REVIEW: Biocatalytic Synthesis of Conducting Polymers and Prospects for Its Application
G. V. Otrokhov1, O. V. Morozova1, I. S. Vasil’eva1, G. P. Shumakovich1, E. A. Zaitseva2, M. E. Khlupova1, and A. I. Yaropolov1*
1A. N. Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, 119071 Moscow, Russia; E-mail: yaropolov@inbi.ras.ru2Lomonosov Moscow State University, Chemical Faculty, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received April 26, 2013
Enzymatic methods of synthesis of conducting polymers, physicochemical properties of the resulting products, and mechanisms of the reactions are considered. The enzymes involved in oxidative polymerization of monomers are briefly characterized. Examples of practical application of enzymatically synthesized conducting polymers are given.
KEY WORDS: conducting polymer, physicochemical properties, enzymatic polymerization, redox mediator, laccase, peroxidase, practical applicationDOI: 10.1134/S0006297913130117
Abbreviations: ABTS, diammonium 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonate); APS, ammonium persulfate; CP, conducting polymers; CSA, camphorsulfonic acid; EDOT, 3,4-ethylenedioxythiophene; HP, horseradish peroxidase; NHE, normal hydrogen electrode; PAMPS, poly(2-acrylamido-2-methyl-1-propanesulfonic acid); PANI, polyaniline; PEDOT, poly(3,4-ethylenedioxythiophene); PP, polypyrrole; SDBS, sodium dodecylbenzenesulfonate.
Virtually all natural polymers including polysaccharides, lignin,
proteins, DNA, etc. are synthesized in vivo with involvement of
enzymes. However, these high molecular weight compounds, as well as the
overwhelming majority of chemically synthesized polymers, are not
conducting. A new class of conductive polymers was discovered in the
end of the last century. The discovery in 1977 of a conducting linear
organic polymer, polyacetylene, stimulated synthesis and study of new
compounds termed “organic metals” [1].
These materials combine such features of organic polymers as low
density, stability, and resistance to corrosion, with conductivity
inherent in metals. And the desired features can be set on the
molecular level through modifications of the initial monomers.
Most frequently, conducting polymers (CP) are synthesized by chemical and electrochemical polymerization. But chemical synthesis is rather unfavorable ecologically because it is performed in medium with high acidity and requires large amounts of an oxidizer, and its reduction products must be utilized. Moreover, chemical oxidation of monomers is often associated with generation of toxic byproducts. Chemical polymerization occurs by an autocatalytic mechanism with a prolonged induction period. Electrochemical polymerization is also performed in medium with high acidity, and, besides, it needs a conducting electrode with geometrically limited dimensions. Therefore, using natural biocatalysts for in vitro synthesis of polymers to decrease the load on the environment (by minimization of byproducts) seems to be very attractive and promising.
An enzymatic approach has been proposed recently for polymerization of monomers, and this approach in many respects corresponds to the requirements of “green” chemistry and can become an alternative to traditional approaches of CP synthesis. This approach can eliminate some troubles unavoidable during chemical and electrochemical syntheses. The biocatalytic synthesis occurs under “soft” conditions (moderately acidic pH values of the reaction mixture, room temperature), its kinetics can be controlled, and the resulting polymer is not polluted with products of disintegration of the oxidizer. Moreover, toxic byproducts of the reaction are also absent. Because polymerization of both natural and artificial monomers with an undivided electron pair goes through a radical mechanism, oxidoreductases catalyzing similar reactions in vivo are used as biocatalysts of such reactions.
In the present review, enzymatic methods of synthesis of conducting polymers, physicochemical properties of the resulting products, and mechanisms of the reactions will be described. The enzymes involved in the oxidative polymerization of monomers will be briefly characterized, and examples of practical application of enzymatically synthesized conducting polymers will be given.
CONDUCTING POLYMERS AND METHODS OF THEIR PRODUCTION
Conducting polymers are highly delocalized π-electron systems with alternating single and double bonds, which can be easily oxidized or reduced [2]. The electron conductivity of these polymers is called “intrinsic” because it is due to the presence in their molecular structure of electric charges capable of translocating along the polymer chain without involvement of other conducting materials such as metals or graphite. In the majority of cases, these materials in the neutral state are dielectrics, and they acquire the ability for conducting only upon the interaction of dopant molecules with the polymer chain regions carrying an unpaired electron. This process is called “doping”. Note that the term doping used for conducting polymers is somewhat different from this term used for inorganic semiconductors. The difference is determined by the amount of the dopant, which in some cases can be 50% of the conducting polymer weight. The dopant interacts with the polymer but does not directly participate in the charge transfer mechanism. The best-known conducting polymers are polyaniline, polypyrrole, and polyethylenedithiophene. Their structural formulas are presented in Fig. 1.
Fig. 1. Structural formulas of the repeated unit of polyaniline and its possible redox states (a), of polypyrrole (b), and of poly(3,4-ethylenedioxythiophene) (c).
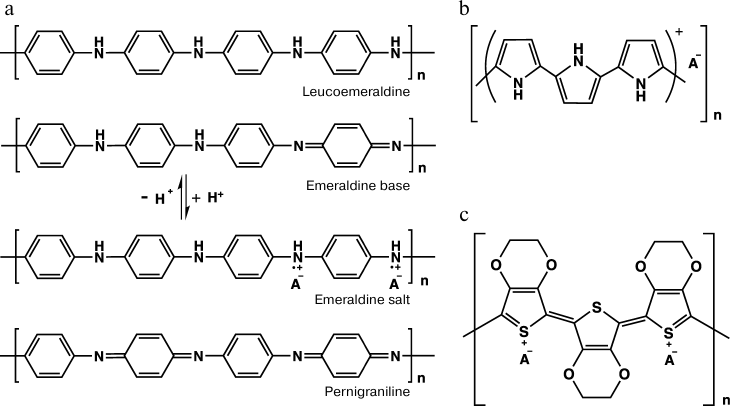
Polyaniline. Among conducting polymers, polyaniline (PANI) attracts attention due to the low price of the monomer, simple preparation, resistance under environmental conditions in both the doped and non-doped states, and also due to the possibility of changing its optoelectronic properties on changes in the medium pH and under the influence of an electric field. The difference in the composition and structure of the repeated unit of PANI results in several redox states of the polymer – from the fully reduced leucoemeraldine to the fully oxidized pernigraniline (Fig. 1a). Different states of PANI can easily transform from one to another via redox transformations. In the semi-oxidized state (emeraldine salt) polyaniline is conducting [3]. Some data suggest that pernigraniline is also conducting [4], but this form is extremely unstable and can be easily hydrolyzed.
There are two main methods of synthesizing PANI: direct oxidation of aniline with chemical oxidizers, and electrooxidation of the monomer on a conducting electrode.
The dark-green emeraldine salt of PANI can be easily prepared by polymerization of aniline in aqueous medium under the influence of such oxidizers as ammonium persulfate (APS), potassium iodate, hydrogen peroxide, potassium dichromate, or ferric perchlorate [5]. The main advantage of the chemical synthesis is its simplicity and the possibility of producing large amounts of PANI with high yield, as well as the low price of the oxidizer. As a rule, the reaction is performed in highly acidic medium at pH values from 0.0 to 2.0. Most frequently, to prevent polymer degradation (overoxidation), the monomer and oxidizer are used in stoichiometrically equal concentrations [5]. To obtain a high molecular weight polymer, aniline is usually chemically polymerized at low temperatures (from 0 to 5°C). Serious shortcomings of this method are the necessity of using large amounts of the oxidizer, the high acidity of the reaction medium, and also the autocatalytic character of the polymerization reaction. The resulting product is essentially unfit for technological application because it is insoluble in common solvents. The poor solubility of PANI is caused by intermolecular hydrogen bonds between amino and imino groups of the polymer chains [6].
To improve the technological applicability of PANI, it was proposed to use “hard” or “soft” templates for PANI synthesis. “Hard” templates are metals, various carbon or plastic materials, and silicon and metal oxides with macro- to nano-dimensions. “Soft” templates include negatively charged polyelectrolytes, micelles of anionic surface-active substances, and also emulsions of electroneutral polymers. Composites based on these materials and PANI are significantly more promising for technological application than pure PANI.
To electrochemically polymerize the monomer, it is oxidized with the anode. The electropolymerization of aniline results in formation of a thin film of the conducting polymer on the electrode surface. The impossibility of covering with polyaniline of a non-conducting surface and limitations because of the electrode area are shortcomings of the method.
Polypyrrole. Another well-studied representative of conducting polymers is polypyrrole (PP), which has a high conductivity, stability in the conducting state, and interesting redox and electromechanical features [7]. The structure of PP is presented in Fig. 1b. As differentiated from PANI, polypyrrole is moderately conducting in neutral media and is a biocompatible polymer [8]. PP has a resonance structure. In the neutral state, PP is a dielectric and acquires conductivity only upon oxidation. The charge is usually delocalized on several pyrrole units and can form a cation-radical (polaron) or a dication (bipolaron), which are charge carriers and are produced upon doping of the polymer [9, 10]. Polarons and bipolarons in PP embrace three or four units of the polymer chain [11]. Depending on conditions of the synthesis, the repeated units of PP can include structural units of 2-pyrrolidinone [12].
