence of these residues some of oxo-bridges can be unstable and destroyed because of attraction by strong positive charges of the five metal ions. Moreover, the other guanidine η-nitrogens of CP43-Arg357 form hydrogen bonds with D1-Asp170 and D1-Ala344. One of the oxygens of the carboxylate group D1-Asp61 forms hydrogen bond with water molecule W1 bound with the Mn4 atom, whereas the other oxygen of carboxylate group D1-Asp61 forms a hydrogen bond with the W2 molecule indirectly, through two other water molecules, W8 and W9. These hydrogen bonds also can be important for stabilization of the Mn4CaO5 cluster [30]. These conclusions are in agreement with many data from mutagenesis and functional studies that have shown the importance of these three amino acid residues for retention of the oxygen evolving function by PS II [31-35].
Binding sites of Cl– in photosystem II. Structural studies on Br–- and I–-substituted PS II [19, 36] revealed two Cl–-binding sites are somewhat remote from the Mn4CaO5 cluster. However, only one of these sites was found in the unmodified PS II structure with resolution of 2.9 Å [7]. Both positions of the halogens are at the distance of 6-7 Å from the Mn4CaO5 cluster; therefore, it seems doubtful that they are natural binding sites of Cl– in PS II [37]. Moreover, although the first Cl–-binding site includes the positively-charged residue D1-Lys317, no similar residues have been detected close to the second binding site of Cl– [19, 36]. This suggested that the anion binding in this site of PS II should be rather weak, which should result in the absence of Cl– in the wild type PS II structure with resolution of 2.9 Å. Moreover, the chlorine ion was supposed to be directly bound with the Mn4CaO5 cluster, because the removal of Cl– was shown to noticeably influence oxygen evolution as well as the properties of the cluster [38, 39].
In the PS II structure with resolution of 1.9 Å, both binding sites of Cl– were clearly seen within the electron density [8], and their locations were the same as earlier described – at a significant distance from the Mn4CaO5 cluster [19, 36]. This was also confirmed on the anomalous difference Fourier map obtained with wavelength of 1.75 Å where Cl– contributed more significantly than the “light” atoms of the amino acid residues. The anomalous difference Fourier map also indicated that there was no other Cl– in the first coordination sphere of the Mn4CaO5 cluster, and this prevented the possibility of a direct binding of Cl– with metal ions of the oxygen-evolving center. And this also was in agreement with the above-presented data indicating that valences of all five metal atoms in the oxygen-evolving center were saturated with oxo-bridges, amino acid residues, and water molecules, and that no additional interactions were needed [30].
The first Cl–-binding site is situated at the distance of 6.7 Å from the Mn4 atom, and the second Cl– is at the distance of 7.4 Å from the Mn2 atom (Fig. 3). Immediately near the first Cl–, at the distance of 3.3 Å, the charged residue D2-Lys317 is located. In addition to lysine, the first Cl– is surrounded by two water molecules and nitrogen of the main chain of D1-Glu333. The other Cl– interacts with nitrogens of the main chain of D1-Asn338 and CP43-Glu354 and two water molecules. Thus, Cl–-binding sites have similar coordination spheres, namely, four groups of atoms surrounding each of two Cl–-binding sites include two water molecules and two amino acid residues. The experimental data indicate that Cl– in PS II could be easily removed by certain procedures that resulted in disorders in oxygen evolution, whereas a simple addition of Cl– into the medium led to re-inclusion of Cl– into PS II and recovery of oxygen production [38, 39]. This can be explained, in particular, by the presence of carboxylate side groups in both amino acid residues D1-Glu333 and CP43-Glu354, and these groups are coordinated with the Mn4CaO5 cluster as bidentate ligands (Table 1), whereas nitrogens of their main chain are bound with two Cl– ions. It seems that the function of Cl– is to fix the structures of these two amino acid residues necessary for stabilization of bonds within the Mn4CaO5 cluster. The absence of Cl– could influence the stable coordination of these residues in the Mn4CaO5 cluster, thus causing disorders in oxygen production. Alternatively, both Cl– ions can be supposed to function for maintaining pathways for proton release due to their location in the beginning of two possible pathways for proton leaving the Mn4CaO5 cluster for the lumen [4, 7, 8, 40-43].
Fig. 3. Positions of chloride ions with respect to the Mn4CaO5 cluster. The bond lengths are indicated in angstroms.
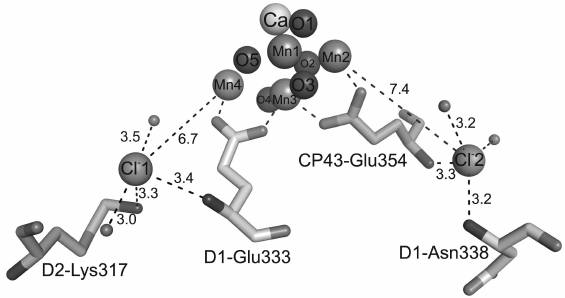
In addition to the presence of one calcium atom in the high resolution structure of the Mn4CaO5 cluster and location of two chloride ions immediately near the cluster, there are also three additional calcium ions and one additional chloride situated on the periphery of the photosynthetic complex.
CALCULATIONS OF PRODUCT/SUBSTRATE CHANNELS IN PS II
The oxygen-evolving complex is located in the depth of the lumenal part of PS II, and water has to find a pathway through the protein towards the manganese cluster [44]. Because of the limited access of competitive water analogs to the decomposition reaction, it was supposed that the site of water oxidation should be protected. It was supposed that protein could guard the oxygen-evolving complex against potentially harmful molecules and minimize side reactions with small molecules brought in with water. Because ions and cellular reducers can compete for binding sites and worsen water decomposition, their entrance seems to be potentially dangerous. Subsequent studies confirmed this hypothesis: in the absence of the “external” subunits, hydrogen peroxide is really produced in PS II, and oxygen release is less [44]. The amount of released oxygen is recovered on addition of substances capable of inducing aggregation of proteins. Access to the oxygen-evolving complex was probed by the electron spin echo envelope modulation method (ESEEM). Binding of alcohols with the Mn4CaO5 cluster was shown to decrease with increase in the size of the alcohol molecule. This promoted the hypothesis that an aqueous channel should exist within PS II [45]. The evolution of O2 molecules and protons from the catalytic center also suggests the existence of channels for their removal [46, 47]. Therefore, different groups of researchers supposed that numerous channels formed by protein subunits of PS II should exist and produce a transporting network responsible for the rapid and effective delivery of initial substrates and removal of products that was necessary for the molecular turnover within PS II. Searching for such channels became possible only after the crystal structure of PS II with resolution higher than 3.8 Å was published [4, 6].
Murray and colleagues used a Caver program to search for cavities in the PS II structure with resolution of 3.5 Å [40]. They found three channels, and they supposed that the widest and least hydrophilic channel (i) was destined for oxygen. The more polar channel (ii) was destined for water and protons, and the most polar channel (iii) was reserved only for protons. It should be noted that the algorithm of the Caver program searched for the shortest pathways from a given point within a protein to the surface with the chosen minimal diameter. Therefore, this program searches for numerous round “pipeline” channels.
