Mitochondria-Targeted Antioxidants Prevent TNFα-Induced Endothelial Cell Damage
I. I. Galkin1,2*, O. Yu. Pletjushkina1,3, R. A. Zinovkin2,3, V. V. Zakharova3,4, I. S. Birjukov4, B. V. Chernyak1,3, and E. N. Popova1,3
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Vorobyevy Gory 1, 119991 Moscow, Russia; fax: +7 (495) 939-0338; E-mail: galkin.ivan.i@gmail.com2Biological Faculty, Lomonosov Moscow State University, Vorobyevy Gory 1, 119991 Moscow, Russia
3Institute of Mitoengineering, Lomonosov Moscow State University, Vorobyevy Gory 1, 119991 Moscow, Russia
4Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Vorobyevy Gory 1, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received September 24, 2013; Revision received October 23, 2013
Increased serum level of tumor necrosis factor α (TNFα) causes endothelial dysfunction and leads to serious vascular pathologies. TNFα signaling is known to involve reactive oxygen species (ROS). Using mitochondria-targeted antioxidant SkQR1, we studied the role of mitochondrial ROS in TNFα-induced apoptosis of human endothelial cell line EAhy926. We found that 0.2 nM SkQR1 prevents TNFα-induced apoptosis. SkQR1 has no influence on TNFα-dependent proteolytic activation of caspase-8 and Bid, but it inhibits cytochrome c release from mitochondria and cleavage of caspase-3 and its substrate PARP. SkQ analogs lacking the antioxidant moieties do not prevent TNFα-induced apoptosis. The antiapoptotic action of SkQR1 may be related to other observations made in these experiments, namely SkQR1-induced increase in Bcl-2 and corresponding decrease in Bax as well as p53. These results indicate that mitochondrial ROS production is involved in TNFα-initiated endothelial cell death, and they suggest the potential of mitochondria-targeted antioxidants as vasoprotectors.
KEY WORDS: endothelium, apoptosis, mitochondria-targeted antioxidant, inflammation, TNFαDOI: 10.1134/S0006297914020059
Abbreviations: C12R1, rhodamine 19 dodecyl ester; C12TPP, dodecyltriphenylphosphonium; NAC, N-acetylcysteine; PARP, poly(ADP-ribose) polymerase; ROS, reactive oxygen species; SkQs, conjugates of plastoquinone and penetrating cations; SkQ1, plastoquinonyl-10(6′-decyltriphenyl)phosphonium; SkQR1, 10-(6′-plastoquinonyl)decylrhodamine 19; TMRM, tetramethylrhodamine methyl ester; TNFα, tumor necrosis factor α; zVAD, N-benzyloxycarbonyl-Val-Ala-Asp-trifluoromethyl ketone.
Vascular endothelium performs numerous functions: it controls the blood
clotting system, vascular tone, tissue perfusion, exchange of fluid and
macromolecules between the blood and tissues, and immune system
development and immune response [1]. Therefore, any
damage or excessive activation of the endothelium leads to serious
pathologies [2]. Various stimuli considered as
cardiovascular risk factors (e.g. tumor necrosis factor alpha
(TNFα)) cause apoptosis of endothelial cells in vitro [3]. Moreover, apoptosis of endothelial cells was
observed in vivo in humans and other animals with various
pathologies and in old animals [3, 4]. One of the significant risk factors for
endothelial dysfunction is the increase in circulatory inflammatory
cytokines, particularly TNFα [5].
TNFα binding to its receptors on the cell surface triggers a series of complex signaling cascades [6, 7] leading to different effects depending on the conditions and type of cell. In endothelial cells, TNFα activates secretion of blood coagulation factors, NO-synthase, expression of tissue factors, adhesion molecules, inflammatory cytokines and chemokines, transendothelial vesicular transport, cytoskeleton reorganization, disassembly of cell–cell contacts, and cell death [5]. TNFα can initiate endothelial cell apoptosis either due to direct activation of caspase cascades [8] or due to release of mitochondrial proapoptotic proteins (cytochrome c, AIF, DIABLO, etc.) [9].