Experimental data on the electrical properties of PP indicate that polypyrrole is not a linear polymer, but it must be considered as a reticulated one containing about 30% of monomeric units in its side chains and interchain crosslinks [13].
The solubility of PP is limited by its rigid structure. There have been attempts to increase PP solubility through modifications of the pyrrole rings in the 3,4-positions with alkyl groups or using N-substituted pyrrole derivatives [14, 15]. The solubility of pyrrole can also be increased by using “soft” templates, such as sodium dodecylbenzenesulfonate (SDBS) [16], sodium di(2-ethylhexyl)sulfocyanate [17], or poly(styrene)sulfonate [18].
Polypyrrole with its block structure can be produced by oxidative polymerization of the monomer in aqueous or anhydrous media [19-22] with involvement of chemical oxidizers or by electrochemical polymerization. As oxidizers, aqueous or anhydrous FeCl3 or other salts of trivalent iron are used [22, 23] as well as APS [21]. The yield and conductivity of the resulting PP is determined by many factors: the choice of solvent and oxidizer, the initial molar ratio between pyrrole and the oxidizer, and the polymerization time and temperature [19, 24]. Decrease in the polymerization time and in the reaction temperature to 0-5°C leads to increase in the PP conductivity [25]. Moreover, the PP conductivity strongly depends on the nature of the solvent and on the redox potential of the reaction medium [26].
Electropolymerization of pyrrole has been performed in both aqueous and anhydrous media (acetonitrile, propylene carbonate, dichloromethane, etc.) [27, 28]. The production of PP by electrochemical polymerization is a complex process depending on many factors. The electrochemical reaction and characteristics of the PP film strongly depend on the nature of the electrolyte, the monomer/electrolyte ratio, the cell construction, the solvent, the electrode material, the applied potential or the rate of its change, and the medium temperature and pH [29-31]. The counter ion also plays an important role.
Poly(3,4-ethylenedioxythiophene). Heteroaromatic thiophene has poor solubility, and therefore its substituted derivatives were synthesized [32]. The thiophene derivative 3,4-ethylenedioxythiophene (EDOT) is most widely used for synthesizing a conducting polymer [33]. The structure of the polymer poly(3,4-ethylenedioxythiophene) (PEDOT) synthesized on its basis is presented in Fig. 1c [7].
Chemical methods of PEDOT synthesis can be subdivided into two groups: oxidizing polymerization of the monomer (3,4-ethylenedioxythiophene, EDOT) and the cross-combination of 2,5-dihalogen derivatives of EDOT [34, 35].
The oxidative polymerization of the monomer results in a doped PEDOT. For the polymerization, FeCl3, Fe(OTs)3, Fe2(SO4)3 [36-38] and APS are used as oxidizers. Various templates are used to produce a resistant aqueous dispersion of PEDOT, similar to the synthesis of PANI and PP [39-42].
The electrochemical polymerization of EDOT produces on the anode a thin light-blue film of the polymer [43, 44]. The polymerization is performed in media of various electrolytes, including polyelectrolytes [45-47], or in an aqueous solution of micelles of surfactants [41, 42, 48, 49]. Using this method, polymeric films with known thickness and high purity can be produced rather rapidly.
ENZYMES INVOLVED IN OXIDATIVE POLYMERIZATION OF MONOMERS
There is now steadily increasing interest in enzymatic production of organic compounds in vitro [50, 51]. This interest is due to the possibility of using many enzymes not only for transformation of their natural substrates, but also for modification of many artificial substances that can be used as the bases for production of various new compounds and materials [52, 53]. Such enzymes, in particular, include oxidoreductases, which play an important role in the synthesis of the natural polymer lignin. The active centers of the majority of these enzymes include metal ions.
Many studies in the field of fine organic synthesis deal with oxidoreductases, in particular with peroxidases and laccases from different sources, because they can catalyze oxidative polymerization of compounds with an undivided electron pair by the radical mechanism with water resulting from the reduction of the oxidizer. In this review, only the main and most important biochemical and catalytic characteristics will be given for the above-mentioned enzymes, those necessary for choosing conditions for synthesis of conducting polymers and understanding of the reaction mechanism.
Peroxidase (EC 1.11.1.7; donor:H2O2 oxidoreductase) is an enzyme widely distributed in plant and animal tissues, fungi, and bacteria. Peroxidases are involved in the generation and subsequent lignification of cell walls, regulation of auxin level through its catabolism, tissue protection against damage and infectious pathogenic microorganisms, and oxidation of indolylacetic acid [54, 55]. However, the physiological role of peroxidases is not clear in detail. Peroxidases isolated from horseradish roots [56], soya beans [57, 58], palm leaves [59], tobacco leaves [60], and tomatoes [61] are the best studied and described in the literature. Peroxidases from various sources usually include several isoenzymes with different biochemical and kinetic parameters.
The prosthetic group ferriprotoporphyrin IX (heme) is in the active center of the enzyme. The Fe3+ ion has four coordination bonds with porphyrin atoms and one bond with the nitrogen atom of the imidazole ring. The sixth coordination position remains vacant, but during catalysis this position is occupied by hydrogen peroxide coordinating the ferric ion.
Peroxidases catalyze oxidation of many reducing substrates. Hydrogen peroxide and some organic peroxides act as oxidizing substrates. Substrates of peroxidases can be divided into two groups [62]. The first group substrates (they mainly include the inorganic ions ferrocyanide, iodide, nitrite, etc. and also ABTS – diammonium 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonate)) interact immediately with the heme. The second group substrates (aromatic amines, phenols, and indoles) do not react directly with the heme. Consequently, an electron transport chain including functional groups of the protein moiety of the molecule has to exist.
The ferric ion of the prosthetic group is able not only to reduce hydrogen peroxide, but it can also activate oxidation of different substrates. The reaction of native peroxidase with an oxidizing substrate results in a spectrophotometrically detectable intermediate, so-called compound I (EI). This compound has been established to be an oxidized derivative of the enzyme that has received two oxidized equivalents from the hydrogen peroxide molecule, i.e. EI formally contains a FeV center [63]. Reducing substrates are oxidized by a one-electron mechanism, and the reaction has two stages through another intermediate, so-called compound II (EII), which contains one oxidized equivalent compared to the native enzyme, i.e. EII is formally the FeIV form of the enzyme. Altogether, for peroxidase five different formal degrees of oxidation of iron are known: FeVI is compound III, FeV is compound I, FeIV is compound II, FeIII is the native enzyme, and FeII is the ferro-enzyme. In the majority of reactions the main roles are played by the native enzyme and compounds I and II, and the mechanism of reactions catalyzed by peroxidases is described by the following scheme:
E + H2O2 → EI + H2O
EI + DH2 → EII + DH•
EII + DH2 → E + DH•,
where DH2 is a donor of hydrogen, and DH• is a radical product of the reaction.
Compound III is produced as a result of the interaction of the native enzyme with an excess of hydrogen peroxide, and it is catalytically inactive (because of prosthetic group destruction). This circumstance has to be taken in consideration when enzymatic reactions are studied.
Laccase (EC 1.10.3.2, p-diphenol:oxygen oxidoreductase) is another representative of oxidoreductases considered in this review. Laccase is a blue copper-containing oxidase found in plants and fungi. The enzyme was first detected in the Japanese lacquer tree Rhus vernicifera [64]. The majority of laccases that have been studied in detail were isolated from different fungi [65, 66]. Fungal laccases are thought to be mainly responsible for generation and degradation of lignin, morphogenesis of fungi, the parasite/host interaction, and also anti-stress protection. The majority of fungi produce several isoforms and isoenzymes of laccases. Although the majority of fungal laccases are monomeric proteins, in the literature there are descriptions of enzymes consisting of several subunits. The molecular weight of the monomer varies from 50 to 130 kDa. Laccases are glycoproteins. The carbohydrate moiety, usually consisting of mannose, N-acetylglucosamine, and galactose, represents from 10 to 45% of the weight of the protein. The carbohydrate moiety is believed to be responsible for the stability of the enzyme [67, 68].
Laccases can catalyze the oxidation by dioxygen of various compounds including ortho- and para-phenols, polyphenols, aminophenols, polyamines, lignins, aryldiamines, and some inorganic ions. This is associated with the four-electron reduction of molecular oxygen to water [68-72]. Inorganic substrates of laccases are electron donors, whereas organic substrates (except ABTS) are donors of hydrogen atoms, which are detached from an organic molecule during the enzymatic catalysis with production of a radical.