Later, the determination of all cavities near the oxygen-evolving center based on the structure with resolution of 3.0 Å, independently of the place of their coming onto the surface, revealed a complicated system of possible transporting channels [41]. An approach used in a work by Ho and Styring was a step forward in the comprehension of delivery of water to the PS II active center through a network of mutually connected cavities and pores within the proteins. Further studies on the transport networks in PS II were continued on a spatial structure with resolution of 2.9 Å, which also was analyzed using the Caver program [7, 42]. Since now this structure represents most completely the protein and lipid composition, except for the solvent molecules, the channels based on it are the most likely ones [42]. Presumptive channels were calculated based on such criteria as the channel diameter, its length and hydrophobicity, and on precessing the structure with the coordination error also taken into account. Altogether, nine putative channels connecting the oxygen-evolving center with the lumen were determined. They were designated as A1, A2, B1, B2, and C-G (for comparison with the earlier and later models, see Table 2). It was supposed that water molecules could be disposed in channels C-G. But channels based on the static structural model were too narrow for providing free passage of H2O or O2 molecules. Hence, it was supposed that channels C-G could act as pathways for proton removal. Note that these channels begin in the Mn4CaO5 cluster on the opposite side from Ca2+ and that the C and G channels are partially united and form an interaction site with Cl–. It was supposed earlier that Cl– could be involved in the proton transfer [38]. Moreover, key amino acid residues believed to be important for proton release, such as D1-Asp61 and D1-Glu65 [48], are situated in these channels. The wider channels begin on the “calcium side” of the Mn4CaO5 cluster. Channels A1 and A2 seem to be involved in supply of water molecules; their minimal radii are, respectively, 1.24 and 1.38 Å. Channels B1 and B2 are somewhat wider, their minimal radius being 1.44 Å, and they have more hydrophobic walls and are likely to act as pathways for removal of oxygen molecules. Channel B2 is really blocked with U-Lys134, which forms a salt bridge with V-Asp79 and thus connects two external subunits. Conformational changes in the side group of U-Lys134 were shown to be capable of enlarging the channel diameter from 2.3 to 3.5 Å and of making B2 a possible pathway for diffusion of molecular oxygen [42].
Table 2. Comparative nomenclature of
product/substrate channels in PS II proposed by different authors

To check the putative pathways of evolution of dioxygen, in some experimental works [7, 42, 49] PS II crystals were treated with xenon (Xe). This noble gas is suitable for determination of oxygen channels in proteins [50-52]. The detection of Xe atoms by X-rays is facilitated by the high density of electrons in these atoms. Because the size of Xe atoms is similar to that of O2 molecules, this gas is used for imitating oxygen in X-ray crystallography. But in PS II, xenon failed to enter the putative oxygen channels, rather filling the membrane part of the complex. Therefore, the experiments were repeated with another noble gas, krypton (Kr), which has a smaller radius [42]. Krypton atoms were detected in the B1 and B2 channels. The availability of these channels for small molecules was also demonstrated by co-crystallization of PS II with dimethyl sulfoxide (DMSO) [42]. Two DMSO molecules were detected in the B1 and B2 channels not far from the Kr-binding sites. Hence, it was supposed that the B1 and B2 channels should be responsible for diffusion of oxygen molecules from the oxygen-evolving complex.
All of the above-described works for detecting aqueous channels in PS II were based on analysis of cavities within the structure obtained by X-ray crystallography. But this approach has two shortages: first, the presence of a cavity within a crystallographic structure does not always means that it is occupied by water; second, channels in the static structure have fixed borders and a rigid base [40, 41]. This problem can be solved, in particular, by assessing the potential of the apparent location of water molecules. This approach was used in a hybrid quantum-molecular study on the oxygen-evolving center that modeled chains of water molecules joined with hydrogen bonds and directed to the Mn4CaO5 cluster along two different trajectories [53, 54]. However, these studies involved only a small region of PS II adjacent to the active site of water decomposition and thus were unable to give a full picture of the positions of the channels.
At physiological temperatures, thermal motions within proteins continuously change the network of channels and can result in transient states of opening/closing of some channels [55]. These processes are likely to play a decisive role in the control of water movement within PS II [56]. Therefore, Vassiliev with colleagues proposed another interesting approach [43]. They modeled water molecules placed in energetically advantageous positions in all cavities of protein constituents of PS II. Then they used molecular dynamics to model the whole PS II complex, including water, under conditions of physiological temperature, thus allowing them to follow the movement of water molecules across the proteins. As a result, water flows were detected in all four channels described in the work by Ho and Styring [41], and they were designated “broad”, “back”, “narrow”, and “large”. The resulting calculations revealed significant differences in PS II conformation in the presence and absence of the Mn4CaO5 cluster. The greatest difference was observed in the region of C-terminal domains of two external proteins, PsbV and PsbU. These regions are involved in formation of a wide opening at the end of the “large” channel in PS II in the absence of the manganese cluster in the model. The possible participation of the PsbU and PsbV proteins in the regulation of ion transport in PS II was confirmed by earlier experimental observations. Mutant strains of cyanobacteria with ΔPsbV and ΔPsbU deletions were characterized by lower evolution of oxygen and the loss of photoautotrophic growth under calcium and chlorine ion limiting conditions [57, 58]. Moreover, a mutant lacking protein PsbV was prone to rapidly lose oxygen-evolving activity in connection with the loss of metal ions in the active center [59], whereas protein PsbU stabilized oxygen evolution at elevated temperatures [60]. All these findings are in agreement with the active role of the C-terminal domains of PsbU and PsbV in the control of water and ion transport observed in the molecular-dynamic modeling.
The appearance of the PS II structure with resolution of 1.9 Å [8] was insufficient for the final determination of the product/substrate channel positions within this system, but it decreased the coordination error of the calculations. Although positions of more than two thousand water molecules in the structure are described, it is difficult to determine water displacements within PS II because the crystal structure is a static model. Therefore, trajectories of the aqueous channel still remain presumptive. Unfortunately, the authors were not successful in identification of dioxygen molecules in the PS II structure, and the question of the “oxygen” channel is still open.
Therefore, new methods for calculation of the channels are still under development. In recent in silico work by Vassiliev et al. [61], constant “injection” of new water molecules into the PS II manganese cluster region was used for accelerating the movements of the molecules using molecular-dynamics calculations. This allowed the authors to follow the trajectory of water movement from PS II under pressure and to determine the energy required for overcoming energy barriers in the channels. Upon analysis of the energy of water movement across the channels, the penetration of water was confirmed through three aqueous channels described in the earlier works: the “narrow” channel, “the large system of channels”, and the “broad” channel [40, 41, 43]. The transudation of water through the earlier revealed “back” channel was not confirmed. Moreover, penetration of water was observed through the channel designated as channel 1 in work [61] that was earlier supposed to be a channel for proton removal [48].
Thus, the exact determination of channel trajectories in such large complexes as PS II is still a nontrivial problem even for modern methods. But it can be expected that the development of an X-ray laser and recording diffraction data with it at physiological temperature will allow researchers to easily determine movements of molecules within any protein and complex. The progress in this field will be considered in the section “Femtosecond diffraction of PS II microcrystals”.