Reactive oxygen species (ROS) are important factors modulating TNFα signaling. TNFα-induced ROS production leads to signal amplification [10]. ROS are produced both by mitochondria and by the NOX family of NADPH oxidases. Overexpression of the mitochondrial superoxide dismutase MnSOD leads to suppression of NFκB and MAPK activities [11] and of apoptosis [12], indicating the important role of mitochondrial ROS. In the present study, we utilized a different approach based on the use of mitochondria-targeted antioxidants of the SkQ family [13]. These compounds belong to a group of penetrating cations. They are accumulated effectively in mitochondria due to the high potential difference on the inner mitochondrial membrane. At nanomolar concentrations, SkQs prevent cell death induced by oxidative stress and modulate several signaling pathways [14-17].
Here we demonstrate that mitochondria-targeted antioxidants prevent TNFα-induced apoptosis. Probably, the vasoprotective action of SkQ1 and SkQR1 explains the therapeutic effect of these compounds observed in renal and brain failure animal models [18, 19].
MATERIALS AND METHODS
Cell line and reagents. Endothelial cell line EaHy926 was cultured in DMEM medium (Dulbecco’s modified Eagle’s medium) (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone, USA) and with hypoxanthine and thymidine (PanEco, Russia) at 37°C in 5% CO2 at 100% humidity [20]. The antioxidants or control compounds were added to the cells and incubated for 4 days. The medium was replaced with DMEM containing 0.2% FBS, and after 24 h human recombinant TNFα (courtesy of L. Shingarova, Institute of Bioorganic Chemistry, Moscow) was added (1-10 ng/ml) for 3-18 h (for details, please refer to figure legends). In some experiments, 15 min before the TNFα treatment 1 µM zVAD-fmk (Sigma, USA) was added to the cells. SkQ1, SkQR1, C12TPP, and C12R1 were synthesized by G. A. Korshunova and N. V. Sumbatyan as described before [21, 22].
Western blot analysis. The cells were harvested, washed with cold PBS, and lysed in sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mM DTT, 0.01% bromophenol blue). The lysates were boiled for 3 min at 95°C. The proteins in the lysates were separated by polyacrylamide gel electrophoresis and transferred to PVDF-membrane (Amersham, USA). The primary antibodies used were to cytochrome c, Bax, p53, Bcl-2, PARP, as well as activated forms of caspase-3, caspase-8, and Bid (Cell Signaling, USA). The secondary anti-rabbit antibody conjugated with horseradish peroxidase (HRP) was from Sigma-Aldrich (USA). ECL substrate kit (Amersham) was used for HRP detection. ImageJ 1.44p software was used for densitometric analysis.
Subcellular fractionation and cytochrome c determination. Subcellular fractions were collected as described earlier [23]. Cells were collected, washed twice with cold PBS, resuspended in homogenizing buffer (8.6% sucrose, 10 mM KCl, 20 mM HEPES, 2 mM EDTA, 1 mM MgCl2, 1 mM DTT, pH 7.4), and incubated for 25 min on ice with occasional shaking. The cells were homogenized with a glass homogenizer, centrifuged (1000g, 10 min), and the supernatant was collected and centrifuged at 16,000g, 15 min. The supernatant was collected and stored at –80°C.
Cytotoxicity test. Two days after TNFα treatment, the cells were incubated in 0.5% solution of MTT (Sigma) for 3 h. The medium was removed, and MTT-formazan crystals were dissolved in DMSO (Sigma). Absorbance was measured using a Thermo Labsystems Multiscan EX plate colorimeter at wavelength 541 nm.
Detection of ROS. ROS production was measured after loading the cells with 5 μM CM-DCF-DA (5-(-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate) (Molecular Probes, USA) for 15 min at 37°C. Flow cytometric analysis was performed using a Beckman Coulter FC500 system as described in [14].
Estimation of apoptosis. Two days after TNFα treatment, the cells were collected, washed with cold PBS, fixed with ice-cold mixture of PBS–ethanol (1 : 2), and incubated overnight at 4°C. The cells were washed with PBS and stained in PBS containing 30 µg/ml propidium iodide (MP Biomedicals, France) and 10 ng/ml RNase A (Fermentas, Lithuania) in the dark for 45 min at 37°C and then analyzed with the Beckman Coulter FC500 flow cytometer.
