REVIEW: Thirty Years of Chlorophyll Modeling
G. R. Seely*
E-mail: seelygns@comcast.net
Received October 6, 2013
This paper summarizes the progress over approximately thirty-year’s work of the author in developing a model of photosynthesis involving chlorophyll on polymeric substrates that would photosensitize endergonic electron transfer. The system finally achieved might, with proper modifications, serve as a means of generating power from solar energy.
KEY WORDS: chlorophyll, polymers, photosensitization, electron transfer, hydrazines, nitro compounds, surfactantsDOI: 10.1134/S000629791403002X
* Editor’s note: The author is a leading researcher in the field of modeling of photosynthesis. He worked for many years in the C. F. Kettering Research Laboratory, Yellow Springs, Ohio, USA (1962-1986), and in the Department of Chemistry and Biochemistry, Arizona State University, Tempe, Arizona, USA (1986-1993).
When I entered Graduate School in the Chemistry Department of the University of California, one of my early jobs was to find a Research Advisor. The one whose work interested me most was Melvin Calvin, who at the time was chiefly engaged in directing a large group studying the fixation of carbon dioxide, work for which he later received a Nobel Prize. But his interests extended beyond that, into the light-sensitized reactions leading to the reduction of NADP. The work that I undertook involved the photoreduction of zinc tetraphenyl porphin, which turned out to have some very interesting aspects.
About the same time (around 1950), there began an outpouring of papers by Prof. Krasnovsky at the Biochemistry Institute, and others, on the photoreduction of chlorophyll and on photosensitized reduction of dyes. Calvin had a great interest in these papers, which he shared with me, because of the obvious possibility that the recently discovered red reduction product of chlorophyll could play a role in the reduction of NADP in vivo. He encouraged me to read these papers, and I even learned enough Russian to do so when they first appeared, before they were translated by the Atomic Energy Commission.
A few years after receiving my Ph.D., I decided to accept an offer from Shell Development Company to join their laboratory in Emeryville, California. There I was assigned to work on elastomers, which were a major interest of the industry at that time, motivated by the new catalysts developed by Ziegler and Natta. Polymers were a new experience for me; I had not learned much about them in college. But there were some interesting aspects to their physical chemistry, and I learned as much about them as I could.
Shell Development was a very interesting place to work, with a number of distinguished chemists there from whom I learned much. But after a few years I felt I was not going anywhere professionally, and that I really wanted to return to photochemistry. Work on chlorophyll model systems had been accelerating, with the aims of learning more about photosynthesis itself, and ultimately converting solar energy into electrochemical form. I conceived the idea that if I combined what I knew about the photochemistry of chlorophyll with what I had learned about polymers (of course without using proprietary information), I could develop more versatile models than those currently being studied. I learned through a conversation with Prof. Calvin that the Charles F. Kettering Research Laboratory in Yellow Springs, Ohio was being upgraded and outfitted for the study of photosynthesis. Prof. Leo P. Vernon of Brigham Young University had been chosen as the Director, and would soon be hiring. I proposed my research plan to him, and was invited to join CFKRL in April, 1962. My work on chlorophyll model systems began there and continued until 1986, when the imminent closure of the Laboratory obliged me to transfer my work to Arizona State University in Tempe, Arizona.
When I had arrived and begun in earnest to investigate the photochemistry of chlorophyll, I came to realize we really did not know the mechanism by which chlorophyll-sensitized redox reactions occur. Two pathways were generally considered; I prefer to call them the “oxidative” and “reductive” pathways, depending on whether the excited state pigment first passes an electron to an oxidant, or receives one from a reductant. Opinion understandably tended to favor the latter, because of the known photoreduction of chlorophyll to a red product, which was reversed by oxidants [1]. Obviously, if I were going to devise model systems for sensitizing redox reactions, I needed to know with reasonable certainty what pathway was involved.
First, an exhaustive investigation of the photoreduction of ethyl chlorophyllide by ascorbic acid suggested that the functional reductant was the ion pair pyridinium ascorbate, and that the transfer of a proton immediately after the electron was necessary to stabilize the reduced chlorophyllide radical. Quantum yields also were rather low.
