Hepatitis B Virus Can Be Inhibited by DNA Methyltransferase 3a via Specific Zinc-Finger-Induced Methylation of the X Promoter
L. Xirong1,2,3, L. Rui1, Y. Xiaoli1, H. Qiuyan1, T. Bikui1, Z. Sibo1, and Z. Naishuo1,2*
1Laboratory of Molecular Immunology, State Key Laboratory of Genetic Engineering, School of Life Sciences, Fudan University, Shanghai 200433, China; E-mail: holyinshanghai@163.com; nzhu@fudan.edu.cn2Shanghai Xinxing Medicine Co., LTD, Shanghai 200135, China; E-mail: naishuoz@sina.com
3Institute of Biomedical Sciences, Fudan University, Shanghai 200433, China; fax: (86) 21-5566-4032; E-mail: lixirong@fudan.edu.cn
* To whom correspondence should be addressed.
Received September 2, 2013; Revision received November 11, 2013
In this work we explored whether DNA methyltransferase 3a (Dnmt3a) targeted to the HBV X promoter (XP) causes epigenetic suppression of hepatitis B virus (HBV). The C-terminus of Dnmt3a (Dnmt3aC) was fused to a six-zinc-finger peptide specific to XP to form a fused DNA methyltransferase (XPDnmt3aC). The binding and methyl-modifying specificity of XPDnmt3aC were verified with an electrophoretic mobility shift assay and methylation-specific PCR, respectively. XP activity and HBV expression were clearly downregulated in HepG2 cells transfected with plasmid pXPDnmt3aC. The injection of XPDnmt3aC into HBV transgenic (TgHBV) mice also showed significant inhibition, leading to low serum HBV surface protein (HBsAg) levels and a reduced viral load. Thus, XPDnmt3aC specifically silenced HBV via site-selective DNA methylation delivered by zinc-finger peptides. This study establishes the foundation of an epigenetic way of controlling HBV-related diseases.
KEY WORDS: hepatitis B virus, X promoter, DNA methyltransferase, zinc finger, specific-site DNA methylation, gene regulationDOI: 10.1134/S0006297914020047
Hepatitis B virus (HBV) chronically infects more than 350 million people worldwide and is a leading cause of liver cirrhosis and hepatocellular carcinoma [1]. Interferon-α and nucleos(t)ide analogs are currently approved for the treatment of chronic HBV infection, although these antiviral agents rarely result in viral clearance or cure [2, 3]. Moreover, the long-term use of nucleos(t)ide analogs, such as lamivudine, adefovir dipivoxil, and entecavir, usually results in drug resistance [3]. The HBV covalently closed circular DNA (cccDNA) persists in the host cell nuclei and drives relapse once the antiviral therapy is discontinued or resistance develops. Therefore, there is an urgent need to design novel antiviral therapies to achieve the goal of silencing HBV.
Hypermethylation at CpG sequences in the promoters of a number of genes commonly leads to transcriptional downregulation [4, 5]. The methyl modification of some viral gene promoters contributes to viral latency and persistence, and it is a common occurrence in human virus-related diseases, including infection with Epstein–Barr virus [6], human immunodeficiency virus type 1 [7], and HBV [8, 9]. It has been reported that increased CpG methylation of cccDNA is associated with low serum HBV DNA levels and suppresses the viral replicative activity [8, 9]. Therefore, exploring the regulatory role of specific DNA methylation in silencing HBV replication is of considerable interest.
The epigenetic reversibility has fueled the development of drugs that target the enzymes that mediate epigenetic modifications in human cancers [10]. Federal Food and Drug Administration (USA) approved epigenetic drugs usually produce genome-wide effects because they are extremely nonspecific. In an early study of targeted DNA methylation, a DNA methyltransferase was genetically fused to the sequences of specific DNA-binding proteins, such as zinc-finger proteins that act as targeting domains, to deliver DNA cytosine methylation to a genomically integrated target promoter. This led to heritable gene silencing, such as the inhibition of herpes simplex virus type 1 [11-16]. To our knowledge, specific DNA methylation has not been used to repress HBV.
The X promoter (XP) of the HBV genome, which overlaps enhancer I and CpG island II, is highly active [17], this supporting the proposition that its activity may be regulated by DNA methylation. In this study, we evaluated the effect of the zinc-finger-induced specific methylation of XP on HBV expression. DNA methyltransferase 3a (Dnmt3a) can specifically methylate the CpG sites in the 5′-flanking region and exons of genes [18]. The NH(2)-terminal regulatory domain of Dnmt3a is responsible for establishing its substrate specificity. To exclude the toxicity of non-specificity, the C-terminal catalytic domain of Dnmt3a (Dnmt3aC) was used to construct the targeted methyltransferase [11-15]. The fused methylase reduced HBV expression in HepG2 cells and in HBV-transgenic (M-TgHBV) mice.
MATERIALS AND METHODS
Construction of plasmid. All primers used in this study were synthesized by Invitrogen (China). The primers are listed in Table 1. The reporter plasmid for XP (nucleotides (nt) 956-1369 of the HBV genome) (GenBank accession No. AY306136.1), namely pXPLuc, was constructed using the pGL3-Basic luciferase reporter vector (Promega, China) with the X promoter primers (Table 1). Six-zinc-finger peptides targeted to XP (XPZF) were designed using ZF Tools [19], which were first selected based on the distance between the Dnmt3a recognition sites and the DNA methylation sites and excluded through blasting on NCBI blastn [16, 19]. XPZFs were synthesized by Jierui Biotech (China). Electrophoretic mobility shift assay (EMSA) was used to determine the final zinc-finger targeting sequence (data not shown). The sequence (nt 1302-1319) to which XPZF specifically binds locates in the overlapping region of XP and CpG island II of the HBV genome (Fig. 1, a-c). The fused methyltransferase is depicted schematically in Fig. 1d. XPZF was amplified by PCR using the labeled sense primer with a Flag tag (DYKDDDDK) and a nuclear localization sequence (PKKKRKV) (Fig. 1d) and then cloned into the pcDNA3.1(+) vector (Invitrogen). The C-terminus of Dnmt3a (nt 2217-3074; GenBank: NM_175629.1) was then generated by PCR using the labeled sense primer with a flexible linker (Gly4Ser)2 and pcDNA3/Flag-DNMT3A (in Arthur D. Riggs’ laboratory) as a template (Fig. 1, d and e). The fragment was then ligated to the C-terminus of XPZF to generate the fused DNA-methyltransferase-expressing construct, pXPDnmt3aC. The replication-competent HBV plasmid was constructed according to the thesis of J. Cui [20] using overlap-PCR to obtain 1.3-genome-length units containing the HBV intrinsic promoter necessary for pre-genomic RNA (pgRNA) transcription.