PLASTOQUINONES
According to the modern concepts, the primary photochemical reaction in the reaction center of the PS II includes energy transfer from an excited molecule of chlorophyll P680 onto a pheophytin molecule. This process takes several picoseconds and results in formation of a state with separated charges P680+Ph– that lives for only 10–8 s due to recombination of charges. However, recombination is prevented due to more rapid transfer of the electron from Ph– onto an electron acceptor molecule, plastoquinone QA. This is accompanied by the loss of about 30% of the energy stored as a result of the primary photoact, but a more stable state of the molecules is produced. Finally, an electron from the chlorophyll P680 molecule is transferred onto the system of quinone electron carriers that results in stabilization of the reaction center state with oxidized chlorophyll P680. In this case, the plastoquinone (PQ) molecules act as fixed one-electron transmitters (QA) and mobile two-electron carriers (QB) in the electron transfer chain from the PS II to PS I through the cyt b6f complex. Plastoquinone Q-binding sites are located in PS II closely to the cytoplasmic side of protein D1, which forms the plastoquinone QB binding site, and to protein D2, which forms the QA binding site. For the electron transfer, a sufficient number of PQ molecules must be present, and rapid exchange is important between the PQ-binding site and the store of free PQs in the thylakoid membrane. Inside PS II a large cavity was detected earlier, which seemed to serve as a reservoir for PQ/PQH2 [6] (Fig. 4a). This cavity is limited by proteins PsbJ, PsbK, D1, D2 and CP43 and also by cyt b559. This cavity is lined mainly with hydrophobic amino acid residues and is filled with phytol chains of chlorophyll and pheophytin molecules and also with acyl chains of lipids. The heme group of the cyt b559 and rings of carotenoid molecules also enter the cavity. This cavity is accessible for intramembrane quinones through a small aperture near cyt b559 (channel I) [6]; also, another aperture was found (channel II) and an additional plastoquinone QC was located [7] (Fig. 4b). The existence of plastoquinone QC is in agreement with the idea of a new PQ-binding site in PS II not far from cyt b559 [62, 63] and about “reserved” plastoquinones located immediately near the QB site [64, 65]. The existence of a separate site for “reserved” plastoquinone is not yet confirmed in the high resolution structure, possibly because of higher purification of the preparation and high mobility of plastoquinone QC [8]. The outlet of channel I has area of about 10 × 20 Å2 and is located between protein PsbJ and cyt b559, whereas the outlet of channel II with area of 12 × 10 Å2 is located between proteins PsbF and D2. Both these channels connect the QB-binding site with the membrane phase. This disposition of the channel fits well a rapid exchange of reduced plastoquinone (PQH2) for fresh plastoquinone from the PQ store in the thylakoid membrane.
Fig. 4. a) Calculated channels I and II for PQ/PQH2 exchange between the plastoquinone pool in the membrane and the QB and QC sites; b) schematic image of PQ/PQH2 exchange cavity within PS II; c) putative mechanisms of PQ/PQH2 exchange between the QB-site and the PQ pool in the thylakoid membrane.
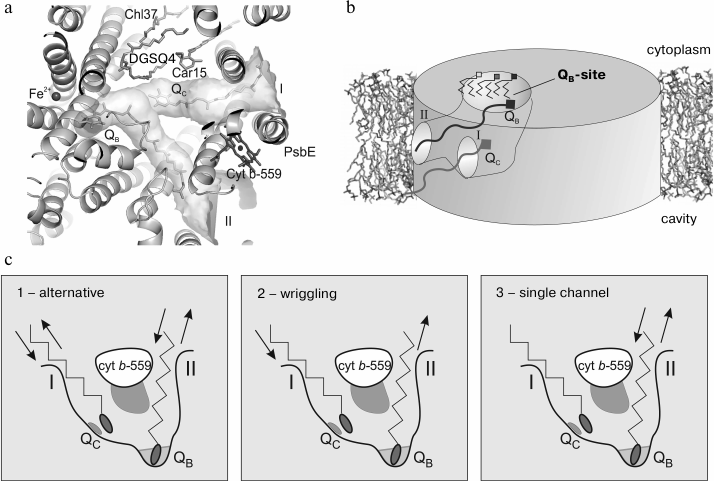
After reduction/protonation, the PQH2 molecule leaves the QB-site and a fresh plastoquinone from the store of PQ molecules enters the QB-site for the next cycle. Three putative mechanisms of PQ/PQH2 exchange were proposed [7] (Fig. 4c). The first two mechanisms suggest that the entrance/removal of PQ and PQH2 should occur either via the same channel (the “alternative” mechanism) or via different channels (the “wriggling” mechanism, because the plastoquinone molecule had to wriggle to change the channel). In the alternative mechanism channels I and II should be used alternatively for the entrance and removal, whereas in the wriggling mechanism channel I should be used only for the entrance and channel II only for the removal of PQ molecules. In the single channel mechanism only channel II should serve both for entrance and removal, whereas channel I is not involved in the PQ/PQH2 exchange.
PS II MONOMER
PS II exists in the thylakoid membrane as both dimer and monomer. The PS II dimer could be isolated and crystallized more easily than the PS II monomer; therefore, before 2010 structural studies concerned only native or modified PS II as dimers. Crystals of the PS II dimer usually belong to orthorhombic spatial group P212121, and the resolution maximum of 2.9 Å could not be overcome for a long time [7]. Moreover, the diffraction picture was anisotropic, i.e. the resolution limit changed in depending on the crystal orientation in the X-rays, which limited the useful resolution of the PS II dimer crystals [6, 66]. Broser and colleagues [67] supposed that crystallization of PS II monomers should promote change in packing in crystals and improve their quality and also to reveal factors influencing PS II oligomerization. As a result of their work, the PS II monomer was crystallized and the first X-ray structural model was obtained of the PS II monomer from T. elongatus with resolution of 3.6 Å [67]. The quality of the data allowed unambiguous determination of the positions of 19 polypeptide subunits, which formed an active photosynthetic complex, but no major changes were found comparative to the PS II dimer structure. The electron density for the peripheral subunit PsbY was absent, although the authors believed that its presence would not cause steric conflicts in the PS II monomer crystal [67]. Nevertheless, protein PsbY was detected by MALDI analysis in 80% of dissolved crystals of the PS II monomer. The authors explained this finding by incomplete occupation of the position of this subunit or by an unordered state of protein PsbY in the PS II monomer. In the PS II dimer, the electron density for this subunit could be observed at the resolution of 3.8 Å [1] and 2.9 Å [7], but it was absent in the structures with resolution of 3.5 Å [4] and 1.9 Å [8]; hence, the PsbY protein was supposed to be freely associated with the photocomplex.
Notwithstanding the low resolution of the spatial structure of the PS II monomer, the electron density quality allowed the researchers to reliably determine positions of pigments such as chlorophyll a and pheophytin. Localizations of all chloride ions and of all 29 molecules of Chla bound with the antenna proteins CP43 and CP47 were also confirmed. Moreover, positions of 11 molecules of β-carotenes were determined. As differentiated from the PS II dimer, there was no β-carotene molecule located on the interaction surface between monomers in the photocomplex dimers; this molecule could be detached during the isolation of the PS II monomer. Electron density fragments were also found for plastoquinones in the binding sites of QA, QB, and QC, but the plastoquinone molecule was unambiguously detected only for QA in the position virtually identical to the position of QA within the PS II dimer [7].