Next, I found that the kinetics of chlorophyll-sensitized photoreduction of the neutral form of Methyl Red and phenosafranine were incompatible with the reductive pathway. Chlorophyll also readily photosensitizes reduction of these dyes by hydrazobenzene, which is inactive in the photoreduction of chlorophyll, at least under these conditions. These facts strongly suggested that the chlorophyll-photosensitized reduction of these and other dyes followed the oxidative pathway. And of course we now know that the primary reactions of photosynthesis follow the oxidative pathway also. By comparing photosensitized reduction chemistry with polarographic half-wave potentials of the oxidant, I then found that if the half-wave potential was higher than –0.6 V vs the SCE (saturated calomel electrode), the reaction followed the oxidative pathway, and if it was lower than –0.9 V, the reductive pathway was followed [2]. In between there was no information. However, it was known that chlorophyll-photosensitized photoreduction of yellow azo dyes and NAD occurred by the reductive pathway.
To improve the estimation of the cross-over potential, I turned to aromatic nitro compounds. These covered a range of reduction potentials from p-dinitrobenzene (–0.458 V vs SCE) to p-nitroaniline (–1.037 V). A semilogarithmic plot of quantum yields for reduction of 29 nitro compounds by hydrazobenzene, sensitized by pyrochlorophyll, showed a break in the slope at about –0.8 V, indicating the beginning of failure of the oxidative pathway [3]. A few experiments with ascorbic acid as reductant showed greater quantum yields in the low potential region, presumably by the reductive pathway. The potential above which the oxidative pathway prevails works out to about –0.6 V vs the Normal Hydrogen Electrode.
We now had two things we needed to proceed with the design and operation of model systems: an insight into the pathways of electron transfer in redox reactions, and a reductant (hydrazobenzene) that efficiently reduces chlorophyll cation but not its triplet excited state.
The first model system to be intensively examined took advantage of what I had learned about the solution properties of polymers and chlorophyll. It was well known that the Mg of chlorophyll is ligated by Lewis bases such as water, alcohols, and especially pyridine. One or two molecules of the last per Mg produces small but characteristic changes in the chlorophyll red band near 665 nm. Vinylpyridines can be prepared and polymerized by free-radical initiators; it was expected that in a non-basic solvent the resulting poly(vinylpyridine) could attach an arbitrary number of chlorophylls, and thus form a model of the antenna system of photosynthetic units, in which the density of chlorophylls per polymer molecule could be changed over a wide range, while keeping the stoichiometric concentration of the pigment nearly constant.
I prepared poly(4-vinylpyridine) by polymerization of the monomer with azobisisobutyronitrile, a standard free-radical initiator, and taking advantage of my experience with polymers, I was able to fractionate it and characterize the fractions by viscosimetry and light-scattering molecular weight determinations. The problem was to find a suitable solvent; it must not be basic, for it would compete with the polymer in ligation of chlorophyll, yet it must dissolve both the polymer and the pigment. The only solvent I could find with the right properties was nitromethane, and that only if it is very dry.
Chlorophylls a and b, bacteriochlorophyll, and even Mg phthalocyanine bind to poly(4-vinylpyridine) in nitromethane, and at a high ratio of polymer to pigment, absorption spectra and fluorescence yields were in the normal range for the pigment in solution. At lower ratios of polymer to pigment, fluorescence becomes progressively quenched, apparently by transfer of excited state energy to quenching centers consisting of associated pigment molecules [4]. Energy transfer by the Förster mechanism was studied by depolarization of fluorescence and by sensitized fluorescence of one pigment by another, using pyrochlorophyll as a stand-in for chlorophyll in many of these experiments in order to avoid allomerization [5, 6]. p-Dinitrobenzene was reduced by hydrazobenzene in a reaction sensitized by chlorophyll on the polymer, but the yield varied linearly with the fluorescence yield, signifying that no triplet states were formed at quenching centers [4].
This model promised to be an excellent one for studying energy transfer in an “antenna” system, and it was susceptible to mathematical treatment, but the existence of energy quenching at rather low pigment concentrations made it unlikely to imitate the efficiency of photosynthesis. Another serious drawback was its sensitivity to water. In the climate of Ohio and in a poorly air-conditioned laboratory it was simply impossible to get and keep nitromethane dry enough during the summer. I could work with it from October to April, then it was necessary to put it away until the drier days of fall. So I abandoned this model in order to divert attention to one, which had a better chance of imitating the photochemistry of photosynthesis.