Fungal laccases usually have a bell-shaped pH optimum in the interval of 3.5-5.0 for substrates that are hydrogen donors [73-76]. The pH optima of plant laccases during oxidation of hydrogen donor substrates are different from those of fungal laccases. Thus, the catalytic activity optimum of laccase from Rhus vernicifera for hydrogen donor substrates is within neutral or weakly alkaline pH values [77].
The temperature optimum for laccases is usually not different from that of other extracellular enzymes of the lignolytic complex and is within the range 50-70°C [65, 68].
The active center of laccases contains four copper ions. One copper ion belongs to the first type (T1), whereas the other ions form a tri-nuclear T2/T3 cluster containing one copper ion of the second type (T2) and two copper ions of the third type (T3) [78, 79]. The distance between the T1 copper ion and the T2/T3 cluster is about 12 Å, and the distance between copper ions T2 and T3 is about 4 Å. The T1 center of laccases is a primary acceptor of electrons from donor substrates. For some laccases, the redox potential of the T1 center has been determined, and for the majority of fungal laccases it is ~750-780 mV (relatively to the normal hydrogen electrode (NHE)), and for plant laccases the potential is 420-440 mV (relatively to the NHE) [80]. The redox potential values of the T1 center of laccases are very important for the catalytic oxidation of various substrates.
Catalysis with laccases has three main stages: reduction of the copper ion of the T1 center by the donor substrate; intramolecular electron transfer (over ~12 Å) from the T1 center onto the T2/T3 cluster; activation of molecular oxygen and its four-electron reduction to water on the T2/T3 cluster [81-83].
Laccases are believed to be capable of catalyzing the oxidation of compounds that have ionization potential close or slightly higher than the redox potential of the primary acceptor of electrons from laccases – the copper ion of the T1 center. However, it has been shown [84] that using redox mediators the enzyme makes possible the oxidation of compounds that cannot be oxidized under the influence laccases alone. Redox mediators are low molecular weight substrates of laccases, and their enzymatic oxidation results in highly reactive products capable of nonenzymatic oxidation of various compounds that are not substrates of the enzyme. This is associated with reduction of the oxidized mediator to its initial form, and thus a closed cycle results [84, 85].
In addition to the above-described oxidoreductases, for synthesis of conducting polymers the copper-containing “blue” oxidase bilirubin oxidase [86], FAD-dependent glucose oxidase [87], and lactate oxidase [88] can also be used.
ENZYMATIC SYNTHESIS OF CONDUCTING POLYMERS AND THEIR
PHYSICOCHEMICAL PROPERTIES
The production of conducting polymers by catalysis with enzymes attracts the attention of researchers in various countries. Many examples of such approaches are considered in reviews [89-93]. The results published during the last years, as well as our own data, will be summarized below.
For synthesis of conducting polymers, especially that of PANI, horseradish peroxidase (HP) is mainly used because this enzyme is commercially available and well known. However, the use of HP is associated with some shortcomings. To produce a conducting polyaniline with linear structure, the polymerization reaction has to be conducted in strongly acidic medium (pH < 3.0). But it is known that horseradish peroxidase is unstable at pH < 4.0 and rapidly loses its initial activity as a result of dissociation into the heme and the apoenzyme [62]. Moreover, it should be noted that this enzyme, like other peroxidases, is extremely sensitive to the concentration of hydrogen peroxide: at H2O2 concentration higher than 1 mM, the enzyme is inactivated. Therefore, during the polymerization hydrogen peroxide must be added into the reaction mixture gradually. To prevent the inactivation of the biocatalyst at acidic pH values under conditions of PANI synthesis, pH-stable peroxidases from soybeans [58, 94] and palm leaves [95] have been used.
During recent years a fungal laccase with high redox potential was proposed for the synthesis of conducting PANI. As differentiated from peroxidase-catalyzed polymerization, during the laccase-catalyzed polymerization of monomers atmospheric oxygen acts as the oxidizer, and this simplifies the synthesis. Moreover, laccases from basidial fungi are active and stable at acidic pH values [96].
Template-free synthesis of conducting polymers. One of the first cases of using enzymes for synthesis of conducting polyaniline has been described in a US patent [97]. Horseradish peroxidase was used as a biocatalyst for the oxidative polymerization and toluene sulfonic acid was used as a dopant. The reaction was performed without a template during 24 h at reaction medium pH 2.2. The reaction product was a dark-green powder that corresponded to PANI emeraldine salt with rather high conductivity of 1-2 S/cm. However, the catalysis of this reaction with native HP seems doubtful because at the solution pH, during the synthesis the holoenzyme dissociates into the apoenzyme and heme and completely loses activity within few minutes. Perhaps under these conditions the reaction can be catalyzed by the heme.
In work [98], the acid-resistant peroxidase from soybeans was used for template-free synthesis of PANI. The reaction scheme is presented in Fig. 2.
Fig. 2. Scheme of enzymatic template-free synthesis of PANI in the presence of peroxidase.
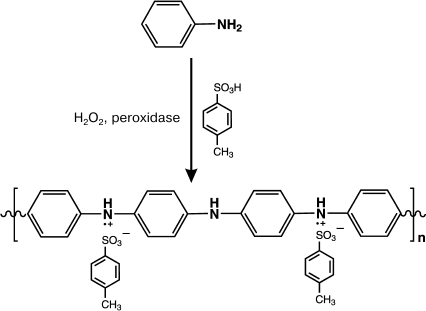
The reaction was performed in aqueous or aqueous–organic solutions (dioxane–water, 30 : 70 vol. %) at pH values from 3.0 to 5.0 with toluene sulfonic acid as an acidic dopant. The enzymatic polymerization at pH 3.0 produced a dark-green precipitate with yield of 71%. The conductivity of the synthesized polymer was 2.4 S/cm. At pH 5.0, the enzymatic polymerization of aniline resulted in a dark-brown water-insoluble product with conductivity <10–6 S/cm. Studies on the resulting PANI samples by Fourier transform IR-spectroscopy revealed that at acidic pH values of the reaction medium a polymer with the linear structure was produced with preferential combination of aniline molecules in the 1,4-position. Polyaniline synthesized under these conditions had characteristic bands in the FTIR spectrum at the frequencies of 1598 and 1500 cm–1, respectively, which are specific for quinoid diimine and phenylene diamine groups of the repeated link of the PANI. The PANI sample synthesized at pH 5.0 had bands in the region of 700-750 cm–1 that corresponded to 1,2-substitutions in the aromatic ring of aniline and characterized branched polymers.
In work [99], glucose oxidase was used for the enzymatic synthesis of PANI. The air-saturated aniline-containing reaction solution was supplemented with glucose as a substrate for glucose oxidase, and the enzymatic oxidation of glucose resulted in production of hydrogen peroxide (that acted as an oxidizer in the reaction of aniline polymerization) and of gluconic acid, which acted as a weak dopant. UV-visible spectra of the PANI synthesized under the influence of glucose oxidase corresponded to a branched polymer.
A similar approach was used for synthesis of polypyrrole catalyzed by lactate oxidase [100]. The enzymatic oxidation of lactate with air oxygen resulted in production of hydrogen peroxide, which acted as an oxidizer in the reaction of pyrrole polymerization.
In work [101], PANI was synthesized with bilirubin oxidase immobilized on various solid surfaces (glass and plastic plates) on which the monomer was polymerized under the influence of dioxygen. As a result, a thin film of conducting PANI was produced. Structural studies revealed that this polymer had partially branched structure.
Synthesis of conducting polymers using “soft” templates. As noted above, polyaniline and other CPs are not thermoplastic, their handling is difficult, and thus they are not technologically convenient for application. Therefore, as a rule, for practice and also for studies on the monomer polymerization the template synthesis of CPs is used. Depending on the particular goal, enzymatic synthesis can be used for production of composites based on hard templates and of CPs by the in situ polymerization of a monomer or for obtaining stable aqueous dispersions of polymers. To produce these dispersions, such “soft” templates are used as polystyrene sulfonate [102-104], poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (PAMPS) [105, 106], polyvinyl sulfonic acid [107], DNA [108, 109], neutral steric stabilizers such as polyvinyl alcohol [110], micelles of surfactants – SDBS [111-113] and sodium dodecyl diphenyl oxide disulfonate [114], anionic vesicles consisting of SDBS and decanoic acid [115].
During polymerization of monomers, templates are responsible for four main functions: 1) they provide the “local” surrounding with pH value, charge density, and monomer concentration different from those in the solution volume; 2) as a result of electrostatic interaction of positively charged protonated aniline molecules with the negatively charged template, the monomer molecules are oriented in an orderly manner on the template, and this provides preferential growth of the polymer chain according to the “head-to-tail” type with minimal branching; 3) the presence of negatively charged polyelectrolytes or surfactant micelles allows researchers to produce highly dispersed CPs in aqueous and aqueous–organic solutions and also to deposit thin layers of PANI onto conducting and non-conducting surfaces of various nature and shape; 4) the template molecules act as charge-compensating anions of the major chain of the polymer.