The electron density of the PS II monomer model was favorable for description of positions of 22 lipid molecules. This result was in agreement with data on the lipid composition in the PS II monomer and dimer from T. elongatus [68]. In the PS II monomer, only five lipid molecules are located on the surface of monomer dimerization near subunits D1, PsbT, and PsbM and forming a stable complex with these proteins. It seems that lipids are important for the structure and possibly also for functioning of the PS II reaction center, are involved in the cycle of subunit D1 recovery, and thus are responsible not only for PS II dimerization as supposed by some authors [7].
Protein–protein contacts between monomers within the PS II dimer are realized by subunit PsbM and its symmetric partner PsbM′. PsbM was shown to be located similarly in both PS II monomer and dimer, but dimerization of PS II monomers did not occur. Some data indicate that upon the removal of protein PsbM from the photosynthesizing complex in the mesophilic cyanobacteria Synechocystis PCC 6803, PS II dimerization continues [69]. All these findings confirm ideas about the secondary character of direct protein–protein contacts on PS II dimerization [7].
FEMTOSECOND DIFFRACTION OF PS II MICROCRYSTALS
To elucidate the structure and functioning of PS II, during the last decades different spectroscopic approaches have been used [70-72], as well as methods of X-ray diffraction using synchrotron radiation [1, 3, 4, 6-8]. Among numerous approaches, X-ray crystallographic analysis is undoubtedly the best for obtaining detailed structure of the Mn4CaO5 cluster and PS II as a whole. Nevertheless, sensitivity to radiation of the redox-active high-valence Mn4CaO5 cluster is a critical problem for X-ray structural studies. X-ray spectroscopic studies revealed that radiation-caused damage in the Mn4CaO5 cluster decreased the oxidation degree of manganese atoms to Mn(III) and Mn(IV), which were present in the undamaged catalytic center as Mn(II) [20, 21]. Based on studies on radiation-induced damage, it was concluded that in all modern X-ray structural studies Mn4CaO5 cluster oxidation was lowered by approximately 25% [8], and sometimes even by ~80% [1, 3, 4, 6, 7]. Such changes occur notwithstanding that all measurements of X-ray diffraction are performed at low temperatures of 100-150K.
It was shown by the extended X-ray absorption fine structure (EXAFS) method that radiation damage increased the distances between Mn-Mn atoms and between Mn-ligand as compared to the undamaged Mn4CaO5 cluster [20]. This resulted not only in a decrease in the oxidation degree of manganese atoms but also in destruction of the cluster structure. Now it is generally adopted that for metal-containing proteins it is difficult to obtain undamaged structures using synchrotron radiation based on X-ray diffraction even at cryogenic temperatures. Note also that the freezing procedure caused conformational changes in some proteins [73].
A new approach in protein crystallography was recently demonstrated in the USA with a linear accelerator, the LCLS, using ultra-short high intensity X-ray impulses in protein crystallography on crystal specimens with sizes from 0.1 to 10 µm [74-81]. Use of very short impulses (less than 70 fs) allowed researchers to obtain diffraction data at room temperature before damage by the radiation occurred [80-83]. This method of obtaining data “probe-before-destroy” not only eliminated the problem of radiation-induced damage, but opened pathways for studies on enzyme systems by X-ray crystallographic analysis at room temperature in the femtosecond time range. This made it possible to use different systems for triggering chemical reactions, such as photoactivation or chemical induction, and this promoted study of the chemical and structural dynamics of different processes. As a result, a group of authors found that PS II microcrystals could be analyzed by X-ray crystallography at room temperature with ultra-short X-ray impulses (shorter than 50 fs) [84]. Individual diffraction points were recorded with the resolution of 5.4 Å, but a complete set of data was obtained with the resolution of 6.5 Å. The electron density map of PS II did not have serious differences from data obtained earlier with synchrotron radiation and truncated to the resolution of 6.5 Å [7]. There was density in the Mn4CaO5 cluster region that indicated that this cluster was not destroyed under the influence of intensive X-ray impulses, which were sometimes stronger than those used in the synchrotron radiation sources. This pilot study was a basis for combining different methods that allowed the researchers to study in detail intermediate states of PS II.
While X-ray crystallography is an important approach for study of the geometric structure of whole complexes, X-ray absorption and emission spectroscopy are powerful methods for studies on chemistry of inorganic systems and metal-containing proteins. X-ray absorption spectroscopy (XAS) has long been used in various systems using synchrotron radiation [85], and recent achievements in this line have allowed researchers to perform experiments with picosecond resolution [86-89]. During recent years, X-ray emission spectroscopy (XES) is used more frequently on synchrotron radiation sources for studies of metal-containing proteins [90-92], geochemical systems [93, 94], coordination complexes [95, 96] and inorganic catalytic centers. In addition to the XAS method, XES measurements of occupation of the electron levels give information about the electronic structure, charge, spin density and the nature of ligands [97-99].
Intermediate chemical states of catalytic centers, in particular in biological systems, were traditionally studied on frozen specimens [90]. However, this approach prevents long-term real-time observations of structural changes. X-ray free-electron laser spectroscopy (XFEL) can overcome these limitations. Because studies based on synchrotron radiation are usually limited to picosecond resolution [89], X-ray spectroscopy in XFEL can be used to study electronic structure within the femtosecond time interval. Moreover, the large flow of XFEL impulses allows researchers to record data on diluted specimens or with weak emission signal for short time intervals.
An experimental installation was developed to concurrently record results of X-ray analysis and X-ray spectroscopy XES on the LCLS [100, 101]. XES was used for determination of the integrity of the Mn4CaO5 cluster electronic state. PS II microcrystals adapted in the dark reflected X-rays to 4.1 Å. The spectrum of the PS II crystals correlated very well with the spectrum of PS II in the initial state, i.e. in state S1. This meant that manganese atoms in the cluster are in the same high-valency state (Mn2IIIMn2IV) in both solution and crystal; thus, the crystallization procedure did not change the S1 state of PS II. Then X-ray diffraction of the S2 state obtained with a visible pumping laser was measured at room temperature, and the results were compared with X-ray crystallographic data obtained in the dark state of S1. The transition of the PS II specimen into the S2 state was tested using labeled water molecules (H218O). Analysis of the labeled O2 produced depending on the number of laser flashes revealed that the S2 state of the reaction center was in ~80% of cases achieved after one light flash. The authors obtained a set of diffraction data with resolution of 5.9 Å from PS II crystals in the S2 state [101]. Comparison of data revealed that the exposure to light did not lead to crystal decomposition or changes in its quality. Moreover, it is suggested that there is no pronounced structural changes in the photosynthesizing complex on the reaction center transition from state S1 to S2.