Visualization of mitochondria. Mitochondria were visualized using MitoTracker Green (100 µM, 30 min) (Molecular Probes), mitochondrial polarization was determined using TMRM (100 µM, 30 min) (Sigma), and accumulation of SkQR1 in mitochondria was also examined [22, 24].
Statistical analysis. Statistical analysis was performed using the Statistics 6.0 software. Data are presented as means ± standard error of the mean (SEM). Statistical significance was calculated using the unpaired Student’s t-test.
RESULTS AND DISCUSSION
The endothelial cell line EAhy926 was initially obtained by fusion of human umbilical vein endothelial (HUVEC) and human lung carcinoma cells (A549) [20]. This cell line is well characterized, retains essential characteristics of endothelial cells, and is widely used to study endothelial cells response to inflammatory stimuli [25].
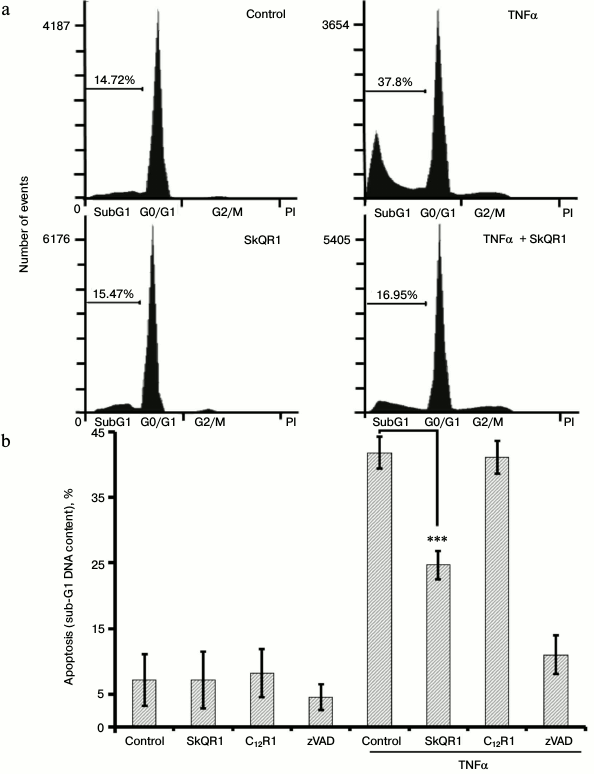
The cytotoxic effect of TNFα on endothelial cells varies considerably among different cell lines and primary cultures. In some cases, TNFα alone can induce apoptosis [26], but in other cases additional cytokines or hydrogen peroxide are required [27, 28]. In our experiments, TNFα caused a pronounced cytotoxic effect that reached a maximum of 35-45% dead cells at the concentration of 5 ng/ml (Fig. 1a). The cell death had all the morphological features of apoptosis: membrane blebbing; chromatin condensation and fragmentation (not shown) accompanied by proteolytic cleavage of Bid, caspase-8, -3, and PARP (Fig. 2, a and b). Flow cytometric analysis revealed a population of cells with reduced DNA content (sub-G1) that allowed us to estimate the percentage of apoptotic cells (Fig. 3a). The broad spectrum caspase inhibitor zVAD-fmk prevented cell death and the appearance of the sub-G1 population (Fig. 3b).
Fig. 1. SkQR1 (4-day pretreatment), but not C12R1, reduces the cytotoxic activity of TNFα (48 h). Cell viability was estimated by the MTT test. a) Effect of SkQR1 (2 nM), N-acetylcysteine (NAC) (5 mM), and Trolox (0.2 mM) on viability of TNFα-treated cells; b) cytotoxic action of TNFα (5 ng/ml) on SkQR1 or C12R1 pretreated cells; * p < 0.05; *** p < 0.0002 compared to control without these substances. Control, vehicle only. This data is representative of eight independent experiments.