Table 1.List of primers used
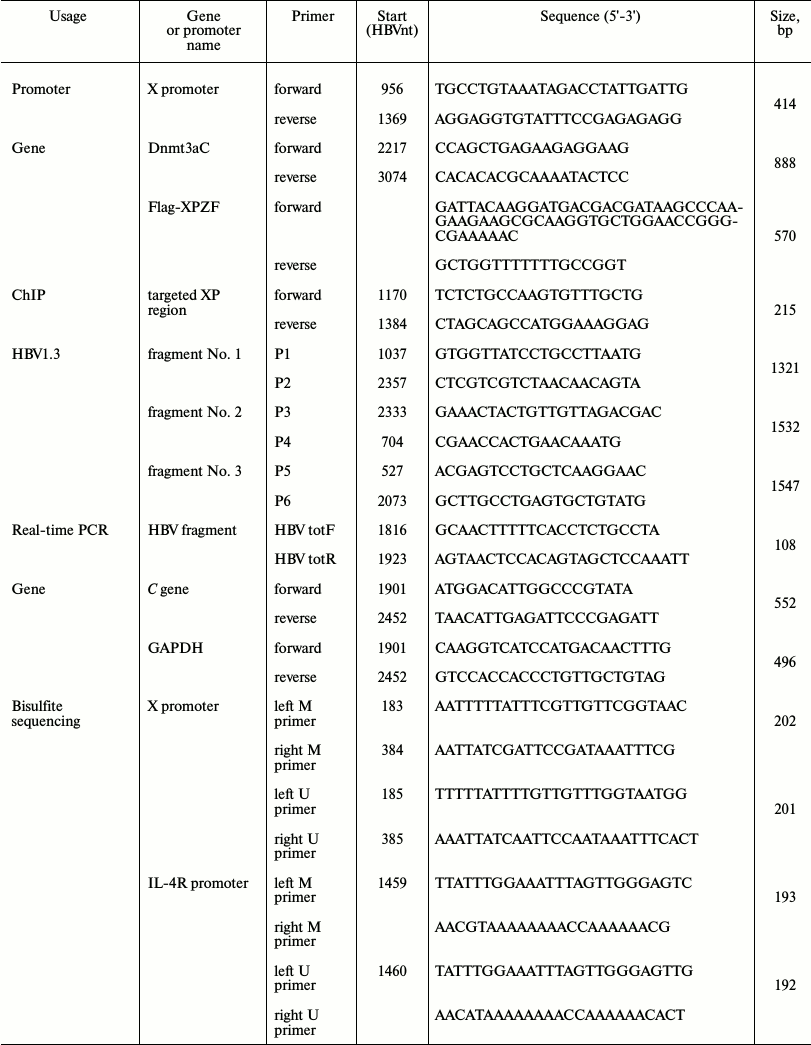
Fig. 1. Methylation pattern of XP and schematic representation of the targeted methyltransferase. a) Computational prediction of CpG islands in X promoter. Criteria used for prediction were island size >100 bp, GC percent >50.0, Obs/Exp >0.6. I, II, and III represented each CpG island. b) Schematic overview of XP region and CpG island II. c) Sequence of the overlapping region between CpG island II and XP. The overlapping region of both is shadowed. CpGs were marked according to the location in the HBV DNA sequence. d) Schematic representation of XPDnmt3aC. e) Mode of methyl-modification of XPDnmt3aC. The N-terminus of the zinc-finger peptide binds to the 3′ terminus of the targeted DNA sequence.
Cell culture and transfection. The cell lines used in this study were obtained from the Cell Bank Center of the Shanghai Institutes for Biological Sciences. HepG2 cells were cultured in RPMI 1640 medium (Gibco, China) supplemented with 10% heat-inactivated fetal bovine serum (Gibco) and 1% penicillin/streptomycin solution (Invitrogen). All cells were transfected with plasmids using Sunma-Sofast (Sunma, China), a cationic polymer gene transfection reagent, according to the manufacturer’s instructions. The transfection efficiency was determined with plasmid pcDNA3.1EGFP (Invitrogen), which was transfected alone or cotransfected with the experimental plasmids in ratio 1 : 1. The transfection efficiency was approximately 55-65% (data not shown).
Transgenic mice. The lineage of transgenic mice, designated M-TgHBV mice, was purchased from Shanghai Research Center for Model Organisms. This HBV is of the adr serotype and belongs to genotype C (GenBank accession No. AF461363.1). It was generated by routine microinjection of linearized HBV DNA isolated from a chronic carrier infected with HBV into fertilized eggs of C57BL/6J mice [21]. Ten-to-15-week-old male transgenic mice with serum HBsAg levels of 291.1 ± 67.61 U/ml and serum HBV DNA levels of (1.31E + 05) ± (1.29E + 05) copies/ml were used. The mice were maintained under specific-pathogen-free conditions and treated according to the Guide for the Care and Use of Laboratory Animals of Fudan University.