Thus, concurrent X-ray crystallographic and XES studies with ultra-short ultra-bright X-ray pulses in the LCLS allowed researchers to examine intact nuclear structures of PS II microcrystals and not damage the electronic structure of the Mn4CaO5 cluster at room temperature. XES also serves as a method for control of the integrity of the metal during the recording of X-ray crystallographic data. This method can be used for future studies on light-induced structural changes within the protein component of PS II and pigments and also on the chemical reaction in the catalytic center in different functional states. It is expected that this method will be used for many metal-containing enzymes, including those that are very sensitive to photo- and radiation-induced damage over a wide time range beginning from femtoseconds.
Structural studies on PS II from cyanobacteria have resulted in creation of a high-resolution spatial model of this huge complex. It has been shown that the photocomplex monomer consists of 20 protein subunits, 54 pigment molecules, and 25 molecules of incorporated lipids; it also has manganese, calcium, chlorine, iron, and bicarbonate ions. Based on the structural data, working mechanisms of mobile electron carriers have been proposed. The structure was determined of the manganese cluster responsible for water decomposition to atmospheric oxygen, protons and electrons. The system of coordination of the cluster atoms by the protein surrounding has been described in detail. However, it is still necessary to elucidate what occurs with the manganese cluster and its surrounding during the oxidation and transition from the S0 to the S4 state. Numerous studies have revealed within the PS II a branched network of channels connecting the Mn4CaO5 cluster with the lumen. This network is supposed to be responsible for transfer of substrates and reaction products during the photo-induced decomposition of water. Nevertheless, the mechanism and direction of the movement in this transfer network are still hypothetical. In the thylakoid membrane of cyanobacteria, the PS II is present as both monomer and dimer. Unfortunately, the resulting crystal structures of the photosystem monomer and dimer did not help to explain the reason for the dimerization. Thus PS II, which is a unique natural photosynthetic complex, still remains a poorly studied object of contemporary fundamental and applied science.
The appearance of the newest devices of synchrotron radiation and their combination with other methods of investigation of chemical and physical properties of atoms gives hope that structural analysis can acquire a novel quantitative level. This will result in the possibility of investigating not only frozen static structures of biomolecules in the initial or final stage of the reaction, but also in intermediate states occurring within femtoseconds at physiological temperatures. Such approaches will help to study in detail atom movements within the photosystem, as well as the overall reaction of the decomposition of water.
This work was supported by the Russian Academy of Sciences Presidium Program “Molecular and Cellular Biology” and by the Russian Foundation for Basic Research (project No. 13-04-01148a).
REFERENCES
1.Zouni, A., Witt, H. T., Kern, J., Fromme, P.,
Krauss, N., Saenger, W., and Orth, P. (2001) Nature, 409,
739-743.
2.Zouni, A., Jordan, R., Schlodder, E., Fromme, P.,
and Witt, H. T. (2000) Biochim. Biophys. Acta, 1457,
103-105.
3.Kamiya, N., and Shen, J. R. (2003) Proc. Natl.
Acad. Sci. USA, 100, 98-103.
4.Ferreira, K. N., Iverson, T. M., Maghlaoui, K.,
Barber, J., and Iwata, S. (2004) Science, 303,
1831-1838.
5.Biesiadka, J., Loll, B., Kern, J., Irrgang, K.-D.,
and Zouni, A. (2004) Phys. Chem. Chem. Phys., 6,
4733-4736.
6.Loll, B., Kern, J., Saenger, W., Zouni, A., and
Biesiadka, J. (2005) Nature, 438, 1040-1044.
7.Guskov, A., Kern, J., Gabdulkhakov, A., Broser, M.,
Zouni, A., and Saenger, W. (2009) Nat. Struct. Mol. Biol.,
16, 334-342.
8.Umena, Y., Kawakami, K., Shen, J. R., and Kamiya,
N. (2011) Nature, 473, 55-60.
9.Adachi, H., Umena, Y., Enami, I., Henmi, T.,
Kamiya, N., and Shen, J. R. (2009) Biochim. Biophys. Acta,
1787, 121-128.
10.Raymond, J., and Blankenship, R. E. (2004)
Biochim. Biophys. Acta, 1655, 133-139.
11.Sakurai, I., Shen, J. R., Leng, J., Ohashi, S.,
Kobayashi, M., and Wada, H. (2006) J. Biochem. (Tokyo),
140, 201-209.
12.Kern, J., and Guskov, A. (2011) J. Photochem.
Photobiol. B, 104, 19-34.
13.Baena-Gonzalez, E., and Aro, E. M. (2002)
Phil. Trans. Roy. Soc. London, Ser. B. Biol. Sci.,
357, 1451-1459.
14.Barbato, R., Friso, G., Rigoni, F., Dalla
Vecchia, F., and Giacometti, G. M. (1992) J. Cell Biol.,
119, 325-335.
15.Rokka, A., Suorsa, M., Saleem, A., Battchikova,
N., and Aro, E. M. (2005) Biochem. J., 388, 159-168.
16.Kok, B., Forbush, B., and McGloin, M. (1970)
Photochem. Photobiol., 11, 457-475.
17.Joliot, P. (2003) Photosynth. Res.,
76, 65-72.
18.Renger, G., and Renger, T. (2008) Photosynth.
Res., 98, 53-80.
19.Kawakami, K., Umena, Y., Kamiya, N., and Shen, J.
R. (2009) Proc. Natl. Acad. Sci. USA, 106, 8567-8572.
20.Yano, J., Kern, J., Irrgang, K. D., Latimer, M.
J., Bergmann, U., Glatzel, P., Pushkar, Y., Biesiadka, J., Loll, B.,
Sauer, K., Messinger, J., Zouni, A., and Yachandra, V. K. (2005)
Proc. Natl. Acad. Sci. USA, 102, 12047-12052.
21.Grabolle, M., Haumann, M., Muller, C., Liebisch,
P., and Dau, H. (2006) J. Biol. Chem., 281,
4580-4588.
22.Yano, J., Kern, J., Sauer, K., Latimer, M. J.,
Pushkar, Y., Biesiadka, J., Loll, B., Saenger, W., Messinger, J.,
Zouni, A., and Yachandra, V. K. (2006) Science, 314,
821-825.
23.Siegbahn, P. E. (2008) Chemistry,
14, 8290-8302.
24.Siegbahn, P. E. (2009) J. Am. Chem. Soc.,
131, 18238-18239.
25.Hoganson, C. W., and Babcock, G. T. (1997)
Science, 277, 1953-1956.
26.Tommos, C., and Babcock, G. T. (2000) Biochim.
Biophys. Acta, 1458, 199-219.
27.Vrettos, J. S., Limburg, J., and Brudvig, G. W.
(2001) Biochim. Biophys. Acta, 1503, 229-245.
28.Renger, G. (2001) Biochim. Biophys. Acta,
1503, 210-228.
29.Hays, A. M., Vassiliev, I. R., Golbeck, J. H.,
and Debus, R. J. (1999) Biochemistry, 38,
11851-11865.
30.Kawakami, K., Umena, Y., Kamiya, N., and Shen, J.
R. (2011) J. Photochem. Photobiol. B, 104, 9-18.
31.Nixon, P. J., and Diner, B. A. (1994) Biochem.