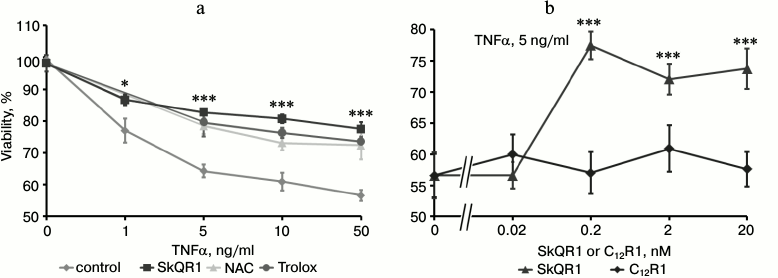
Fig. 2. SkQR1 (20 nM, 4 days) suppresses TNFα-induced (50 ng/ml, 8 h) release of cytochrome c and cleavage of caspase 3 and PARP but does not affect the cleavage of caspase 8 and Bid. a) Detection of components of cleaved proapoptotic proteins and PARP in total lysates. The result of a typical experiment is shown; b) densitometric analysis of Western blot experiments; * p < 0.05; ** p < 0.002; c) detection of cytochrome c in cytoplasmic fraction. Tubulin amount was measured as a load control. The result of a typical experiment is shown.
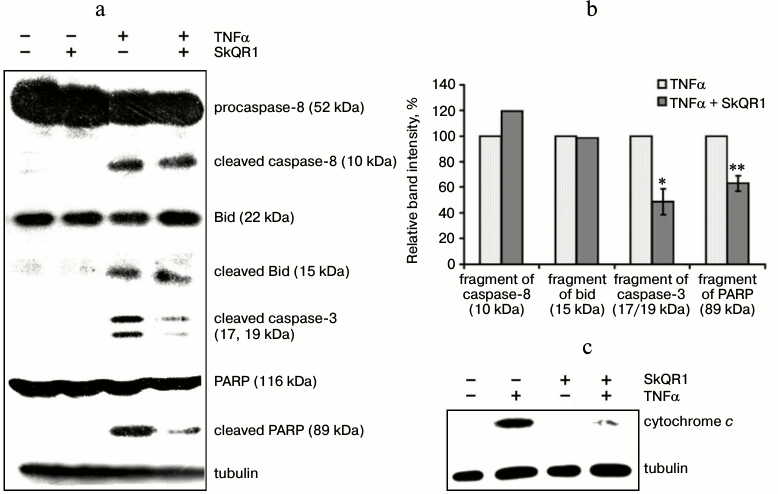
Fig. 3. SkQR1 (20 nM, 4-day pretreatment) suppresses TNFα-induced (10 ng/ml, 48 h) apoptosis. The percentage of apoptotic cells with hypodiploid (sub-G1) DNA content is indicated. C12R1 (20 nM) was added analogously to SkQR1, and zVAD-fmk (50 µM) – 15 min before TNFα addition. a) FACS analysis of cell cycle, a typical experiment being presented. b) Statistical analysis of at least seven separate experiments; *** p < 0.0001 compared to control without SkQR1. Control, vehicle.
SkQR1 inhibited TNFα-induced apoptosis in EAhy926 cells, being most effective at the concentration of 0.2 nM (Fig. 1b). Another mitochondria-targeted antioxidant SkQ1 also possessed antiapoptotic activity (data not shown). The SkQR1 analog devoid of the plastoquinone antioxidant group (C12R1) did not protect cells from TNFα-induced apoptosis (Figs. 1b and 3b). The same effect was observed with SkQ1, while its analog devoid of plastoquinone (C12TPP) did not prevent apoptosis (data is not shown). The antioxidants NAC and Trolox prevented cell death at concentrations of 5 and 0.2 mM, respectively (Fig. 1a).
In various cellular models, TNFα induces a rapid (during 5-15 min) transient increase in ROS production. Then ROS level decreases and increases again in few hours after TNFα treatment [29-31]. ROS measurement using the fluorescent probe CM-DCF-DA revealed only the late phase of ROS increase (~30% after 15 h of TNFα treatment, data not shown). In this case the mitochondria-targeted antioxidants, unlike NAC, did not reduce ROS level. Perhaps mitochondrial ROS accumulated only in the first phase of TNFα response and did not contribute significantly to the steady-state level of hydrogen peroxide. However, the antiapoptotic action of SkQ indicates that TNFα-induced apoptosis is largely dependent on mitochondrial ROS.