Western blot analysis. The expression of XPDnmt3aC was analyzed 48 h after transfection of plasmid pXPDnmt3aC into HepG2 cells in six-well plates. Total cell lysates were prepared with RIPA reagent (Shenneng Bo Cai, China). Proteins were resolved on 10% SDS-PAGE and transferred onto nitrocellulose membranes. Mouse monoclonal antibody directed against Dnmt3a (Abcam) and the corresponding horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody were incubated with the membranes. The membranes were then visualized with enhanced chemiluminescence (Thermo) and overexposure (30 s) to film. GAPDH was used as the endogenous control.
XPDnmt3aC, the HBV surface antigen (HBsAg), and X protein (HBx) in liver tissue samples from the treated transgenic mice were monitored at the indicated times. The mouse anti-Dnmt3a antibody, goat anti-HBsAg antibody (Abcam), and mouse anti-HBx antibody (Abcam) were used for the immunoblotting analysis. The specific bands on the Western blots were quantified with the Image J software (NIH). The levels of proteins were calculated with densitometry and normalized to that of β-actin.
Immunofluorescence assay. HepG2 cells were seeded at a density of 5·105 cells per well on cover glasses in six-well plates. Forty-eight hours after their transfection with pXPDnmt3aC, the cells were fixed in 4% formaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 10% FBS for 30 min at room temperature. The cells were then incubated with mouse anti-Dnmt3a antibody and fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG secondary antibody. The nucleic acid was stained with 1 µg/ml 4′,6-diamidino-2-phenylindole (DAPI) for 1 min. The stained coverslips were observed under a laser scanning confocal microscope (Leica, Germany).
EMSA. Nuclear protein extracts were prepared using the Cell Plasma and Nuclear Protein Extract Kit (Viagene, China) 48 h after 1·107 HepG2 cells were transfected with pXPDnmt3aC according to manufacturer’s instruction. The EMSA probes labeled with biotin at the 5′ end were as follows: ttgctcgCAGCCGGTCTGGAGCGAAacttatc, which contained an XPZF binding site in upper-case font. The mutated sites in the labeled mutant probes are underlined, as follows: ttgctcgCGGCTGTTGTGGATCGATacttatc. The EMSA was performed using the LightShift Chemiluminescent EMSA Kit (Thermo Scientific). Each 15 µg of nuclear protein sample was incubated with the 5′-biotin-end-labeled duplex described above (50 nM) in reaction buffer (binding buffer, 2.5% glycerol, 50 ng/μl poly(dI·dC), 0.5 mg/ml BSA) at room temperature for 20 min to form the protein–DNA complex. The protein–DNA complexes were separated in a 5.5% nondenaturing acrylamide gel, transferred to positive nylon membrane, and then detected by chemiluminescence.
XPDnmt3aC binding capacity was also examined using EMSA in M-TgHBV mice. Nuclear protein extracts were isolated from liver samples excised 48 h post-injection of pXPDnmt3aC. EMSA was performed as described before.
Chromatin immunoprecipitation (ChIP) assays. ChIP assays were performed as described previously [22]. pXPDnmt3aC plasmids were transfected into 2·107 HepG2 cells that had been transfected with pHBV1.3 plasmids for 24 h. After 40 h, formaldehyde was added to the media to cross-link protein to DNA. The cells were lysed in SDS lysis buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 150 mM NaCl, 1% SDS) and then sonicated to shear DNA to an average fragment size of 200-1000 bp. Dnmt3a antibody and Protein G (Thermo Pierce) was used for immunoprecipitation. PCR amplifications were performed using the targeted XP fragment primers (Table 1). Samples from three independent immunoprecipitations were analyzed.
Luciferase reporter assay. HepG2 cells were transfected with 0.3 µg of the reporter plasmid pXPLuc 24 h after their transfection with pXPDnmt3aC in 24-well plates. Luciferase activity was assayed 48 h after cotransfection. Firefly and Renilla luciferase activities were measured with a GloMax 20/20 luminometer (Promega, USA) 48 h after transfection using the Dual-Luciferase Reporter Assay System (Promega). The firefly luciferase activity was normalized to the Renilla luciferase activity, and the promoter activity was calculated as the relative luciferase activity [23].
Detection of HBV expression and replication. The ability of pHBV1.3 to produce independent replication-competent viruses was analyzed first. The cell supernatants and cells were collected 48 h after HepG2 cells were transfected with plasmid pHBV1.3 and stored at −80°C until analysis. Cells were lysed with lysis buffer (20 mM Tris-HCl, pH 8.0, 137 mM NaCl, 1% Nonidet P-40 and 2 mM EDTA) containing 1× proteinase inhibitor mixture (Roche Applied Science). The HBsAg in the culture and lysis supernatant was measured with a sandwich enzyme-linked immunosorbent assay (ELISA) kit (KHB, China). Cell genomes were isolated using Multisource Genomic DNA Miniprep Kit (AXYGEN, China). The exogenous DNA and RNA and the free HBV DNA and RNA in the culture supernatant and cell genomes were removed by treatment with DNase I and ribonuclease A. The viral core DNA was exposed by digestion with proteinase K and extracted twice with phenol–chloroform (25 : 24 v/v). The standard plasmid was constructed by cloning a fragment comprising 0.9 of the HBV genome (nt 1037-3215, 1-704) into the pMD-T19 vector (TaKaRa, China). Amplification for core DNA was performed with the SYBR® Green Real-time PCR Master Mix (Toyobo, China) and primers of HBVtotF and HBVtotR (Table 1) according to Henning et al. [24]. The data were analyzed with the Stratagene MxPro software. The numbers of viral copies were calculated based on the standard curve and are presented as logarithms to base 10 (LOG). Total RNA was isolated using TRIzol Reagent (Invitrogen). The C gene was tested as representative of the HBV RNAs using reverse transcription (RT)-PCR because HBV pgRNAs act as the mRNAs of the open reading frames of pre-C, C, and P.