Soc. Trans., 22, 338-343.
32.Chu, H. A., Nguyen, A. P., and Debus, R. J.
(1995) Biochemistry, 34, 5839-5858.
33.Hwang, H. J., Dilbeck, P., Debus, R. J., and
Burnap, R. L. (2007) Biochemistry, 46, 11987-11997.
34.Debus, R. J. (2008) Coord. Chem. Rev.,
252, 244-258.
35.Service, R. J., Hillier, W., and Debus, R. J.
(2010) Biochemistry, 49, 6655-6669.
36.Murray, J. W. (2008) Energy Environ. Sci.,
1, 161-166.
37.Guskov, A., Gabdulkhakov, A., Broser, M.,
Glockner, C., Hellmich, J., Kern, J., Frank, J., Muh, F., Saenger, W.,
and Zouni, A. (2010) Chemphyschem., 11, 1160-1171.
38.Olesen, K., and Andreasson, L. E. (2003)
Biochemistry, 42, 2025-2035.
39.Popelkova, H., and Yocum, C. F. (2007)
Photosynth. Res., 93, 111-121.
40.Murray, J. W., and Barber, J. (2007) J.
Struct. Biol., 159, 228-237.
41.Ho, F. M., and Styring, S. (2008) Biochim.
Biophys. Acta, 1777, 140-153.
42.Gabdulkhakov, A., Guskov, A., Broser, M., Kern,
J., Muh, F., Saenger, W., and Zouni, A. (2009) Structure,
17, 1223-1234.
43.Vassiliev, S., Comte, P., Mahboob, A., and Bruce,
D. (2010) Biochemistry, 49, 1873-1881.
44.Klimov, V. V., Ananyev, G. M., Zastryzhnaya, O.
M., Wydrzynski, T., and Renger, G. (1993) Photosynth. Res.,
38, 409-416.
45.Force, D. A., Randall, D. W., Lorigan, G. A.,
Clemens, K. L., and Britt, R. D. (1998) J. Am. Chem. Soc.,
120, 13321-13333.
46.Anderson, J. M., and Chow, W. S. (2002) Phil.
Trans. Roy. Soc. London Ser. B. Biol. Sci., 357,
1421-1430.
47.Wraight, C. A. (2006) Biochim. Biophys.
Acta, 1757, 886-912.
48.Ishikita, H., Saenger, W., Loll, B., Biesiadka,
J., and Knapp, E. W. (2006) Biochemistry, 45,
2063-2071.
49.Murray, J. W., Maghlaoui, K., Kargul, J.,
Sugiura, M., and Barber, J. (2008) Photosynth. Res., 98,
523-527.
50.Prange, T., Schiltz, M., Pernot, L., Colloch, N.,
Longhi, S., Bourguet, W., and Fourme, R. (1998) Proteins –
Structure Function and Genetics, 30, 61-73.
51.Cohen, J., Arkhipov, A., Braun, R., and Schulten,
K. (2006) Biophys. J., 91, 1844-1857.
52.Luna, V. M., Chen, Y., Fee, J. A., and Stout, C.
D. (2008) Biochemistry, 47, 4657-4665.
53.Sproviero, E. M., Gascon, J. A., McEvoy, J. P.,
Brudvig, G. W., and Batista, V. S. (2008) Coord. Chem. Rev.,
252, 395-415.
54.Sproviero, E. M., McEvoy, J. P., Gascon, J. A.,
Brudvig, G. W., and Batista, V. S. (2008) Photosynth. Res.,
97, 91-114.
55.Cohen, J., Kim, K., King, P., Seibert, M., and
Schulten, K. (2005) Structure, 13, 1321-1329.
56.Ho, F. M. (2008) Photosynth. Res.,
98, 503-522.
57.Shen, J. R., Ikeuchi, M., and Inoue, Y. (1997)
J. Biol. Chem., 272, 17821-17826.
58.Shen, J. R., Vermaas, W., and Inoue, Y. (1995)
J. Biol. Chem., 270, 6901-6907.
59.Li, Z., Andrews, H., Eaton-Rye, J. J., and
Burnap, R. L. (2004) Biochemistry, 43, 14161-14170.
60.Nishiyama, Y., Los, D. A., Hayashi, H., and
Murata, N. (1997) Plant Physiol., 115, 1473-1480.
61.Vassiliev, S., Zaraiskaya, T., and Bruce, D.
(2012) Biochim. Biophys. Acta, 1817, 1671-1678.
62.Kruk, J., and Strzalka, K. (2001) J. Biol.
Chem., 276, 86-91.
63.Kaminskaya, O., Shuvalov, V. A., and Renger, G.
(2007) Biochemistry, 46, 1091-1105.
64.Kern, J., Loll, B., Luneberg, C., DiFiore, D.,
Biesiadka, J., Irrgang, K. D., and Zouni, A. (2005) Biochim.
Biophys. Acta, 1706, 147-157.
65.Krivanek, R., Kern, J., Zouni, A., Dau, H., and
Haumann, M. (2007) Biochim. Biophys. Acta, 1767,
520-527.
66.Kern, J., Loll, B., Biesiadka, J., Zouni, A.,
Irrgang, K.-D., and Saenger, W. (2005) Photosynth. Res.,
84, 153-159.
67.Broser, M., Gabdulkhakov, A., Kern, J., Guskov,
A., Muh, F., Saenger, W., and Zouni, A. (2010) J. Biol. Chem.,
285, 26255-26262.
68.Loll, B., Kern, J., Saenger, W., Zouni, A., and
Biesiadka, J. (2007) Biochim. Biophys. Acta, 1767,
509-519.
69.Bentley, F. K., Luo, H., Dilbeck, P., Burnap, R.
L., and Eaton-Rye, J. J. (2008) Biochemistry, 47,
11637-11646.
70.Yano, J., and Yachandra, V. K. (2008) Inorg.
Chem., 47, 1711-1726.
71.Haumann, M., Liebisch, P., Muller, C., Barra, M.,
Grabolle, M., and Dau, H. (2005) Science, 310,
1019-1021.
72.Messinger, J., Badger, M., and Wydrzynski, T.
(1995) Proc. Natl. Acad. Sci. USA, 92, 3209-3213.
73.Fraser, J. S., van den Bedem, H., Samelson, A.
J., Lang, P. T., Holton, J. M., Echols, N., and Alber, T. (2011)
Proc. Natl. Acad. Sci. USA, 108, 16247-16252.
74.Chapman, H. N., Fromme, P., Barty, A., White, T.
A., Kirian, R. A., Aquila, A., Hunter, M. S., Schulz, J., DePonte, D.