I had earlier pointed out what I thought were the differences between “essentially liquid” model systems of chlorophyll and “essentially solid” ones. In the former, such as micelles, vesicles, and poly(vinylpyridine) solutions, chlorophyll and other reactants diffused freely, and reactivity depended on diffusion. Fluorescence was strong but associations of chlorophyll usually led to trapping and quenching of singlet excited state energy. Essentially solid systems such as suspensions or layers containing chlorophyll showed strong evidence of association, but were generally non- or weakly fluorescent with little evidence of photochemical activity. Perhaps if models could be constructed with limited or selective mobility, conditions might be found under which fluorescence and photoactivity are retained even at high chlorophyll concentrations. Photosynthetic units are such systems; although membrane structure is responsible for much of their success, at least some of the reactants are able to diffuse.
Again, my experience with polymers suggested a way. High molecular weight unbranched polyethylene is not soluble in any solvent that I know of at room temperature, because it contains a network of microcrystalline condensations of chain segments that act in effect as crosslinks for the intervening randomly directed chain segments. I had watched my colleagues measure intrinsic viscosity of polyethylene samples; to do so, they had to heat the polymer in a suitable hydrocarbon solvent to almost 100°C in a thermostatted viscometer, at which point it dissolves. On cooling below the cloud point, polymer particles separated, but are swollen with solvent.
Particles swollen with tetradecane were prepared in this way and collected by filtration, rinsed with methanol, and dried. The tetradecane content was about 75%. Chlorophyll was applied to them in suspension in aqueous methanol. Attachment to the surface required the presence of the phytyl group. Surfactants could be added along with the chlorophyll, or directly to the dried green particles in a volatile solvent such as an ether or methanol. Oxidants and reductants could be added similarly in a small amount of volatile solvent.
To perform photochemistry and to record spectra, the particles were suspended in an aqueous phase thickened physically by a polysaccharide such as guar gum, and optically by powdered cellulose so as to reduce the “sieve effect”, and take better advantage of the Kubelka–Munk theory of light transport through scattering media. Thus, structural rigidity is enhanced without unduly reducing diffusion.
When chlorophyll is applied to particles without a surfactant, or with one that does not ligate the Mg, the pigment is found mostly in the form called “Chl 740”, which probably consists of sheets of chlorophylls linked by water molecules, and it has little if any photochemistry. If a ligating surfactant is present, Chl 740 is absent and the pigment appears as the monomer and in associated species, which are characteristic of the surfactant. These species are fluorescent at room temperature as well as at lower temperatures down to 90 K. Absorption and fluorescence spectra can be resolved into Gaussian bands and correlated, but the relationship is complex and suggests extensive energy transfer. Useful ligating surfactants include those with amide functional groups, sometimes grafted with pyridine or imidazole functions.
The by now familiar photosensitized reduction of p-dinitrobenzene by hydrazobenzene was used to compare photoactivity of chlorophyll in solution and on particles [7]. Ethyl chlorophyllide smoothly reduced p-dinitrobenzene to p-nitrophenyl hydroxylamine in 1-pentanol solution, and after that there is no further change in the spectrum. On particles with chlorophyll and dimethylmyristamide as ligating surfactant, however, the reaction does not stop at the hydroxylamine stage, but continues, much more slowly, to form products, which were not readily identified. The second stage perhaps starts with photoreduction of the remaining nitro group, followed by condensation of nitroso with hydroxylamine.
In a related series of experiments, quantum yields for photosensitized reduction of a number of nitro compounds by chlorophyll with dimethylmyristamide on particles were plotted semilogarithmically against the potential of their cyclic voltammetric one-electron reduction in acetonitrile [8]. Also plotted, for comparison, are quantum yields for triplet state sensitized photoreduction in 1-pentanol solution, and Stern–Volmer constants for quenching of pyrochlorophyll fluorescence by nitro compounds, obtained earlier, considered to be an indicator of singlet state reduction. Although there was much scattering of data, the slope of the line representing decline of quantum yield with potential for chlorophyll on particles agreed better with reactivity of the singlet state or the ion radical than with triplet state reactivity.
A useful model for energy generation simulating photosynthesis should demonstrate five of its features: (1) the density of pigment should be high, for efficient capture and transport of light energy; (2) use should be made of excited singlet state energy, or at least that of ion pairs derived from it; (3) reductants and oxidants should be readily available to the photoactivated pigment to maximize the probability of electron transfer; (4) net products of electron transfer reactions should be partitioned into different phases, to minimize back reactions; (5) the net reaction should be endergonic, for power generation. All of these features appear to have been demonstrated in the present particulate system.