To assess the effect of XPDnmt3aC on HBV replication and expression, culture supernatants were collected 24, 48, and 72 h after HepG2 cells were cotransfected with pXPDnmt3aC and pHBV1.3. HBsAg, viral core DNA, and C gene were measured as described before. Furthermore, a mixture of 20 µg of pXPDnmt3aC or empty pcDNA3.1(+) vector and 20 µl of EntransterTM-in vivo Transfection Reagent (Engreen Biosystem Co, Ltd., China) was hydrodynamically injected into the tail veins of M-TgHBV mice in a volume of saline equivalent to 8% of the body mass of the mouse. All mice were injected repeatedly on day 4 and 8. Livers and sera were collected on days 0, 4, 8, 12, and 20. Tissues were fixed in 4% paraformaldehyde and embedded in paraffin. The sections were stained with hematoxylin and eosin to examine for histopathological abnormalities. Immunohistochemistry was performed using mouse monoclonal anti-Dnmt3a, anti-HBx, and anti-HBsAg antibodies and the corresponding biotin-conjugated secondary antibodies. The staining was then visualized with avidin-conjugated HRP with diaminobenzidine (DAB) as the substrate. The C gene in tissue samples and serum viral core DNA were measured as described previously. Serum HBsAg was quantified with a chemiluminescent microparticle immunoassay on an Abbott Architect i2000 instrument.
Methylation-specific PCR (MSP) assay. HepG2 cells were cotransfected with plasmid pXPDnmt3aC and pHBV1.3 for 40 h. The cell genomes containing the HBV genome were extracted. The promoter of interleukin 4 receptor (IL4R) was also analyzed as a nonspecific control. The bisulfite reaction was performed on 500 ng of DNA. Sodium bisulfite conversion and purification of DNA were performed with the EpiTech Plus Bisulfite Conversion Kit (Qiagen, China), according to the manufacturer’s recommendations. A fragment of 202 bp, corresponding to the HBV genomic sequence at nucleotide 1138-1339, was amplified with primers designed using the Methprimer software for methylated DNA (Table 1) [23]. The amplified fragment was cloned into the pMD19-T vector and sequenced. The data quality was checked using the BiQ Analyzer software, a standard tool for processing DNA methylation data from bisulfite sequencing (http://biqanalyzer.bioinf.mpi-inf.mpg.de/) [25].
The de novo methylation of the HBV genome was also examined in M-TgHBV mice. Liver tissue was resected, and cell genomes were isolated 48 h after pXPDnmt3aC was injected. Methylation assay was then performed.
Statistical analysis. Statistical tests were performed with the SPSS software (version 16.0, 2007; SPSS Inc.). The data are reported as means ± SD. Different experimental groups are compared with Student’s t-test. A value of P < 0.05 is considered statistically significant.
RESULTS
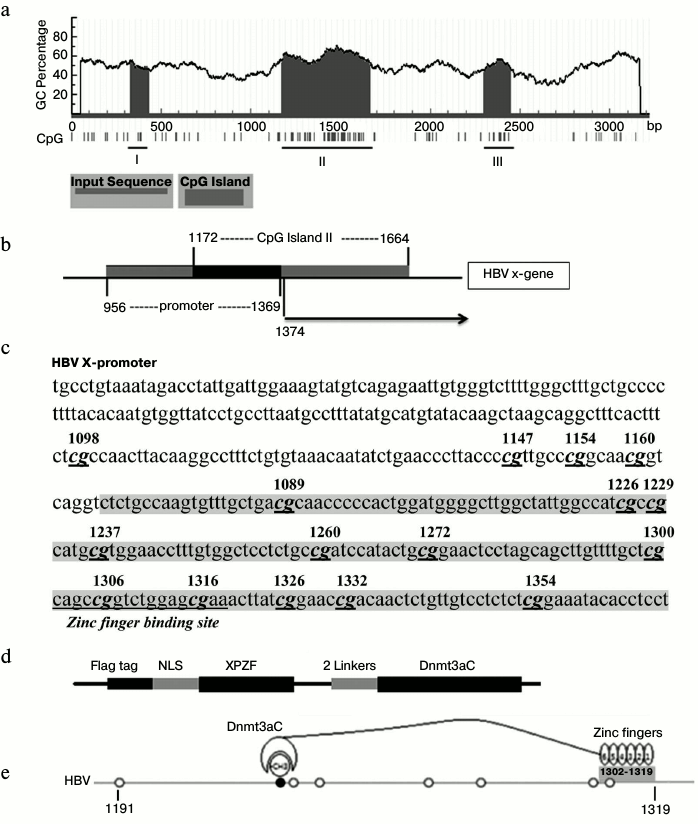
Fused methyltransferase bound to a specific DNA sequence leading to de novo methylation. The expression of XPDnmt3aC was assayed by Western blotting 48 h after the transfection of HepG2 cells. A specific XPDnmt3aC band of 53.9 kDa was clearly visible (Fig. 2A). As shown in Fig. 2D, the intensity of immunofluorescently stained Dnmt3a was significantly elevated in cells transfected with pXPDnmt3aC compared with that in cells treated with the empty vector and untreated. The overlapping green signals from XPDnmt3aC and the blue signals from nucleic acid demonstrated that XPDnmt3aC localized predominantly in the cell nuclei (Fig. 2D). The capacity of the recombinant pHBV1.3 for independent replication was verified using ELISA of supernatant HBsAg (Table 2), quantitative PCR for viral copies in supernatants and cells (Fig. 2B), and RT-PCR amplification of the C gene (Fig. 2C) 48 h after pHBV1.3 was transfected into HepG2 cells. HBsAg expression was considered positive when the sample optical density at 450 nm (OD450) was ≥2.1 times the value for the cells transfected with the empty vector pcDNA3.1. In supernatant and cells, HBsAg was 0.374 ± 0.049 and 0.282 ± 0.044, and the viral titers were 6.532·108 and 5.312·107 copies/μl, respectively. The C gene showed the expected molecular size of 552 bp.