TNFα-induced apoptosis starts with the autocatalytic proteolysis of caspase-8, which is associated with TNFα receptor and adapter proteins [32]. Cleavage of caspase-8 was detected in EAhy926 cells after TNFα treatment, and it was not affected by SkQR1 (Fig. 2, a and b). Antioxidants also did not inhibit the next steps, i.e. caspase-8-mediated cleavage and activation of the Bid protein (Fig. 2, a and b). Activated Bid binds to mitochondria and triggers the release of proapoptotic proteins from the intermembrane space [33]. Data presented in Fig. 2c show that SkQR1 inhibited TNFα-induced cytochrome c release. SkQR1 also inhibited the next signaling steps: cleavage of caspase-3 and its substrate PARP (Fig. 2, a and b). Thus, it can be assumed that the protective effect of SkQ compounds is directed at the components of mitochondria responsible for release of proapoptotic proteins.
The most likely candidate as an SkQ target is mitochondrial lipid cardiolipin, which is very prone to peroxidation. Its oxidation may serve as a trigger to start mitochondrial membrane lipid peroxidation and ROS generation [13, 34]. Cardiolipin acts as an anchor for cytochrome c at the outer surface of the inner mitochondrial membrane [35]. Cardiolipin oxidation promotes release of cytochrome c into the cytoplasm during apoptosis [36]. In turn, solubilized cytochrome c functions as cardiolipin peroxidase, promoting further cardiolipin oxidation [37]. In isolated mitochondria, mitochondria-targeted antioxidants effectively inhibit cardiolipin peroxidation during oxidative stress [13, 24]. Thus, cardiolipin appears to be a very likely candidate for the primary mitochondrial ROS target in TNFα-induced apoptosis.
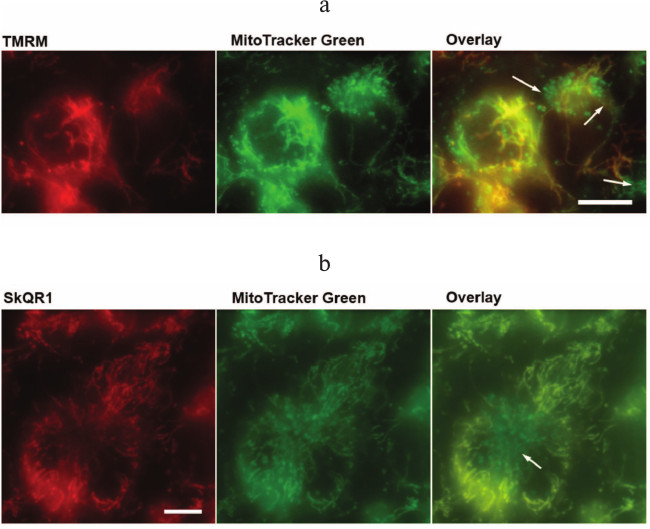
It is noteworthy that contrary to NAC and Trolox, the antiapoptotic action of SkQR1 is developed only after 4-day preincubation with cells (data not shown). However, fluorescent SkQR1 accumulated in cells within 1-2 h [38]. The delay in the appearance of the protective effect may be due to the existence of a small fraction of mitochondria with low potential and hence slow accumulation of SkQ cationic antioxidants [39]. Using a combination of MitoTracker Green and TMRM, we revealed a significant fraction of depolarized mitochondria located near the cell nucleus in EAhy926 cells (Fig. 4a; see color insert). It can be suggested that these mitochondria produce excessive ROS, which leads to the increased oxidation of cardiolipin that in turn accelerates and simplifies cytochrome c release from mitochondria under various stimuli. Presumably these mitochondria indeed determine the fate of cells when exposed to TNFα or other apoptotic stresses. The in vitro studies with SkQR1 revealed the population of mitochondria with absence of SkQR1 (Fig. 4b; see color insert). Upon prolonged incubation, the mitochondria-targeted antioxidants may accumulate in the mitochondria due to fluctuations of the membrane potential and fusion/fission of mitochondrial reticulum.
Fig. 4. Mitochondria with low potential in EAhy926 cells. Accumulation of TMRM (100 µM, 30 min) (a) and SkQR1 (20 nM, 2 h) (b) in mitochondria stained with Mitotracker Green (1 µM, 30 min). The arrows indicate mitochondria with low membrane potential and SkQR1 content. The bar size is 15 µm. The result of a typical experiment is shown.