P., Weierstall, U., Doak, R. B., Maia, F. R., Martin, A. V.,
Schlichting, I., Lomb, L., Coppola, N., Shoeman, R. L., Epp, S. W.,
Hartmann, R., Rolles, D., Rudenko, A., Foucar, L., Kimmel, N.,
Weidenspointner, G., Holl, P., Liang, M., Barthelmess, M., Caleman, C.,
Boutet, S., Bogan, M. J., Krzywinski, J., Bostedt, C., Bajt, S.,
Gumprecht, L., Rudek, B., Erk, B., Schmidt, C., Homke, A., Reich, C.,
Pietschner, D., Struder, L., Hauser, G., Gorke, H., Ullrich, J.,
Herrmann, S., Schaller, G., Schopper, F., Soltau, H., Kuhnel, K. U.,
Messerschmidt, M., Bozek, J. D., Hau-Riege, S. P., Frank, M., Hampton,
C. Y., Sierra, R. G., Starodub, D., Williams, G. J., Hajdu, J.,
Timneanu, N., Seibert, M. M., Andreasson, J., Rocker, A., Jonsson, O.,
Svenda, M., Stern, S., Nass, K., Andritschke, R., Schroter, C. D.,
Krasniqi, F., Bott, M., Schmidt, K. E., Wang, X., Grotjohann, I.,
Holton, J. M., Barends, T. R., Neutze, R., Marchesini, S., Fromme, R.,
Schorb, S., Rupp, D., Adolph, M., Gorkhover, T., Andersson, I.,
Hirsemann, H., Potdevin, G., Graafsma, H., Nilsson, B., and Spence, J.
C. (2011) Nature, 470, 73-77.
75.Kirian, R. A., White, T. A., Holton, J. M.,
Chapman, H. N., Fromme, P., Barty, A., Lomb, L., Aquila, A., Maia, F.
R., Martin, A. V., Fromme, R., Wang, X., Hunter, M. S., Schmidt, K. E.,
and Spence, J. C. (2011) Acta Crystallogr. A, 67,
131-140.
76.Hunter, M. S., DePonte, D. P., Shapiro, D. A.,
Kirian, R. A., Wang, X., Starodub, D., Marchesini, S., Weierstall, U.,
Doak, R. B., Spence, J. C., and Fromme, P. (2011) Biophys. J.,
100, 198-206.
77.Johansson, L. C., Arnlund, D., White, T. A.,
Katona, G., Deponte, D. P., Weierstall, U., Doak, R. B., Shoeman, R.
L., Lomb, L., Malmerberg, E., Davidsson, J., Nass, K., Liang, M.,
Andreasson, J., Aquila, A., Bajt, S., Barthelmess, M., Barty, A.,
Bogan, M. J., Bostedt, C., Bozek, J. D., Caleman, C., Coffee, R.,
Coppola, N., Ekeberg, T., Epp, S. W., Erk, B., Fleckenstein, H.,
Foucar, L., Graafsma, H., Gumprecht, L., Hajdu, J., Hampton, C. Y.,
Hartmann, R., Hartmann, A., Hauser, G., Hirsemann, H., Holl, P.,
Hunter, M. S., Kassemeyer, S., Kimmel, N., Kirian, R. A., Maia, F. R.,
Marchesini, S., Martin, A. V., Reich, C., Rolles, D., Rudek, B.,
Rudenko, A., Schlichting, I., Schulz, J., Seibert, M. M., Sierra, R.
G., Soltau, H., Starodub, D., Stellato, F., Stern, S., Struder, L.,
Timneanu, N., Ullrich, J., Wahlgren, W. Y., Wang, X., Weidenspointner,
G., Wunderer, C., Fromme, P., Chapman, H. N., Spence, J. C., and
Neutze, R. (2012) Nat. Methods, 9, 263-265.
78.Koopmann, R., Cupelli, K., Redecke, L., Nass, K.,
Deponte, D. P., White, T. A., Stellato, F., Rehders, D., Liang, M.,
Andreasson, J., Aquila, A., Bajt, S., Barthelmess, M., Barty, A.,
Bogan, M. J., Bostedt, C., Boutet, S., Bozek, J. D., Caleman, C.,
Coppola, N., Davidsson, J., Doak, R. B., Ekeberg, T., Epp, S. W., Erk,
B., Fleckenstein, H., Foucar, L., Graafsma, H., Gumprecht, L., Hajdu,
J., Hampton, C. Y., Hartmann, A., Hartmann, R., Hauser, G., Hirsemann,
H., Holl, P., Hunter, M. S., Kassemeyer, S., Kirian, R. A., Lomb, L.,
Maia, F. R., Kimmel, N., Martin, A. V., Messerschmidt, M., Reich, C.,
Rolles, D., Rudek, B., Rudenko, A., Schlichting, I., Schulz, J.,
Seibert, M. M., Shoeman, R. L., Sierra, R. G., Soltau, H., Stern, S.,
Struder, L., Timneanu, N., Ullrich, J., Wang, X., Weidenspointner, G.,
Weierstall, U., Williams, G. J., Wunderer, C. B., Fromme, P., Spence,
J. C., Stehle, T., Chapman, H. N., Betzel, C., and Duszenko, M. (2012)
Nat. Methods, 9, 259-262.
79.Aquila, A., Hunter, M. S., Doak, R. B., Kirian,
R. A., Fromme, P., White, T. A., Andreasson, J., Arnlund, D., Bajt, S.,
Barends, T. R., Barthelmess, M., Bogan, M. J., Bostedt, C., Bottin, H.,
Bozek, J. D., Caleman, C., Coppola, N., Davidsson, J., DePonte, D. P.,
Elser, V., Epp, S. W., Erk, B., Fleckenstein, H., Foucar, L., Frank,
M., Fromme, R., Graafsma, H., Grotjohann, I., Gumprecht, L., Hajdu, J.,
Hampton, C. Y., Hartmann, A., Hartmann, R., Hau-Riege, S., Hauser, G.,
Hirsemann, H., Holl, P., Holton, J. M., Homke, A., Johansson, L.,
Kimmel, N., Kassemeyer, S., Krasniqi, F., Kuhnel, K. U., Liang, M.,
Lomb, L., Malmerberg, E., Marchesini, S., Martin, A. V., Maia, F. R.,
Messerschmidt, M., Nass, K., Reich, C., Neutze, R., Rolles, D., Rudek,
B., Rudenko, A., Schlichting, I., Schmidt, C., Schmidt, K. E., Schulz,
J., Seibert, M. M., Shoeman, R. L., Sierra, R., Soltau, H., Starodub,
D., Stellato, F., Stern, S., Struder, L., Timneanu, N., Ullrich, J.,
Wang, X., Williams, G. J., Weidenspointner, G., Weierstall, U.,
Wunderer, C., Barty, A., Spence, J. C., and Chapman, H. N. (2012)
Opt. Express, 20, 2706-2716.
80.Barty, A., Caleman C., and Chapman, H. N., et al.
(2012) Nat. Photonics, 6, 35-40.