Fig. 2. Expression of XPDnmt3aC and HBV expression. A) The whole protein extract was prepared 48 h after transfection of pXPDnmt3aC into HepG2 cells. Immunoblotting assay was performed using anti-Dnmt3a antibody. B) Real-time PCR assay for HBV. Viral copies were measured 48 h after HepG2 cells were transfected with pHBV1.3. C) RT-PCR of the C gene 48 h after HepG2 cells were transfected with pHBV1.3. D) Immunofluorescence localization of XPDnmt3aC in HepG2 cells transiently transfected with the pXPDnmt3aC plasmid for 48 h. Panels (a), (d), and (g) were stained with anti-Dnmt3a antibody; (b), (e), and (h) were stained with DAPI; (c), (f), and (i) are the merged images of the two earlier images on the same line. Original magnification, ×400.
Table 2. ELISA for hepatitis B virus surface
antigen expressed by pHBV1.3 plasmid
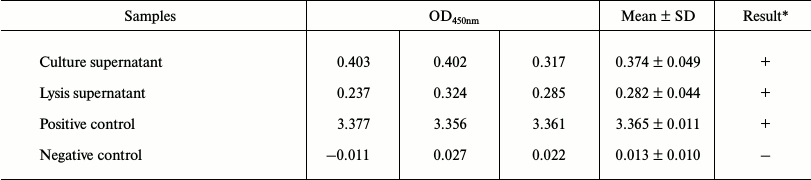
* Positive standard: sample values of OD450 nm ≥ 2.1 times
that of negative control values.
To determine the binding ability of the chimeric methylase, nuclear extract was used for EMSA 48 h after the transfection of pXPDnmt3aC plasmids into HepG2 cells. Bands apparently representing complexes of XPDnmt3aC bound to specific DNA probes were observed after the specific reactions or the mutant–probe competition reactions (Fig. 3a). However, the shift in the band was very weak after the unlabeled wild-type probe competition reaction, and no specific band was visible in the negative vector control or the blank control reaction. The supershifted bands were so intense that the shifted bands became very weak. The binding capacity of XPDnmt3aC was also confirmed using EMSA in M-TgHBV mice, the map of which is similar to that in cells (data not shown). Next, ChIP assays were performed using anti-Dnmt3a antibodies 40 h after cotransfection of pXPDnmt3aC and pHBV1.3 into HepG2 cells. A 251-bp region covering the zinc-finger-binding site was amplified by PCR using immunoprecipitated DNA as template, and a specific band was observed (Fig. 3b). The input DNA showed the same band, whereas no signal was observed in the normal IgG control. Collectively, these results provided evidence that XPDnmt3aC specifically bound to the target sequence.
Fig. 3. Determination of binding specificity and methyl-modifying characteristics of XPDnmt3aC. a) EMSA was performed with nuclear extract from HepG2 cells transfected with the pXPDnmt3aC plasmid (lanes 2, 4-7) as test groups (T) or the empty pcDNA3.1(+) vector (lane 3) as control (C) for 48 h. b) ChIP assays. Cell lysates were prepared 40 h after pXPDnmt3aC and pHBV1.3 plasmids were cotransfected into HepG2 cells. Gel electrophoresis of the PCR product using the XPZF-ChIP primer pair and various templates. c) Representative targeted methylation of XP induced by XPDnmt3aC. MSP assay was performed 40 h after cotransfection of pHBV1.3 and XPDnmt3aC into HepG2 cells. The sites of methylated CpGs are marked by black filled circles and numbered for the location in the HBV DNA sequence.
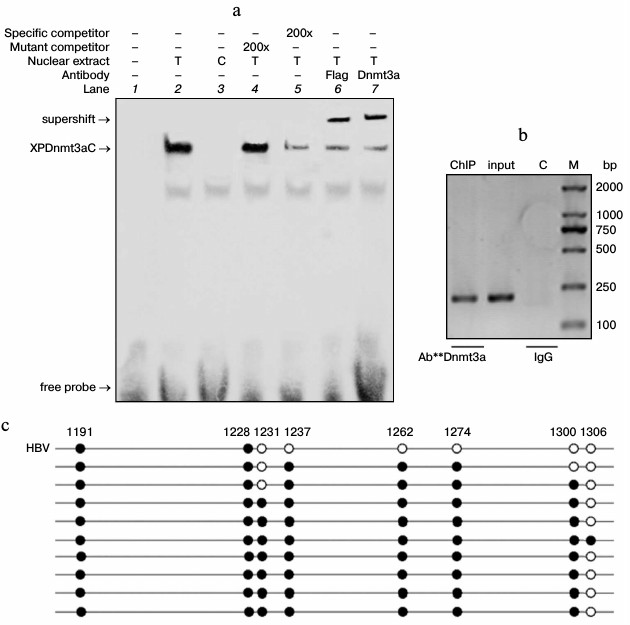
To examine the specific methylation, the cell genome including HBV was analyzed with MSP 40 h after cotransfection of pHBV1.3 and pXPDnmt3aC into HepG2 cells. The result revealed that the targeted methylation chiefly occurred within the region extending from the zinc-finger-binding site to –111 bp upstream (Fig. 3c). In 10 clones tested, the CpGs at sites –74 and –111 in seven clones were all methylated de novo, whereas the methylation of CpGs in the zinc-finger-binding region only appeared in one of these seven clones. No targeted methylation occurred in the corresponding region in the untreated HBV genome and IL4R promoter (data not shown). The result of MSP in vivo was similar to that in cells. These findings indicated that XPDnmt3aC specifically methyl-modified the target region.