Properties of the resulting composite materials greatly depend on the template on which the monomer is enzymatically polymerized. In work [116], the influence of the template on conducting PANI synthesis and properties was studied in detail. As a catalyst, HP was used and aniline was polymerized at pH 4.3 where the enzyme remained stable and catalytically active. PANI synthesized using a polycationic (poly-diallyl dimethylammonium chloride) or a neutral (polyethylene glycol) template had physicochemical properties similar to those of a non-conducting branched polymer produced at pH > 4.0 in the absence of template. Using other cationic and neutral templates such as polyvinyl alcohol and polyvinylamine gave the same result in the absence of strongly acidic dopants.
On using polysulfonic acids, DNA, RNA, and various micellar solutions of anionic surfactants, conducting PANIs were produced with different physicochemical properties and conductivities.
For enzymatic template synthesis of PANIs, acid-resistant peroxidases from soybeans [117], palm leaves [118], and chloroperoxidase [119, 120] have been used. A conducting PANI was also produced using a high redox potential fungal laccase from Trametes hirsuta (E0T1 = 0.78 V with respect to the NHE) on PAMPS, polystyrene sulfonate, and SDBS templates [106, 121]. It should be noted that the low redox potential laccase from lacquer tree Rhus vernicifera latex (E0T1 = 0.42 V with respect to the NHE) did not catalyze aniline polymerization because of the great difference in ionization potentials of aniline and the primary acceptor of electrons – the plant laccase T1 center.
Comparison of the physicochemical properties of PANI synthesized using the templates under the influence of peroxidase from soybeans [98] and fungal laccase [106] with properties of the polymer produced under the same conditions by the traditional chemical method with APS revealed similarity of their physicochemical properties, including the conductivity.
To repeatedly use the enzymes, in some works it was proposed to synthesize the conducting PANI under the influence of peroxidase [122] and laccase [123] immobilized on solid carriers or in a biphasic system based on an ionic fluid [124]. In all cases, a conducting PANI was produced very similar in properties to the polymer synthesized under the influence of a homogenous biocatalyst.
During recent years, great interest has been given to synthesis of chiral conducting polymers because they are very promising for practical application in different fields, e.g. for creating surface-modified electrodes, electrochemical asymmetric synthesis, chiral chromatography, and chiral membrane separating technology [125, 126]. On synthesizing chiral conducting polymers, enzymes can provide not only the ecological purity of the process but also the obtaining of a final product with unusual properties. In work [127], a pseudo-soluble chiral PANI was produced by the template approach using HP as the catalyst. A weak polyacrylic acid was used as the template and S or R optical isomers of camphorsulfonic acid (CSA) were used as dopants. Because the PANI molecule does not have asymmetric carbon atoms, a spiral-like conformation of the polymer is due to the interaction with a corresponding CSA enantiomer. The resulting conducting PANI had very interesting optoelectronic properties. HP played an important role in the polymerization of aniline, and PANI produced under its catalysis displayed the same optical activity without depending on the optical activity of the CSA isomers used.
The biocatalytic synthesis of chiral PANI with involvement of another enzyme, the fungal high redox potential fungal laccase from T. hirsuta and the same CSA enantiomers as low molecular weight dopants, gave different results [96]. Depending on the dopant used, the resulting PANI had different optical activities (Fig. 3). The treatment of the enzymatically synthesized PANI doped with S-CSA (curve 1) by a solution of R-CSA changed the optical activity of the polymer (curve 3). The subsequent treatment of PANI with S-CSA solution produced an optically active polymer PANI/S-CSA (curve 4). Thus, the oxidative polymerization of aniline catalyzed by laccase, as differentiated from the catalysis by HP, resulted in PANI/CSA products with optical activities depending on the optical activity of the chiral dopant. A similar effect was observed if PANI was enzymatically synthesized in the presence of R-CSA and redoped under the influence of another dopant isomer.
Fig. 3. CD spectra of doped polyaniline dispersion: PANI/S-CSA as a product of enzymatic synthesis (1); polymers treated upon synthesis with solutions of S-CSA (2) and R-CSA (3); the polymer from (3) after treatment with R-CSA solution (4).
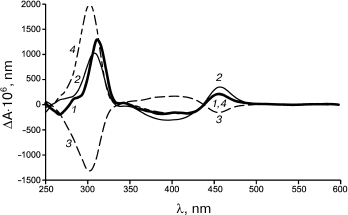
Note that upon the chemical synthesis of PANI doped with CSA optical isomers, the chirality of the synthesized polymer was also different [128, 129].
Optically active conducting PANI can be also produced by enzymatic synthesis on a chiral template of DNA. Work [108] describes the production of an optically active PANI on a long-chain DNA double helix at pH 4.3 catalyzed by HP. It was shown by circular dichroism (CD) spectroscopy that the chiral PANI doped with phosphate groups of DNA had positive optical activity, and in the dedoped state at pH 9.0 the optical activity of the polymer disappeared.
Chiral PANI was also synthesized on a short-chain DNA template using HP and myeloperoxidase-11 as biocatalysts of the oxidative polymerization of aniline [130]. CD spectroscopy showed that in conducting inter-polymer PANI/DNA complexes, PANI had different optical activity in the region of 370-450 nm depending on the biocatalyst used. The difference in the stoichiometry of the PANI/DNA complexes suggests an important role of the biocatalyst in the direction of twisting the conducting PANI spiral on the DNA template. The binding of the electrically active PANI with the biological template and a possibility of controlling its conformation seems fundamentally promising for basic studies on biomolecules and also for constructing unique biosensors, nanoconductors, and creating an interface between electronic devices and biological objects.
As noted above, the chemical synthesis of the conducting PANI goes autocatalytically with a long induction period [130]. And the kinetics of enzymatic synthesis of this polymer can be controlled, and the emeraldine salt of PANI is produced immediately after initiating the polymerization with peroxidases or with fungal laccases.
In work [132], template polymerization of aniline dimer (N-phenyl-1,4-phenylene diamine) on SDBS micelles was studied to identify intermediate products of the aniline polymerization under the influence of laccase. It was shown by MALDI TOF that the main products of the reaction were oligomers with m/z ratio lower than 2180, which corresponded to the polymerization degree of 24 (in terms of aniline subunits). Enzymatically synthesized oligomers mainly consisted of para-substituted units represented as the emeraldine salt. The rate of aniline dimer polymerization under the influence of laccase was several times higher than the rate of aniline monomer polymerization under the same conditions.
To elucidate differences in the mechanisms of chemical and enzymatic syntheses of PANI and also to study specific features of the template polymerization of aniline using laccase from T. hirsuta, the reaction medium redox potential was measured in our laboratory during the synthesis under conditions of disconnected chain with concurrent recording of UV-visible spectra of the resulting products.
On initiation of the chemical polymerization of aniline with APS, the redox potential (pH 3.5) of the solution containing aniline and PAMPS decreased by ~15-20 mV and reached a minimum in a few minutes. This was associated with appearance in the electronic spectra of the reaction medium of an absorption band at wavelength 320-330 nm characteristic for π–π*-transition in aromatic rings on the oxidation of aniline. Then the redox potential of the medium sharply increased without changes in the optical density in the region of 500-1100 nm. Fifty minutes later the optical density in the electronic spectra of the reaction medium increased in the region of 600 nm, which corresponded to the oxidized chains of aniline oligomers, whereas the rate of medium redox-potential growth significantly decreased and soon the reaction medium potential became maximal and then decreased. Concurrently, the absorption band of the inter-polymer complex PANI/PAMPS displaced to longer wavelength (from 600 to ~730 nm) that suggested the generation of the emeraldine salt of PANI.
In the enzymatic polymerization of aniline in the presence of dioxygen, the redox potential of the reaction medium sharply increased immediately after initiating the reaction with laccase, and then for ~20 min it decreased monotonously. Immediately after the enzyme addition, the absorption increased in the region of 700 nm in the solution electronic spectra, which suggested the generation of the conducting inter-polymer complex PANI/PAMPS.
It is known that APS gradually decomposes in acidic medium, and the oxidation potential of aniline oligomers is significantly lower than the oxidation potential of the monomer. Therefore, it can be reasonably concluded that during the polymerization of aniline with APS as an oxidizer the growing PANI chains are constantly in the oxidized state, whereas the reaction mixture always contains a fraction of unoxidized aniline. After the complete decomposition of APS, oxidized chains of PANI are reduced by the unreacted monomer to the conducting emeraldine salt. This explains the presence of the induction period in the generation of the PANI emeraldine salt during the chemical synthesis. On the contrary, the enzymatic synthesis lacks the induction period, and the PANI chains grow due to generation of half-oxidized forms of aniline oligomers, which have been identified using MALDI TOF spectrometry [132].
The formal mechanism of initial stages of the polymer chain growth during the chemical and laccase-catalyzed synthesis of PANI can be presented as follows (Fig. 4).
Fig. 4. Formal scheme of initial stages of aniline polymerization in chemical (a) and laccase-catalyzed (b) syntheses.