81.Lomb, L., Barends, T. R. M., Kassemeyer, S.,
Aquila, A., Epp, S. W., Erk, B., Foucar, L., Hartmann, R., Rudek, B.,
Rolles, D., Rudenko, A., Shoeman, R. L., Andreasson, J., Bajt, S.,
Barthelmess, M., Barty, A., Bogan, M. J., Bostedt, C., Bozek, J. D.,
Caleman, C., Coffee, R., Coppola, N., DePonte, D. P., Doak, R. B.,
Ekeberg, T., Fleckenstein, H., Fromme, P., Gebhardt, M., Graafsma, H.,
Gumprecht, L., Hampton, C. Y., Hartmann, A., Hauser, G., Hirsemann, H.,
Holl, P., Holton, J. M., Hunter, M. S., Kabsch, W., Kimmel, N., Kirian,
R. A., Liang, M., Maia, F. R. N. C., Meinhart, A., Marchesini, S.,
Martin, A. V., Nass, K., Reich, C., Schulz, J., Seibert, M. M., Sierra,
R., Soltau, H., Spence, J. C. H., Steinbrener, J., Stellato, F., Stern,
S., Timneanu, N., Wang, X., Weidenspointner, G., Weierstall, U., White,
T. A., Wunderer, C., Chapman, H. N., Ullrich, J., Struder, L., and
Schlichting, I. (2011) Phys. Rev. B, 84,
214111-214116.
82.Caleman, C., Huldt, G., Maia, F. R., Ortiz, C.,
Parak, F. G., Hajdu, J., van der Spoel, D., Chapman, H. N., and
Timneanu, N. (2011) ACS Nano, 5, 139-146.
83.Neutze, R., Wouts, R., van der Spoel, D.,
Weckert, E., and Hajdu, J. (2000) Nature, 406,
752-757.
84.Kern, J., Alonso-Mori, R., Hellmich, J., Tran,
R., Hattne, J., Laksmono, H., Glockner, C., Echols, N., Sierra, R. G.,
Sellberg, J., Lassalle-Kaiser, B., Gildea, R. J., Glatzel, P.,
Grosse-Kunstleve, R. W., Latimer, M. J., McQueen, T. A., DiFiore, D.,
Fry, A. R., Messerschmidt, M., Miahnahri, A., Schafer, D. W., Seibert,
M. M., Sokaras, D., Weng, T. C., Zwart, P. H., White, W. E., Adams, P.
D., Bogan, M. J., Boutet, S., Williams, G. J., Messinger, J., Sauter,
N. K., Zouni, A., Bergmann, U., Yano, J., and Yachandra, V. K. (2012)
Proc. Natl. Acad. Sci. USA, 109, 9721-9726.
85.Yano, J., and Yachandra, V. K. (2009)
Photosynth. Res., 102, 241-254.
86.Chen, L. X. (2005) Annu. Rev. Phys. Chem.,
56, 221-254.
87.Bressler, C., and Chergui, M. (2010) Annu.
Rev. Phys. Chem., 61, 263-282.
88.March, A. M., Stickrath, A., Doumy, G., Kanter,
E. P., Krassig, B., Southworth, S. H., Attenkofer, K., Kurtz, C. A.,
Chen, L. X., and Young, L. (2011) Rev. Sci. Instrum., 82,
073110-073118.
89.Vanko, G., Glatzel, P., Pham, V. T., Abela, R.,
Grolimund, D., Borca, C. N., Johnson, S. L., Milne, C. J., and
Bressler, C. (2010) Angew. Chem. Int. Ed. Engl., 49,
5910-5912.
90.Messinger, J., Robblee, J. H., Bergmann, U.,
Fernandez, C., Glatzel, P., Visser, H., Cinco, R. M., McFarlane, K. L.,
Bellacchio, E., Pizarro, S. A., Cramer, S. P., Sauer, K., Klein, M. P.,
and Yachandra, V. K. (2001) J. Am. Chem. Soc., 123,
7804-7820.
91.Pushkar, Y., Long, X., Glatzel, P., Brudvig, G.
W., Dismukes, G. C., Collins, T. J., Yachandra, V. K., Yano, J., and
Bergmann, U. (2010) Angew. Chem. Int. Ed. Engl., 49,
800-803.
92.Lancaster, K. M., Roemelt, M., Ettenhuber, P.,
Hu, Y., Ribbe, M. W., Neese, F., Bergmann, U., and DeBeer, S. (2011)
Science, 334, 974-977.
93.Mori, R. A., Paris, E., Giuli, G., Eeckhout, S.
G., Kavcic, M., Zitnik, M., Bucar, K., Pettersson, L. G., and Glatzel,
P. (2010) Inorg. Chem., 49, 6468-6473.
94.Mori, R. A., Paris, E., Giuli, G., Eeckhout, S.
G., Kavcic, M., Zitnik, M., Bucar, K., Pettersson, L. G. M., and
Glatzel, P. (2009) Analyt. Chem., 81, 6516-6525.
95.Smolentsev, G., Soldatov, A. V., Messinger, J.,
Merz, K., Weyhermuller, T., Bergmann, U., Pushkar, Y., Yano, J.,
Yachandra, V. K., and Glatzel, P. (2009) J. Am. Chem. Soc.,
131, 13161-13167.
96.Beckwith, M. A., Roemelt, M., Collomb, M. N.,
DuBoc, C., Weng, T. C., Bergmann, U., Glatzel, P., Neese, F., and
DeBeer, S. (2011) Inorg. Chem., 50, 8397-8409.
97.Glatzel, P., and Bergmann, U. (2005) Coord.
Chem. Rev., 249, 65-95.
98.Bergmann, U., Horne, C. R., Collins, T. J.,
Workman, J. M., and Cramer, S. P. (1999) Chem. Phys. Let.,
302, 119-124.
99.Bergmann, U., and Glatzel, P. (2009)
Photosynth. Res., 102, 255-266.
100.Alonso-Mori, R., Kern, J., Gildea, R. J.,
Sokaras, D., Weng, T. C., Lassalle-Kaiser, B., Tran, R., Hattne, J.,
Laksmono, H., Hellmich, J., Glockner, C., Echols, N., Sierra, R. G.,
Schafer, D. W., Sellberg, J., Kenney, C., Herbst, R., Pines, J., Hart,
P., Herrmann, S., Grosse-Kunstleve, R. W., Latimer, M. J., Fry, A. R.,
Messerschmidt, M. M., Miahnahri, A., Seibert, M. M., Zwart, P. H.,
White, W. E., Adams, P. D., Bogan, M. J., Boutet, S., Williams, G. J.,
Zouni, A., Messinger, J., Glatzel, P., Sauter, N. K., Yachandra, V. K.,
Yano, J., and Bergmann, U. (2012) Proc. Natl. Acad. Sci. USA,
109, 19103-19107.
101.Kern, J., Alonso-Mori, R., Tran, R., Hattne,
J., Gildea, R. J., Echols, N., Glockner, C., Hellmich, J., Laksmono,
H., Sierra, R. G., Lassalle-Kaiser, B., Koroidov, S., Lampe, A., Han,
G., Gul, S., Difiore, D., Milathianaki, D., Fry, A. R., Miahnahri, A.,
Schafer, D. W., Messerschmidt, M., Seibert, M. M., Koglin, J. E.,
Sokaras, D., Weng, T. C., Sellberg, J., Latimer, M. J.,
Grosse-Kunstleve, R. W., Zwart, P. H., White, W. E., Glatzel, P.,
Adams, P. D., Bogan, M. J., Williams, G. J., Boutet, S., Messinger, J.,
Zouni, A., Sauter, N. K., Yachandra, V. K., Bergmann, U., and Yano, J.
(2013) Science, 340, 491-495.