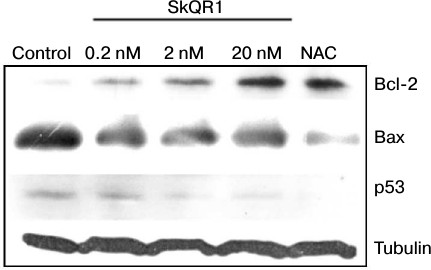
Along with the proposed explanation, we cannot exclude the possibility of induction of protective systems by mitochondria-targeted compounds. We have observed an increase in antiapoptotic protein Bcl-2 content with corresponding decrease in proapoptotic proteins Bax and p53 in cells treated with SkQR1 (Fig. 5) or SkQ1 (data not shown). NAC induced the same changes at 5 mM (Fig. 5), while the analogs of SkQR1 and SkQ1 devoid of the plastoquinone did not cause these effects (not shown). The Bcl-2 family proteins control cytochrome c release from mitochondria [40]. It was shown that inhibition of MnSOD resulting in mtROS production makes cells more sensitive to apoptotic stimuli via regulation of Bcl-2 family proteins [41]. ROS-dependent increase in proapoptotic protein Bcl-2 family members (e.g. Bax) and decrease in antiapoptotic ones (e.g. Bcl-2) were inhibited by various antioxidants, as shown before [42-44]. ROS-induced upregulation of p53 is well documented [41]. SkQ probably decreased the ratio of Bax/Bcl-2 and downregulated p53 due to lowering of basal mtROS level and, as a consequence, suppressed the TNFα-induced apoptosis.
Fig. 5. SkQR1 (20 nM, 4 days) and NAC (5 mM, 4 days) induce Bcl-2 and decrease Bax and p53 expression in EAhy926 cells. Control, vehicle. The result of a typical experiment is shown.
The high efficiency of mitochondria-targeted antioxidants of the SkQ family in protection of endothelial cells against TNFα-induced apoptosis allow considering these substances as potential vasoprotectors.
We would like to thank V. P. Skulachev for the critical discussion of the manuscript and providing valuable suggestions.
This work was supported by the Russian Foundation for Basic Research (grant Nos. 12-04-01563-a, 12-04-00538-a and 13-04-40309H).
REFERENCES
1.Pober, J. S., Min, W., and Bradley, J. R. (2009)
Annu. Rev. Pathol., 4, 71-95.
2.Verrier, E. D., and Boyle, E. M., Jr. (1996)
Ann. Thorac. Surg., 62, 915-922.
3.Winn, R. K., and Harlan, J. M. (2005) J. Thromb.
Haemost., 3, 1815-1824.
4.Tham, D. M., Martin-McNulty, B., Wang, Y. X.,
Wilson, D. W., Vergona, R., Sullivan, M. E., Dole, W., and Rutledge, J.
C. (2002) Physiol. Genom., 11, 21-30.
5.Zhang, H., Park, Y., Wu, J., Chen, X., Lee, S.,
Yang, J., Dellsperger, K. C., and Zhang, C. (2009) Clin. Sci.
(Lond.), 116, 219-230.
6.Madge, L. A., and Pober, J. S. (2001) Exp. Mol.
Pathol., 70, 317-325.
7.Wajant, H., Pfizenmaier, K., and Scheurich, P.
(2003) Cell Death Differ., 10, 45-65.
8.Wang, L., Du, F., and Wang, X. (2008) Cell,
133, 693-703.
9.Walter, D. H., Haendeler, J., Galle, J., Zeiher, A.
M., and Dimmeler, S. (1998) Circulation, 98,
1153-1157.
10.Goossens, V., De Vos, K., Vercammen, D.,
Steemans, M., Vancompernolle, K., Fiers, W., Vandenabeele, P., and
Grooten, J. (1999) Biofactors, 10, 145-156.
11.Li, J. J., Oberley, L. W., Fan, M., and Colburn,
N. H. (1998) FASEB J., 12, 1713-1723.
12.Zhao, Y., Kiningham, K. K., Lin, S. M., and St
Clair, D. K. (2001) Antioxid. Redox Signal., 3,
375-386.