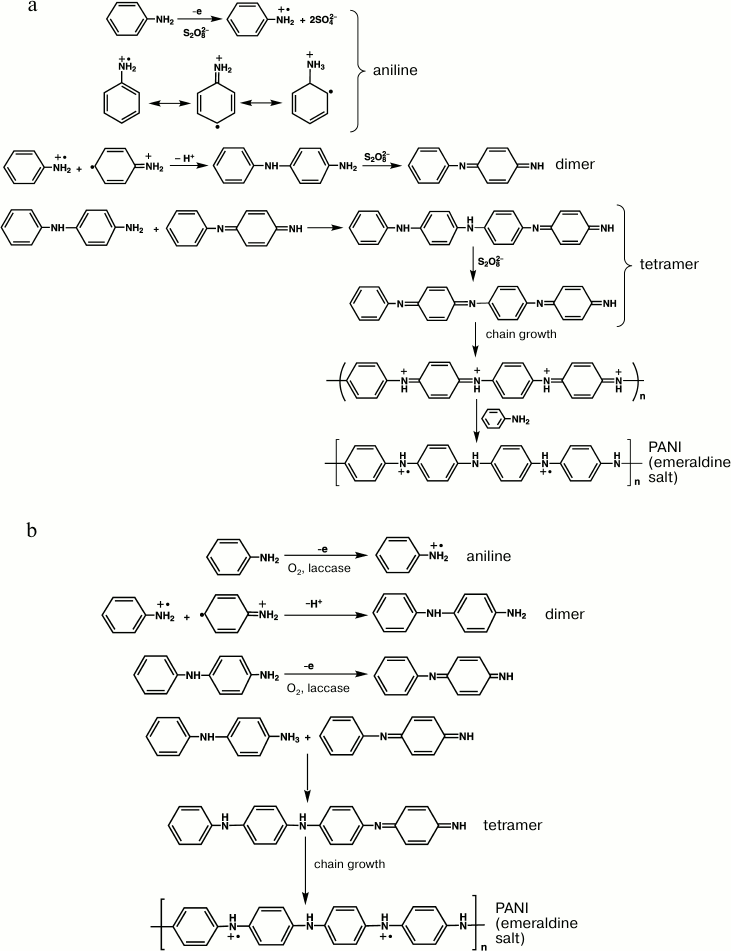
It should be noted that using laccases and peroxidases for synthesis of PANI is associated with oxidation of aniline (or its low molecular weight oligomers) in the active centers of these enzymes and generation of reactive products, whereas the polymer chain grows within the reaction solution volume.
Although the kinetics of the enzymatic oxidative polymerization of aniline can be controlled and there is no induction period in the generation of the final product, the PANI emeraldine salt, the reaction rate is relatively low and high concentrations of the enzymes must be used to increase this rate. Redox mediators of the enzymes can significantly accelerate the monomer polymerization, which seems promising for synthesis of conducting polymers having high molecular weight and high conductivity. The scheme of the enzyme–mediator system functioning is shown in Fig. 5.
Fig. 5. Scheme presenting the role of a redox mediator during the enzymatic synthesis of conducting polymers.
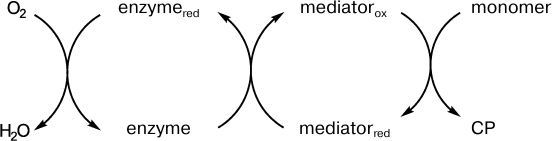
As differentiated from aniline, EDOT, having a high ionization potential, cannot be oxidized with enzymes alone. In the literature there is only one description of PEDOT synthesis on a polysulfostyrene template using HP [133]. The conductivity of the resulting PEDOT product was 2·10–3 S/cm. The reaction was performed for 24 h at pH 2.0. But HP at this pH value is known to completely lose its activity within a few minutes. It seems that under the experimental conditions at strongly acidic pH values of the reaction medium low molecular weight 3,4-ethylenedioxythiophene oligomers are produced similarly to pyrrole polymerization under the influence of acids in the absence of an oxidizer [21, 134]. The oxidation potential of EDOT oligomers is lower than that of the monomer, and their oxidation can be catalyzed by the heme produced upon the dissociation of the holoenzyme at acidic pH of the solution. This is indirectly confirmed by data presented in work [135] on the template synthesis of PEDOT initiated by thiophene trimer using soybean peroxidase. But this hypothesis needs additional confirmation.
The enzymatic oxidation of EDOT with production of the radical and subsequent growth of the polymer chain can occur in the presence of the corresponding redox mediator of the enzyme. In work [136], template oxidative polymerization of EDOT was performed with a laccase–mediator system. Potassium octacyanomolybdate(4+) was used as the redox mediator, and its enzymatic oxidation with dioxygen resulted in octacyanomolybdate(5+) anion. This anion nonenzymatically oxidized EDOT in a diffusion-controlled regimen to the corresponding radical with subsequent growth of the polymer chain. PAMPS was used as a template. As a result, a dark-blue aqueous dispersion of the inter-polymer complex PEDOT/PAMPS was produced. The complex was studied by UV-visible and FTIR spectroscopies. The conductivity of the PEDOT/PAMPS complex was 10–5 S/cm.
The enzyme–mediator systems based on laccase [137] and HP [138] and ABTS as a mediator were used for synthesis of conducting polypyrrole (PP). The enzymatic oxidation of the redox mediator produces the cation-radical ABTS•+, which nonenzymatically oxidizes pyrrole. The redox mediator is concurrently reduced to the initial form. These publications revealed an extremely low rate of monomer oxidation under the influence of the enzyme in the absence of the redox mediator and the production of low molecular weight pyrrole oligomers. In work [110], the spectral characteristics and conductivity of PP produced with the peroxidase/ABTS system and of PP traditionally synthesized chemically through pyrrole oxidation with ferric chlorate were compared. PP products synthesized by these two methods have similar characteristics of their FTIR spectra, whereas the conductivity of the enzymatically synthesized PP was slightly lower than that of the chemically prepared polymer.
In work [139], physicochemical properties of PANI synthesized with laccase and by the laccase–mediator approach on “soft” templates (PAMPS and straight micelles of SDBS) were compared. Potassium octacyanomolybdate(4+) was used as a redox mediator. On laccase–mediator-based synthesis, the rate of template polymerization of aniline was approximately an order of magnitude higher than the rate of the laccase-catalyzed synthesis at the same concentrations of the enzyme. The spectral characteristics (UV-visible and FTIR spectra) of PANI specimens synthesized by both methods were similar. However, the conductivity of the PANI/PAMPS complex prepared by the laccase–mediator approach was approximately fivefold higher than the conductivity of the complex produced in the absence of the mediator and was 4.8-5.9 mS/cm. Moreover, the laccase–mediator synthesis gave threefold higher yield of the polymer. Aniline oligomers extracted with tetrahydrofuran from the PANI samples were identified by MALDI TOF, and the laccase–mediator system was shown to result in aniline oligomers with higher molecular weight.
PROSPECTS OF PRACTICAL APPLICATION OF ENZYMATICALLY SYNTHESIZED
CONDUCTING POLYMERS
Composites based on conducting polymers attract the attention of specialists in different fields of science and technology due to prospects of their application as materials for electrodes in devices for accumulation and storage of energy (supercondensers), in “light” galvanic elements, as anticorrosion and antistatic coatings, for separation of optical isomers of physiologically active substances, and for creating flexible solar batteries, light-emitting diodes, chemo/biosensors, biomedical systems, etc. [140-143]. Conducting polymers for these purposes are synthesized mainly chemically or electrochemically. In this section of the review, only applications of enzymatically synthesized CPs will be presented.
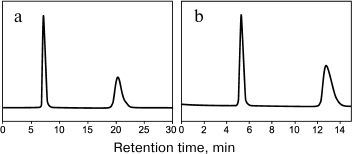
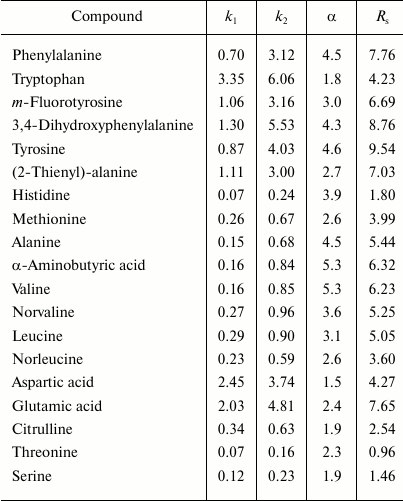
Russian Federation patent No. 2348455 of 10.10.2006 describes a method for production of a chiral sorbent based on enzymatically synthesized PANI for separation of physiologically active substances using high-performance liquid chromatography [144]. The high redox potential laccase from the basidial fungus T. hirsuta was used as a biocatalyst. Because PANI is practically insoluble in eluents traditionally used in liquid chromatography, the polymer can be effectively and rather simply used as a coating due to the synthesis immediately on the sorbent surface by the so-called in situ method. The resulting chiral sorbent was used for filling a 4 × 250-mm chromatographic column. Optical isomers of amino acids and their derivatives are separated within a few minutes, and the separation occurs virtually to the baseline (Fig. 6 and table). For the majority of racemates, the enantioselectivity factors are high – from 1.8 to 4.5.