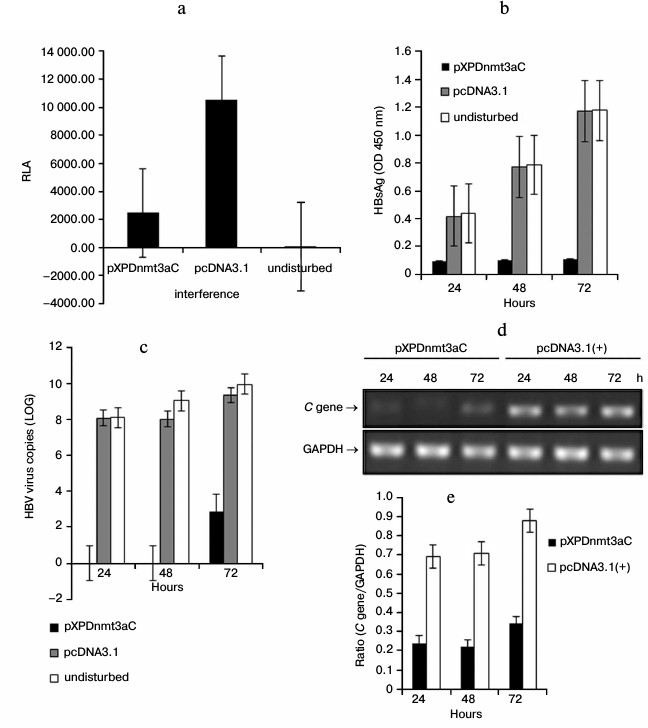
Targeted methylation of XP led to gene repression in HepG2 cells. To assess the influence of XPDnmt3aC on the activity of XP, HepG2 cells were cotransfected with the reporter pXPLuc and pXPDnmt3aC for 48 h. The luciferase activity decreased significantly (by 77%) due to exogenous methylation compared with that of pXPLuc alone (t-test, P < 0.01; Fig. 4a). To further evaluate the influence of XPDnmt3aC on HBV replication and expression, HBsAg, viral load, and C gene expression were measured 24, 48, and 72 h after cotransfection of HepG2 cells with pHBV1.3 and pXPDnmt3aC (Fig. 4, b-e). At 24 and 48 h, the HBsAg levels in the disturbance group had clearly decreased, by 78 and 87%, respectively (t-test, P < 0.01; Fig. 4b). At 72 h, the HBsAg levels were still reduced by more than 90% compared with those in the cells transfected with the undisturbed pHBV1.3 control (OD450, 1.175 ± 0.006; t-test, P < 0.01; Fig. 4b). The virus in the disturbance group was below the limit of detection at 24 and 48 h after cotransfection (Fig. 4c). However, the viral load rebounded slightly to about 29% of that in the pHBV1.3-transfected cells lacking pXPDnmt3aC (LOG, 9.989) at 72 h (t-test, P < 0.01; Fig. 4c). The expression of the C gene was analyzed using relative quantitative RT-PCR and declined to about 30-40% between 24 and 72 h after cotransfection compared with that in cells transfected with the negative vector (t-test, P < 0.05; Fig. 4, d and e). A lower bound of C gene expression was also observed 72 h after cotransfection. These findings demonstrated that XPDnmt3aC inhibited XP activity and HBV expression and replication.
Fig. 4. Inhibitory effects of XPDnmt3aC on XP activity and HBV expression in HepG2 cells. a) HepG2 cells were cotransfected with the reporter pXPLuc and pXPDnmt3aC for 48 h. The luciferase activity decreased sharply compared with that in HepG2 cells transfected with the negative control vector (t-test, P < 0.01). Levels of HBsAg expression (b), virus (c), and C gene expression (d and e) were measured at 24, 48, and 72 h after HepG2 cells were cotransfected with pHBV1.3 and pXPDnmt3aC. All three were clearly repressed by XPDnmt3aC compared with their levels in the negative control (t-test, P < 0.01). The levels of virus and C gene expression rebounded slightly at 72 h.
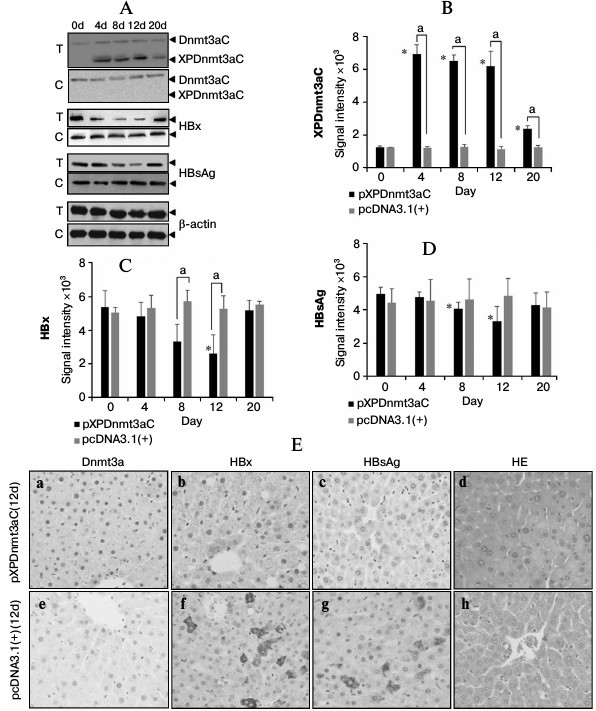
Targeted XP methylation repressed virus expression in M-TgHBV mice. To further access the inhibitory effect of XPDnmt3aC on HBV expression, we tested the effects of XPDnmt3aC in M-TgHBV mice. The expression of recombinant XPDnmt3aC, HBsAg, and HBx was tested using Western blotting, as shown in Fig. 5, A-D. XPDnmt3aC was expressed strongly from day 4 to day 12, but it decreased sharply on day 20. In contrast, HBx and HBsAg expression was reduced significantly on days 8 and 12 (t-test, P < 0.01), but rebounded to the level of the control (t-test, P > 0.05). The immunohistochemical signals for HBx and HBsAg were far weaker in the liver tissue sections on day 12 after the injection of pXPDnmt3aC than in the vector control in the M-TgHBV mice (Fig. 5E).