13.Skulachev, V. P., Antonenko, Y. N., Cherepanov,
D. A., Chernyak, B. V., Izyumov, D. S., Khailova, L. S., Klishin, S.
S., Korshunova, G. A., Lyamzaev, K. G., Pletjushkina, O. Y., Roginsky,
V. A., Rokitskaya, T. I., Severin, F. F., Severina, I. I., Simonyan, R.
A., Skulachev, M. V., Sumbatyan, N. V., Sukhanova, E. I., Tashlitsky,
V. N., Trendeleva, T. A., Vyssokikh, M. Y., and Zvyagilskaya, R. A.
(2010) Biochim. Biophys. Acta, 1797, 878-889.
14.Chernyak, B. V., Izyumov, D. S., Lyamzaev, K. G.,
Pashkovskaya, A. A., Pletjushkina, O. Y., Antonenko, Y. N., Sakharov,
D. V., Wirtz, K. W., and Skulachev, V. P. (2006) Biochim. Biophys.
Acta, 1757, 525-534.
15.Pletjushkina, O. Y., Lyamzaev, K. G., Popova, E.
N., Nepryakhina, O. K., Ivanova, O. Y., Domnina, L. V., Chernyak, B.
V., and Skulachev, V. P. (2006) Biochim. Biophys. Acta,
1757, 518-524.
16.Popova, E. N., Pletjushkina, O. Y., Dugina, V.
B., Domnina, L. V., Ivanova, O. Y., Izyumov, D. S., Skulachev, V. P.,
and Chernyak, B. V. (2010) Antioxid. Redox Signal., 13,
1297-1307.
17.Agapova, L. S., Chernyak, B. V., Domnina, L. V.,
Dugina, V. B., Efimenko, A. Y., Fetisova, E. K., Ivanova, O. Y.,
Kalinina, N. I., Khromova, N. V., Kopnin, B. P., Kopnin, P. B.,
Korotetskaya, M. V., Lichinitser, M. R., Lukashev, A. L., Pletjushkina,
O. Y., Popova, E. N., Skulachev, M. V., Shagieva, G. S., Stepanova, E.
V., Titova, E. V., Tkachuk, V. A., Vasiliev, J. M., and Skulachev, V.
P. (2008) Biochemistry (Moscow), 73, 1300-1316.
18.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. B., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashook, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasilyeva, A. K., Victorov, I. V., Zorov, D. B., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1288-1299.
19.Plotnikov, E. Y., Silachev, D. N., Chupyrkina, A.
A., Danshina, M. I., Jankauskas, S. S., Morosanova, M. A., Stelmashook,
E. V., Vasileva, A. K., Goryacheva, E. S., Pirogov, Y. A., Isaev, N.
K., and Zorov, D. B. (2010) Biochemistry (Moscow), 75,
145-150.
20.Edgell, C. J., McDonald, C. C., and Graham, J. B.
(1983) Proc. Natl. Acad. Sci. USA, 80, 3734-3737.
21.Lyamzaev, K. G., Pustovidko, A. V., Simonyan, R.
A., Rokitskaya, T. I., Domnina, L. V., Ivanova, O. Y., Severina, I. I.,
Sumbatyan, N. V., Korshunova, G. A., Tashlitsky, V. N., Roginsky, V.
A., Antonenko, Y. N., Skulachev, M. V., Chernyak, B. V., and Skulachev,
V. P. (2011) Pharm. Res., 28, 2883-2895.
22.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L.
E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Y.,
Izyumov, D. S., Khailova, L. S., Klishin, S. S., Korshunova, G. A.,
Lyamzaev, K. G., Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A.
A., Pletjushkina, O. Y., Pustovidko, A. V., Roginsky, V. A.,
Rokitskaya, T. I., Ruuge, E. K., Saprunova, V. B., Severina, I. I.,
Simonyan, R. A., Skulachev, I. V., Skulachev, M. V., Sumbatyan, N. V.,
Sviryaeva, I. V., Tashlitsky, V. N., Vassiliev, J. M., Vyssokikh, M.
Y., Yaguzhinsky, L. S., Zamyatnin, A. A., Jr., and Skulachev, V. P.
(2008) Biochemistry (Moscow), 73, 1273-1287.