Fig. 6. Separation chromatograms of enantiomers of 3,4-dihydroxy-phenylalanine (a) and of phenylalanine (b) [144].
Values of retention factors (k), enantioselectivity (α),
and resolving power (RS) on a hybrid sorbent [144]
Due to their stability and the absence of toxicity, CPs can be used for removing static electricity. In our laboratory the inter-polymer PANI/PAMPS complex synthesized using laccase was shown to have conductivity of ~10 mS/cm determined by the double-point method. The rate of positive electric charge removal from wool tissue surface treated with the PANI/PAMPS complex synthesized enzymatically and chemically increased 300- and 280-fold, respectively, compared to the control in the absence of PANI.
The enzymatically synthesized composite based on conducting PANI and multi-wall carbon nanotubes (MWCNT) is also used for making electrodes of a supercondenser, an apparatus for energy accumulation and storage. PANI was synthesized in situ on the surface of functionalized MWCNTs using the laccase–mediator system. By scanning electron microscopy, the PANI was shown to cover the carbon nanomaterial surface as a uniform thin layer. The resulting composite had high specific capacity (~440 F/g) measured by cyclic volt-amperometry and (~390 F/g) in the galvanostatic regimen in charge/discharge cycles. On the basis of this composite, a flexible and thin (300-400 µm) supercondenser was elaborated with a gel electrolyte that is simultaneously a separator for separation of the electrodes of the supercondenser.
Work [145] describes the production of a composite based on carboxylated MWCNTs with covalently bound enzymes (HP and glucose oxidase) and with PANI precipitated on this modified surface. The enzymatic oxidation of glucose catalyzed by glucose oxidase produced hydrogen peroxide, which, in turn acted as an oxidizer in the peroxidase-catalyzed polymerization of aniline. The synthesized biocomposite material was used for production of a biosensor for quantitative determination of glucose. The detection limit of the analyst was 0.02 mM at signal/noise ratio of 3 and analysis sensitivity of 0.94 µA/mM. The linear range of glucose concentration measurement with the biosensor was up to 12 mM, which fully covers the clinical range of glucose determination (3.5-6.5 mM) [146].
In this review, achievements in the synthesis of conducting polymers catalyzed by oxidoreductases are described. The use of enzymes for synthesis of new functional materials is an alternative for traditional chemical and electrochemical approaches for producing both conducting polymers and composites based on them.
Among oxidoreductases, peroxidases and fungal laccases having high redox potential are the most promising for this purpose. Owing to advances in gene engineering, large amounts of recombinant oxidoreductases with improved properties and acceptable price can be produced, and this will make the biocatalytic production of CPs commercially justified. Enzymatic synthesis is an ecologically favorable approach for producing CPs under “soft” conditions without generation of toxic byproducts, and kinetics of the monomer polymerization can be controlled. Templates of different nature and structure are promising for the improvement of the operational characteristics of CPs.
Redox mediators of enzymes allow increased reaction rate and polymerization of monomers that cannot be oxidized with oxidoreductases alone. Moreover, enzymatic and enzyme–mediator syntheses allow researchers to control molecular weight and some physicochemical parameters of synthesized CPs. However, it should be noted that the growth mechanism of the polymer chain during enzymatic synthesis of CPs, as well as the interaction of the resulting oligomers with active centers of the enzymes, are still not clear and need further studies.
REFERENCES
1.Syed, A. A., and Dinesan, M. K. (1991)
Talanta, 38, 815-837.
2.Diaz, A. F., Rubinson J. F., and Mark, H. B. (1988)
Adv. Polym. Sci., 84, 113-139.
3.Rao, P. S., Sathyanarayana, D. N., and Jeevananda,
T. (2001) Advanced Functional Molecules and Polymers, Gordon and
Breach, Tokyo.
4.Chandrakanthi, N., and Careem, M. A. (2000)
Polym. Bull., 45, 113-120.
5.Genies, E. M., Boyle, A., Lapkowski, M., and
Tsintavis, C. (1990) Synth. Met., 36, 139-182.
6.Pron, A., and Rannou, P. (2002) Prog. Polym.
Sci., 27, 135-190.
7.Machida, S., Miyata, S., and Techagumpuch, A.
(1989) Synth. Met., 31, 311-318.
8.George, P. M., Lyckman, A. W., LaVan, D. A., Hegde,
A., Leung, Y., Avasare, R., Testa, C., Alexander, P. M., Langer, R.,
and Sur, M. (2005) Biomaterials, 26, 3511-3519.
9.Bredas, J. L., Chance, R. R., and Silbey, R. (1982)
Phys. Rev. B., 26, 5843-5854.
10.Bredas, J. L., Themans, B., Andre, J. M., Chance,
R. R., and Silbey, R. (1984) Synth. Met., 9, 265-274.
11.Bredas, J. L., and Street, G. B. (1985) Acc.
Chem. Res., 18, 309-315.
12.Bocchi, V., Chierici, L., Gardini, G. P.,
Mondelli, R., Bocchi, V., Chierici, L., Gardini, G. P., and Mondelli,
R. (1970) Tetrahedron, 26, 4073-4082.
13.Joo, J., Lee, J. K., Baeck, J. S., Kim, K. H.,
Oh, E. J., and Epstein, J. (2001) Synth. Met., 117,
45-51.
14.Gursoy, S. S., Uygun, A., and Tilki, T. (2010)
J. Macromol. Sci. Pure Appl. Chem., 47, 681-688.
15.Zheng, H., Shi, Q., Du, K., Mei, Y., and Zhang,
P. (2013) Mol. Divers., doi: 10.1007/s11030-013-9426-1.
16.Lee, G. J., Lee, S. H., Ahn, K. S., and Kim, K.
H. (2002) J. Appl. Polym. Sci., 84, 2583-2590.
17.Oh, E. J., and Jang, K. S. (2001) Synth.
Met., 119, 109-110.
18.Qi, Z., and Pickup, P. G. (1997) Chem.
Mater., 9, 2934-2939.
19.Armes, S. P. (1987) Synth. Met.,
20, 365-371.
20.Machida, S., Miyata, S., and Techagumpuch, A.
(1989) Synth. Met., 31, 311-318.
21.Rapi, S., Bocchi, V., and Gardini, G. P. (1988)
Synth. Met., 24, 217-221.
22.Chao, T. H., and March, J. (1988) J. Polym.
Sci., Part A: Polym. Chem., 26, 743-753.
23.Menshikova, A. Y., Shabsels, B. M., and Evseeva,
T. G. (2003) Zh. Prikl. Khim., 76, 851-855.
24.Dhawan, S. K., and Trivedi, D. C. (1993) Bull.
Mater. Sci., 16, 371-380.
25.Sun, B., Jones, J. J., Burford, R. P., and
Skyllas-Kazacos, M. (1989) J. Mater. Sci., 24,
4024-4029.
26.Whang, Y. E., Han, J. H., Motobe, T., Watanabe,
T., and Miyata, S. (1991) Synth. Met., 45, 151-161.
27.Scharifker, B. R., Garcia-Pastoriza, E., and
Marino, W. (1991) J. Electroanal. Chem., 300, 85-98.
28.Yamaura, M., Sato, K., and Hagiwara, T. (1991)
Synth. Met., 41, 439-442.
29.Bradner, F. P., and Shapiro, J. S. (1988)
Synth. Met., 26, 69-77.
30.Imisides, M. D., John, R., Riley, P. J., and
Wallace, G. G. (1991) Electroanalysis, 3, 879-884.
31.John, R., and Wallace, G. G. (1992) Polym.
Int., 27, 255-260.
32.Roncali, J., Blanchard, Ph., and Frere, P. (2005)
J. Mater. Chem., 15, 1589-1610.
33.Czardybon, A., and Lapkowski, M. (2001)
Synth. Met., 119, 161-162.
34.Pyshkina, O., Kubarkov, A., and Sergeyev, V.
(2010) Sci. J. Riga Tech. Univ., 21, 51-54.
35.Baika, W., Luana, W., Zhaoa, R. H., Kooa, S., and
Kimb, K.-S. (2009) Synth. Met., 159, 1244-1246.
36.Kumar, A., and Reynolds, J. (1996)
Macromolecules, 29, 7629-7630.
37.Aasmundtveit, K. E., Samuelsen, E. J.,
Pettersson, L. A. A., Inganas, O., Johansson, T., and
Feidenhans’l, R. (1999) Synth. Met., 101,
561-564.
38.Wu, C.-H., Don, T.-M., and Chiu, W.-Y. (2011)
Polymer, 52, 1375-1384.
39.Gok, A., Omastova, M., and Yavuz, A. G. (2007)
Synth. Met., 157, 23-29.