Fig. 5. Representative expression assay of XPDnmt3aC, HBsAg, and HBx in the livers of M-TgHBV mice. A) Western blotting analysis at appointed times after injection of pXPDnmt3aC or vector pcDNA3.1(+). Relative levels of XPDnmt3aC (B), HBx (C), and HBsAg (D) were quantified by densitometry and normalized to that of β-actin. Data are shown as means ± SD of measurements made in three mice. *P < 0.05, significantly different from day 0 control in the same group; aP < 0.05, significantly different from negative control at the same time point. “T” represents test groups and “C” the negative control. E) Representative immunohistochemical assays. Staining was performed 12 day after injection of plasmids. Panels (a) and (e) show Dnmt3a stained with specific antibodies, (b) and (f), HBx; (c) and (g), HBsAg; and (d) and (h), hematoxylin–eosin (HE) stained sections. Original magnification, ×400.
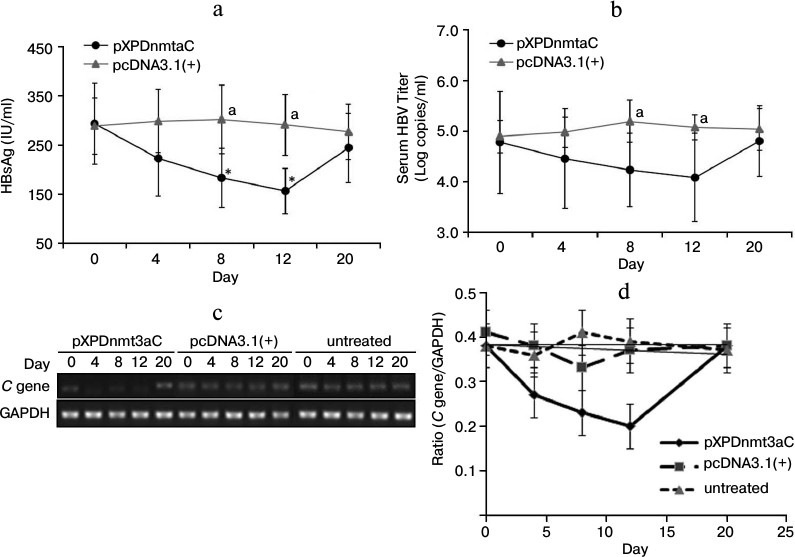
The serum levels of HBsAg were determined using a chemiluminescent microparticle immunoassay. There was a sharp decline from day 4 after the injection of pXPDnmt3aC, and on day 12, HBsAg was only about 58% of the day 0 level (t-test, P < 0.05; Fig. 6a). The number of viral copies varied in a similar way to HBsAg levels and was significantly reduced (by 19%) on day 12 compared with the number in the negative vector control (t-test, P < 0.05; Fig. 6b). The expression of the C gene was downregulated from day 4 and had declined to approximate 56% of the vector control value on day 12 (t-test, P < 0.05; Fig. 6, c and d). However, the expression of all three molecules had rebounded on day 20 (t-test, P > 0.05), which is consistent with the results observed in cells. These results further indicated that XPDnmt3aC silenced HBV to some extent.
Fig. 6. Effect of XPDnmt3aC on HBV expression in M-TgHBV mice. Data are shown as means ± SD (n = 5). a-d) Serum HBsAg and virus, and C gene expression in liver tissues were measured at appointed times after the mice were injected with plasmid pXPDnmt3aC or vector pcDNA3.1(+). Levels of virus had rebounded slightly on day 20. *P < 0.05, significantly different from the same group on day 0; aP < 0.05, significantly different at the same time point from the group treated with the control vector.
DISCUSSION
Prior studies have established that the high incidence of drug resistance to nucleos(t)ide analogs and persistent cccDNA increased the risk of hepatitis relapse [2-4, 26]. Both HBx and HBsAg have transactivational potential to stimulate transcription from HBV’s own promoters and from some oncogene promoters, including the c-myc promoter, and contribute to hepatocellular carcinoma [27]. It has been widely assumed that viral clearance is chiefly mediated by the destruction of infected cells by viral-antigen-specific cytotoxic T lymphocytes and that the pathogenesis of persistent hepadnavirus infection is also mediated by these cells [28]. Therefore, it is important to find a means of silencing HBV to prevent and treat chronic HBV-related diseases. Specific DNA methylation mediated by zinc-finger protein had been studied for ten years and showed obviously inhibitory role in the expression of the target gene [11-16]. Given that XP activity can be potentially regulated by methylation and HBx is important for HBV replication [29], we explored whether the recombinant methylase targeted to XP could silence HBV by site-specific DNA methylation. In our current study, we provided evidence that XPDnmt3aC specifically methyl-modified the target sequence in XP, which resulted in repression of HBV expression.
To achieve good specificity, the Cys2-His2-type zinc-finger DNA-binding proteins have been used to bind specifically to many different DNA sequences [12-15, 30]. In past studies, it has been well established that using three, four, or six zinc fingers as the target domain can lead to specific methylation [12-15]. In this study, we devised a six-zinc-finger-inducing DNA methyltransferase targeted to XP to achieve higher specificity. The specifically methylated CpGs were primarily located in the region upstream from the zinc-finger-binding sites, which may be attributable to the binding of the N-terminus of zinc fingers of XPDnmt3aC to the 3′ terminus of the targeted DNA sequence. The CpG sites in the region ranging from the zinc-finger-binding site to –111 bp upstream were frequently methylated, which might be attributable to the folding pattern of the targeted DNA. Even though we did not distinguish various forms of HBV DNA, nude cccDNA can be specifically methylated, whereas the cccDNA that forms a mini-chromosome with other proteins might be difficult to be methyl-modified because of steric hindrance.
Some N-terminal domains, such as the cysteine-rich zinc-finger DNA-binding motif and the polybromo-homology domain, might contribute to the usual off-target effects of Dnmt3a [10]. Therefore, its C-terminal catalytic domain was used to construct the fused DNA methyltransferase in this study. XPDnmt3aC clearly suppressed HBV expression and replication in vivo and in vitro. However, the expression of HBx, HBsAg, the virus, and the C gene rebounded slightly at a later stage, accompanied by a reduction in XPDnmt3aC, which implies the possible loss of de novo methylation and replication of some unmodified virus. Prolonging the administration of XPDnmt3aC might help to achieve a cure. Therefore, a method to increase the stability of the methylation warrants further study.