23.Ethell, D. W., and Green, D. R. (2002) in
Apoptosis Techniques and Protocols (LeBlanc, A., ed.) Springer,
New York, pp. 21-34.
24.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) Biochim. Biophys. Acta, 1787, 437-461.
25.Ahn, K., Pan, S., Beningo, K., and Hupe, D.
(1995) Life Sci., 56, 2331-2341.
26.Polunovsky, V. A., Wendt, C. H., Ingbar, D. H.,
Peterson, M. S., and Bitterman, P. B. (1994) Exp. Cell Res.,
214, 584-594.
27.Ohmori, Y., Schreiber, R. D., and Hamilton, T. A.
(1997) J. Biol. Chem., 272, 14899-14907.
28.Luo, T., and Xia, Z. (2006) Anesth.
Analg., 103, 110-116, table of contents.
29.Woo, C. H., Eom, Y. W., Yoo, M. H., You, H. J.,
Han, H. J., Song, W. K., Yoo, Y. J., Chun, J. S., and Kim, J. H. (2000)
J. Biol. Chem., 275, 32357-32362.
30.Corda, S., Laplace, C., Vicaut, E., and
Duranteau, J. (2001) Am. J. Respir. Cell Mol. Biol., 24,
762-768.
31.Deshpande, S. S., Angkeow, P., Huang, J., Ozaki,
M., and Irani, K. (2000) FASEB J., 14, 1705-1714.
32.Martin, D. A., Siegel, R. M., Zheng, L., and
Lenardo, M. J. (1998) J. Biol. Chem., 273, 4345-4349.
33.Luo, X., Budihardjo, I., Zou, H., Slaughter, C.,
and Wang, X. (1998) Cell, 94, 481-490.
34.Antonenko, Y. N., Roginsky, V. A., Pashkovskaya,
A. A., Rokitskaya, T. I., Kotova, E. A., Zaspa, A. A., Chernyak, B. V.,
and Skulachev, V. P. (2008) J. Membr. Biol., 222,
141-149.
35.Tuominen, E. K., Wallace, C. J., and Kinnunen, P.
K. (2002) J. Biol. Chem., 277, 8822-8826.
36.Choi, S. Y., Gonzalvez, F., Jenkins, G. M.,
Slomianny, C., Chretien, D., Arnoult, D., Petit, P. X., and Frohman, M.
A. (2007) Cell Death Differ., 14, 597-606.
37.Kagan, V. E., Tyurin, V. A., Jiang, J., Tyurina,
Y. Y., Ritov, V. B., Amoscato, A. A., Osipov, A. N., Belikova, N. A.,
Kapralov, A. A., Kini, V., Vlasova, I. I., Zhao, Q., Zou, M., Di, P.,
Svistunenko, D. A., Kurnikov, I. V., and Borisenko, G. G. (2005)
Nat. Chem. Biol., 1, 223-232.
38.Izyumov, D. S., Domnina, L. V., Nepryakhina, O.
K., Avetisyan, A. V., Golyshev, S. A., Ivanova, O. Y., Korotetskaya, M.
V., Lyamzaev, K. G., Pletjushkina, O. Y., Popova, E. N., and Chernyak,
B. V. (2010) Biochemistry (Moscow), 75, 123-129.
39.Kuznetsov, A. V., and Margreiter, R. (2009)
Int. J. Mol. Sci., 10, 1911-1929.
40.Thomenius, M. J., and Distelhorst, C. W. (2003)
J. Cell Sci., 116, 4493-4499.
41.Li, D., Ueta, E., Kimura, T., Yamamoto, T., and
Osaki, T. (2004) Cancer Sci., 95, 644-650.
42.Luanpitpong, S., Chanvorachote, P., Nimmannit,
U., Leonard, S. S., Stehlik, C., Wang, L., and Rojanasakul, Y. (2012)
Biochem. Pharmacol., 83, 1643-1654.
43.Nakamura, T., and Sakamoto, K. (2001) Biochem.
Biophys. Res. Commun., 284, 203-210.
44.Aoki, M., Nata, T., Morishita, R., Matsushita,
H., Nakagami, H., Yamamoto, K., Yamazaki, K., Nakabayashi, M., Ogihara,
T., and Kaneda, Y. (2001) Hypertension, 38, 48-55.