40.Groenendaal, L. B., Jonas, F., Freitag, D.,
Pielartzik, H., and Reynolds, J. R. (2000) Adv. Mater.,
12, 481-494.
41.Groenendaal, L. B., Zotti, G., Aubert, P.,
Waybright, S. M., and Reynolds, J. R. (2003) Adv. Mater.,
15, 855-879.
42.Randrimahazaka, H., Noel, C., and Chevrot, C.
(1999) J. Electroanal. Chem., 472, 103-111.
43.Yamato, H., Ohwa, M., and Wernet, W. (1995) J.
Electroanal. Chem., 397, 163-170.
44.Yamato, H., Kai, K., Ohwa, M., Asakura, T.,
Koshiba, T., and Wernet, W. (1996) Synth. Met., 83,
125-130.
45.Yamato, H., Kai, K.-I., Ohwa, M., Wernet, W., and
Matsumura, M. (1997) Electrochim. Acta, 42,
2517-2523.
46.Lima, A., Schottland, P., Sadki, S., and Chevrot,
C. (1998) Synth. Met., 93, 33-41.
47.Sakmeche, N., Aaron, J. J., Fall, M., Aeiyach,
S., Jouini, M., Lacroix, J. C., and Lacaze, P. C. (1996) Chem.
Commun., 24, 2723-2724.
48.Lee, Y., Park, S., and Son, Y. (1999) Mol.
Cryst. Liq. Cryst., 327, 237-240.
49.Kudoh, Y., Akami, K., and Matsuya, Y. (1998)
Synth. Met., 98, 65-70.
50.Witayakran, S., and Ragauskas, A. J. (2009)
Adv. Synth. Catal., 351, 1187-1209.
51.Kunamneni, A., Camarero, S., Garcia-Burgos, C.,
Plou, F. J., Ballesteros, A., and Alcalde, M. (2008) Microb. Cell
Factories, 7, 32; doi: 10.1186/1475-2859-7-32.
52.Santaniello, E., Ferraboschi, P., Grisenti, P.,
and Manzocchi, A. (1992) Chem. Rev., 92, 1071-1140.
53.Seoane, G. (2000) Curr. Org. Chem.,
4, 283-304.
54.Farrell, R. L., Murtagh, K. E., Tien, M., Mozuch,
M. D., and Kirk, T. K. (1989) Enzyme Microb. Technol.,
11, 322-328.
55.Wakamatsu, K., and Takahama, U. (1993)
Physiol. Plants, 88, 167-171.
56.Dunford, H. B. (1991) Peroxidases in Chemistry
and Biology, Vol. 2, CRC Press, Inc., pp. 1-24.
57.McEldoon, J. P., and Dordick, J. S. (1996)
Biotechnol. Prog., 12, 555-558.
58.Nissum, M., Schodt, C. B., and Welinder, K. G.
(2001) Biochim. Biophys. Acta, 1545, 339-348.
59.Sakharov, I. Y. (2004) Biochemistry
(Moscow), 69, 823-829.
60.Gazaryan, I. G., and Lagrimini, L. M. (1996)
Phytochemistry, 41, 1029-1034.
61.Kokkinakis, D. M., and Brooks, J. L. (1979)
Plant Physiol., 63, 93-99.
62.Chance, B. (1949) Science, 109,
204-208.
63.Jones, P., and Dunford, H. B. (1977) Dokl.
Bolg. Akad. Nauk, 5, 121-134.
64.Yoshida, H. (1883) J. Chem. Soc. Trans.,
43, 472-486.
65.Baldrian, P. (2006) FEMS Microbiol. Rev.,
30, 215-242.
66.Thurston, C. F. (1994) Microbiology,
140, 19-26.
67.Claus, H. (2004) Micron, 35,
93-96.
68.Morozova, O. V., Shumakovich, G. P., Gorbacheva,
M. A., Shleev, S. V., and Yaropolov, A. I. (2007) Biochemistry
(Moscow), 72, 1136-1150.
69.Lee, S.-K., George, S. D., Antholine, W. E.,
Hedman, B., Hodgson, K. O., and Solomon, E. I. (2002) J. Am. Chem.
Soc., 124, 6180-6193.
70.Mayer, A. M., and Staples, R. C. (2002)
Phytochemistry, 60, 551-565.
71.Riva, S. (2006) Trends Biotechnol.,
24, 219-226.
72.Yaropolov, A. I., Skorobogat’ko, O. V.,
Vartanov, S. S., and Varfolomeyev, S. D. (1994) Appl. Biochem.
Biotechnol., 49, 257-280.
73.Baldrian, P. (2004) Appl. Microbiol.
Biotechnol., 63, 560-563.
74.Kurniawati, S., and Nicell, J. A. (2008)
Bioresour. Technol., 99, 7825-7834.
75.Shin, K. S., and Lee, Y. J. (2000) Arch.
Biochem. Biophys., 384, 109-115.
76.Shleev, S. V., Morozova, O. V., Nikitina, O. V.,
Gorshina, E. S., Rusinova, T. V., Serezhenkov, V. A., Burbaev, D. S.,
Gazaryan, I. G., and Yaropolov, A. I. (2004) Biochimie,
86, 693-703.
77.Shiba, T., Xiao, L., Miyakoshi, T., and Chen,
C.-L. (2000) J. Mol. Catal. B: Enzym., 10,
605-615.
78.Bertrand, T., Jolivalt, C., Caminade, E., Joly,
N., Mougin, C., and Briozzo, P. (2002) Acta Cryst., D58,
319-321.
79.Polyakov, K. M., Fedorova, T. V., Stepanova, E.
V., Cherkashin, E. A., Kurzeev, S. A., Strokopytov, B. V., Lamzin, V.
S., and Koroleva, O. V. (2009) Acta Cryst., D65,
611-617.
80.Morozova, O. V., Shumakovich, G. P., Shleev, S.
V., and Yaropolov, A. I. (2007) Prikl. Biochim. Mikrobiol.,
43, 583-597.
81.Solomon, E. I., Sundaram, U. M., and Machonkin,
T. E. (1996) Chem. Rev., 96, 2563-2606.
82.Bento, I., Martins, L. O., Gato, L. G., Armenia,
C. M., and Lindley, P. F. (2005) Dalton Transact., 21,
3507-3513.
83.Solomon, E. I., Chen, P., Metz, M., Lee, S. K.,
and Palmer, A. E. (2001) Angew. Chem. Int. Ed. Engl., 40,
4570-4590.
84.Bourbonnais, R., and Paice, M. G. (1990)
FEBS, 267, 99-102.
85.Fabbrini, M., Galli, C., and Gentili, P. (2002)
J. Mol. Catal. B: Enzym., 16, 231-240.
86.Shimizu, A., Kwon, J.-H., Sasaki, T., Satoh, T.,
Sakurai, N., Sakurai, T., Yamaguchi, S., and Samejima, T. (1999)
Biochemistry, 38, 3034-3042.
87.Bankar, S. B., Bule, M. V., Singhal, R. S., and
Ananthanarayan, L. (2009) Biotech. Adv., 27, 489-501.
88.Sztajer, H., Wang, W., Lunsdorf, H., Stocker, A.,
and Schmid, R. D. (1996) Appl. Microbiol. Biotechnol.,
45, 600-606.
89.Kobayashi, S., and Makino, A. (2009) Chem.
Rev., 109, 5288-5353.
90.Bouldin, R., Kokil, A., Ravichandran, S.,
Nagarajan, S., Kumar, J., Samuelson, L. A., Bruno, F. F., and
Nagarajan, R. (2010) Green Polymer Chem. Biocatal. Biomater. ACS
Symp. Ser., 1043, 315-341.
91.Walde, P., and Guo, Z. (2011) Soft Matter,
7, 316-331.
92.Xu, P., Singh, A., and Kaplan, D. (2006) Adv.
Polym. Sci., 194, 69-94.
93.Hollmann, F., and Arends, I. W. C. E. (2012)
Polymers, 4, 759-793.
94.McEldoon, J. P., and Dordick, J. S. (1996)
Biotechnol. Prog., 12, 555-558.
95.Sakharov, I. Y., and Sakharova, I. V. (2002)
Biochim. Biophys. Acta, 1598, 108-114.
96.Vasil’eva, I. S., Morozova, O. V.,
Shumakovich, G. P., Shleev, S. V., Sakharov, I. Y., and Yaropolov, A.
I. (2007) Synth. Met., 157, 684-689.
97.USA Patent No. 5,420,237 (1995).
98.Cruz-Silva, R., Romero-Garcia, J.,
Angulo-Sanchez, J. L., Ledezma-Perez, A., Arias-Marin, E., Moggio, I.,
and Flores-Loyola, E. (2005) Eur. Polym. J., 41,
1129-1135.
99.Kausaite, A., Ramanaviciene, A., and
Ramanavicius, A. (2009) Polymer, 50, 1846-1851.