As a conclusion, the data obtained in this study demonstrate that this fused DNA methyltransferase can inhibit HBV expression and replication. This study established a valuable foundation for an epigenetic regulatory technique to control HBV-related diseases.
We thank Arthur D. Riggs (Beckman Research Institute of the City of Hope) for kindly providing the original plasmid (pcDNA3/Flag-DNMT3A).
This work was supported by the Major National Projects for Infectious Diseases (2008ZX10002002 and 2012ZX10002-006) and for New Drug Research and Development (2009ZX0913-710), the National Natural Science Foundation of China (30901315 and 30970144), and the Major National Projects for Infectious Diseases (2012zx10002006-002-003).
REFERENCES
1.Gonzalez, S. A., and Keeffe, E. B. (2011) Front.
Biosci., 16, 225-250.
2.Janssen, H. L., Gerken, G., Carreno, V., Marcellin,
P., Naoumov, N. V., Craxi, A., Ring-Larsen, H., Kitis, G., van Hattum,
J., de Vries, R. A., Michielsen, P. P., ten Kate, F. J., Hop, W. C.,
Heijtink, R. A., Honkoop, P., and Schalm, S. W. (1999)
Hepatology, 30, 238-243.
3.Warner, N., and Locarnini, S. (2008)
Hepatology, 48, 88-98.
4.Kwon, H., and Lok, A. S. (2011) Nat. Rev.
Gastroenterol. Hepatol., 8, 275-284.
5.Huang, J., Wang, Y., Guo, Y., and Sun, S. (2010)
Hepatology, 52, 60-70.
6.Bergbauer, M., Kalla, M., Schmeinck, A., Gobel, C.,
Rothbauer, U., Eck, S., Benet-Pages, A., Strom, T. M., and
Hammerschmidt, W. (2010) PLoS Pathog., 6, e1001114.
7.Jones, B., and Chen, J. (2006) EMBO J.,
25, 2443-2452.
8.Kauder, S. E., Bosque, A., Lindqvist, A.,
Planelles, V., and Verdin, E. (2009) PLoS Pathog., 5,
e1000495.
9.Guo, Y., Li, Y., Mu, S., Zhang, J., and Yan, Z.
(2009) J. Med. Virol., 81, 1177-1183.
10.Kim, J. W., Lee, S. H., Park, Y. S., Hweng, J.
H., Jeong, S. H., Kim, N., and Lee, D. H. (2011) Intervirology,
54, 316-325.
11.Turek-Plewa, J., and Jagodzinski, P. P. (2005)
Cell Mol. Biol. Lett., 10, 631-647.
12.Xu, G. L., and Bestor, T. (1997) Nat.
Genet., 17, 376-378.
13.Carvin, C. D., Parr, R. D., and Kladde, M. P.
(2003) Nucleic Acids Res., 31, 6493-6501.
14.Smith, A. E., and Ford, K. (2007) Nucleic
Acids Res., 35, 740-754.
15.Li, F., Papworth, M., Minczuk, M., Rohde, C.,
Zhang, Y., Ragozin, S., and Jeltsch, A. (2007) Nucleic Acids
Res., 35, 100-112.
16.Meister, G. E., Chandrasegaran, S., and
Ostermeier, M. (2010) Nucleic Acids Res., 38,
1749-1759.
17.Smith, A. E., Hurd, P. J., Bannister, A. J.,
Kouzarides, T., and Ford, K. G. (2008) J. Biol. Chem.,
283, 9878-9885.
18.Oka, M., Rodic, N., Graddy, J., Chang, L. J., and
Terada, N. (2006) J. Biol. Chem., 281, 9901-9908.
19.Mandell, J. G., and Barbas, C. F. (2006)
Nucleic Acids Res., 34, W516-523.
20.Cui, J. (2009) A New Strategy for Constructing
in vitro Replication-Competent Replicon of Hepatitis B Virus and Its
Preliminary Application, in Medicine, Chongqing Medical
University, Chongqing.
21.Ren, J., Wang, L., Chen, Z., Ma, Z. M., Zhu, H.
G., Yang, D. L., Li, X. Y., Wang, B. I., Fei, J., Wang, Z. G., and Wen,
Y. M. (2006) J. Med. Virol., 78, 551-560.
22.Pollicino, T., Belloni, L., Raffa, G., Pediconi,
N., Squadrito, G., Raimondo, G., and Levrero, M. (2006)
Gastroenterology, 130, 823-837.
23.Tang, B., Zhao, R., Sun, Y., Zhu, Y., Zhong, J.,
Zhao, G., and Zhu, N. (2011) Mol. Immunol., 48,
1001-1008.
24.Hennig, H., Puchta, I., Luhm, J., Schlenke, P.,
Goerg, S., and Kirchner, H. (2002) Blood, 100,
2637-2641.
25.Bock, C., Reither, S., Mikeska, T., Paulsen, M.,
Walter, J., and Lengauer, T. (2005) Bioinformatics, 21,
4067-4068.
26.Caselmann, W. H., Meyer, M., Kekule, A. S.,
Lauer, U., Hofschneider, P. H., and Koshy, R. (1990) Proc. Natl.
Acad. Sci. USA, 87, 2970-2974.
27.Hildt, E., Saher, G., Bruss, V., and
Hofschneider, P. H. (1996) Virology, 225, 235-239.
28.Chisari, F. V., and Ferrari, C. (1995) Annu.
Rev. Immunol., 13, 29-60.
29.Lucifora, J., Arzberger, S., Durantel, D.,
Belloni, L., Strubin, M., Levrero, M., Zoulim, F., Hantz, O., and
Protzer, U. (2011) J. Hepatol., 55, 996-1003.
30.Liu, Q., Xia, Z., Zhong, X., and Case, C. C.
(2002) J. Biol. Chem., 277, 3850-3856.