100.Cui, X., Li, C. M., Zang, J., Zhou, Q., Gan,
Y., Bao, H., Guo, J., Lee, V. S., and Moochhala, S. M. (2007) J.
Phys. Chem. C, 111, 2025-2031.
101.Aizawa, M., Wang, L., Shinohara, H., and
Ikariyama, Y. (1990) J. Biotechnol., 14, 301-309.
102.Samuelson, L. A., Anagnostopoulos, A., Alva, K.
S., Kumar, J., and Tripathy, S. K. (1998) Macromolecules,
31, 4376-4378.
103.Liu, W., Kumar, J., Tripathy, S., Senecal, K.
J., and Samuelson, L. (1999) J. Am. Chem. Soc., 121,
71-78.
104.Karamyshev, A. V., Shleev, S. V., Koroleva, O.
V., Yaropolov, A. I., and Sakharov, I. Y. (2003) Enzyme Microb.
Technol., 33, 556-564.
105.Mazhuto, Y. M., Karamyshev, A. V., Shleev, S.
V., Sakharov, I. Y., and Yaropolov, A. I. (2005) Prikl. Biokhim.
Mikrobiol., 41, 247-250.
106.Shumakovich, G. P., Vasil’eva, I. S.,
Morozova, O. V., Khomenkov, V. G., Staroverova, I. N., Budashov, I. A.,
Kurochkin, I. N., Boyeva, J. A., Sergeyev, V. G., and Yaropolov, A. I.
(2010) J. Appl. Polym. Sci., 117, 1544-1550.
107.Shen, Y., Sun, J., Wu, J., and Zhon, Q. (2005)
J. Appl. Polym. Sci., 96, 814-817.
108.Nagarajan, R., Liu, W., Kumar, J., Tripathy, S.
K., Bruno, F. F., and Samuelson, L. A. (2001) Macromolecules,
34, 3921-3927.
109.Nickels, P., Dittmer, W. U., Beyer, S.,
Kotthaus, J. P., and Simmel, F. C. (2004) Nanotechnology,
15, 1521-1529.
110.Cruz-Silva, R., Amaro, E., Escamilla, A.,
Nicho, M. E., Sepulveda-Guzman, S., Arizmendi, L., Romero-Garcia, J.,
Castillon-Barraza, F. F., and Farias, M. H. (2008) J. Colloid
Interface Sci., 328, 263-269.
111.Sahoo, S. K., Nagarajan, R., Roy, S.,
Samuelson, L. A., Kumar, J., and Cholli, A. L. (2004)
Macromolecules, 37, 4130-4138.
112.Samuelson, L., Liu, W., Nagarajan, R., Kumar,
J., Bruno, F. F., Cholli, A., and Tripathy, S. (2001) Synth.
Met., 119, 271-272.
113.Streltsov, A. V., Shumakovich, G. P., Morozova,
O. V., Gorbacheva, M. A., and Yaropolov, A. I. (2008) Appl. Biochem.
Microb., 44, 264-270.
114.Rumbau, V., Pomposo, J. A., Alduncin, J. A.,
Grande, H., Mecerreyes, D., and Ochoteco, E. (2007) Enzyme Microb.
Technol., 40, 1412-1421.
115.Guo, Z., Ruuegger, H., Kissner, R., Ishikawa,
T., Willeke, M., and Walde, P. (2009) Langmuir, 25,
11390-11405.
116.Liu, W., Cholli, A. L., Nagarajan, R., Kumar,
J., Tripathy, S., Bruno, F. F., and Samuelson, L. (1999) J. Am.
Chem. Soc., 121, 11345-11355.
117.Cruz-Silva, R., Ruiz-Flores, C., Arizmendi, L.,
Romero-Garcia, J., Arias-Marin, E., Moggio, I., Castillon, F. F., and
Farias, M. H. (2006) Polymer, 47, 1563-1568.
118.Caramyshev, A. V., Evtushenko, E. G., Ivanov,
V. F., Barcelo, A. R., Roig, M. G., Shnyrov, V. L., van Huystee, R. B.,
Kurochkin, I. N., Vorobiev, A. Kh., and Sakharov, I. Y. (2005)
Biomacromolecules, 6, 1360-1366.
119.Longoria, A. M., Hu, H., and Vazquez-Duhalt, R.
(2010) Appl. Biochem. Biotech., 162, 927-934.
120.Roman, P., Cruz-Silva, R., and Vazquez-Duhalt,
R. (2012) Synth. Met., 162, 794-799.
121.Streltsov, A. V., Morozova, O. V., Arkharova,
N. A., Klechkovskaya, V. V., Staroverova, I. N., Shumakovich, G. P.,
and Yaropolov, A. I. (2009) J. Appl. Pol. Sci., 114,
928-934.
122.Jin, Z., Su, Y., and Duan, Y. (2001) Synth.
Met., 122, 237-242.
123.Vasil’eva, I. S., Morozova, O. V.,
Shumakovich, G. P., and Yaropolov, A. I. (2009) Prikl. Biokhim.
Mikrobiol., 45, 27-30.
124.Rumbau, V., Marcilla, R., Ochoteco, E.,
Pomposo, J. A., and Mecerreyes, D. (2006) Macromolecules,
39, 8547-8549.
125.Guo, H., Knobler, Ch. M., and Kaner, R. B.
(1999) Synth. Met., 101, 44-47.
126.Moutet, J.-C., Saint-Aman, E., Tran-Van, F.,
Angibeaud, P., and Utille, J.-P. (1992) Adv. Mater., 4,
511-513.
127.Thiyagarajan, M., Samuelson, L. A., Kumar, J.,
and Cholli, A. L. (2003) J. Am. Chem. Soc., 125,
11502-11503.
128.Ashraf, S. A., Kane-Maguire, L. A. P., Majidi,
M. R., Pyne, S. G., and Wallace, G. G. (1997) Polymer,
38, 2627-2631.
129.Kane-Maguire, L. A. P., MacDiarmid, A. G.,
Norris, I. D., Wallace, G. G., and Zheng, W. (1999) Synth. Met.,
106, 171-176.
130.Zeifman, Y. S., Maiboroda, I. O., Grishchenko,
Y. V., Morozova, O. V., Vasil’eva, I. S., Shumakovich, G. P., and
Yaropolov, A. I. (2012) Prikl. Biokhim. Mikrobiol., 48,
169-174.
131.Tzou, K., and Gregory, R. V. (1992) Synth.
Met., 47, 267-277.
132.Shumakovich, G., Streltsov, A., Gorshina, E.,
Rusinova, T., Kurova, V., Vasil’eva, I., Otrokhov, G., Morozova,
O., and Yaropolov, A. (2011) J. Mol. Catal. B: Enzym.,
69, 83-88.
133.Rumbau, V., Pomposo, J. A., Eleta, A.,
Rodriguez, J., Grande, H., Mecerreyes, D., and Ochoteco, E. (2007)
Biomacromolecules, 8, 315-317.
134.Hawkins, S. L., and Ratcliffe, N. M. (2000)
J. Mat. Chem., 10, 2057-2062.
135.Nagarajan, S., Kumar, J., Bruno, F. F.,
Samuelson, L. A., and Nagarajan, R. (2008) Macromolecules,
41, 3049-3052.
136.Shumakovich, G., Otrokhov, G., Vasil’eva,
I., Pankratov, D., Morozova, O., and Yaropolov, A. (2012) J. Mol.
Catal. B: Enzym., 81, 66-68.
137.Song, H.-K., Tayhas, G., and Palmore, R. (2005)
J. Phys. Chem. B, 109, 19278-19287.
138.Kupriyanovich, Y. N., Sukhov, B. G., Medvedeva,
S. A., Mikhaleva, A. I., Vakul’skaya, T. I., Myachina, G. F., and
Trofimov, B. A. (2008) Mendel. Commun., 18, 56-58.
139.Shumakovich, G., Kurova, V., Vasil’eva,
I., Pankratov, D., Otrokhov, G., Morozova, O., and Yaropolov, A. (2012)
J. Mol. Catal. B: Enzym., 77, 105-110.
140.Riul, A., Jr., Malmegrim, R., Fonseca, F. J.,
and Mattoso, L. H. C. (2003) Biosens. Bioelect., 18,
1365-1369.
141.Wessling, B., and Posdorfer, J. (1999)
Synth. Met., 102, 1400-1401.
142.Soto-Oviedo, M. A., Araujo, O. A., Faez, R.,
Rezende, M. C., and De Paoli, M.-A. (2006) Synth. Met.,
156, 1249-1255.
143.Carpi, F., and De Rossi, D. (2006) Opt.
Laser Technol., 38, 292-305.
144.Russian Federation Patent No. 2348455 of
10.10.2006.
145.Sheng, Q., and Zheng, J. (2009) Biosens.
Bioelect., 24, 1621-1628.
146.Heider, G. H., Sasso, S. V., Huang, K.,
Yacynych, A. M., and Wieck, H. J. (1990) Anal. Chem., 62,
1106-1110.