As to the first two, the density of chlorophyll is high because it is confined to the surface layer of the hydrocarbon particles, and the ligating surfactants play the role of the protein in suppressing concentration quenching of fluorescence. As to the third, one way to achieve this is to choose an electron acceptor that has little solubility in either water or hydrocarbon, and so remains on the surface of the particles onto which it is deposited. Or, if the acceptor is charged, it may be attracted to the surface by a surfactant of opposite charge. A third approach is to ligate the pigment with a surfactant to which an oxidant is grafted that serves as the primary electron acceptor. Surfactants grafted with quinone or nitro functions have been used with some success [9].
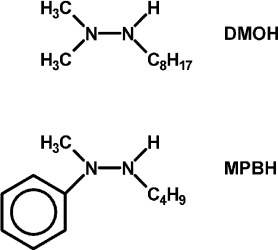
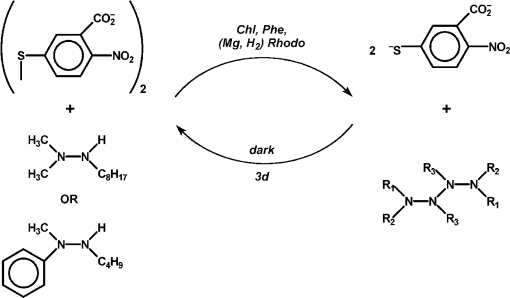
In hopes of demonstrating the fourth and fifth features, some changes were made in the list of reagents. First, the tetramethylammonium salt of 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB, Ellman’s reagent for protein SH) was used as the electron acceptor. DTNB is reduced reversibly by one electron, but when the second nitro group is reduced, it splits into two 2-nitro-5-thiolatobenzoate ions, which are bright yellow and convenient to follow spectroscopically. Dodecylpyridinium iodide was added to the particles to attract the anionic oxidant to them. Instead of hydrazobenzene, two trisubstituted hydrazines, 1,1-dimethyl-2-octylhydrazine (DMOH) and 2-butyl-1-methyl-1-phenylhydrazine (MPBH), were prepared and used as reductants (Fig. 1). These are oxidized to free radicals that can dimerize to tetrazanes, which should be soluble in the hydrocarbon particles.
Fig. 1. Structures of 1,1-dimethyl-2-octylhydrazine (DMOH) and 2-butyl-1-methyl-1-phenylhydrazine (MPBH).
Three new surfactants were introduced, aminoethylmyristamide (AEMA), tridecylimidazoline, and tridecylimidazole, representing the synthetic pathway to the last (Fig. 2). The imidazoline, if not neutralized, is a strong enough base to allomerize chlorophyll partially or completely to a cyclic lactone derivative of purpurin 7, or rhodochlorin (Mg Rhodo). Also, small amounts of Mg-free pigment sometimes formed under these conditions.
Nineteen photosensitized reductions of DTNB were performed with various combinations of pigments, surfactants, and reductants. In every case, reaction proceeded to or near to completion, though the quantum yield was not especially high, as followed by build-up of absorbance of the thiolate [10]. In every case, the thiolate band regressed on standing in the dark. In four cases, regression exceeded 90%, but it took three days. This was taken to signify that the photoreaction was endergonic. The slowness of the regression supports the belief that products were concentrated into different phases.
Almost as interesting as the observation of reversibility was the observation that most reactions appeared to go in two stages. The first, faster stage ended in the loss in the difference spectra of one or more associated forms of pigment. In a reaction with chlorophyll ligated with AEMA, there is apparently loss of a small band on the long-wavelength side of the main absorption band accompanied by a blue shift of a second band. In this case, it might represent the loss of a small amount of pheophytin closely associated with chlorophyll, and which is responsible for the faster phase of reduction.
To better represent results of the experiments with the reversible system, I have collected some of them into a few Figures. Figures 3-5 concerns Mg Rhodo sensitized reduction of DTNB by DMOH under three conditions.
Fig. 2. N-methylmyristamide and the structures of three surfactants used in these experiments.
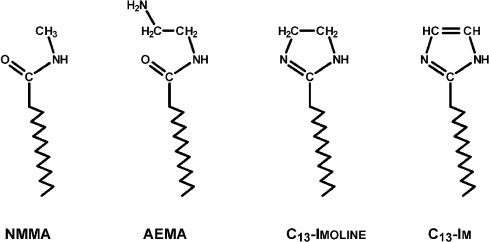
Fig. 3. Reversibility of the sensitized reduction of DTNB by DMOH. Traces from the spectral record of reaction recorded before (1) and after 1000 (2) and 5000 s (3) of 670 nm irradiation, and then after 66 h in the dark (4). The sensitizer is Mg Rhodo. This reaction shows almost complete reversion in 66 h.
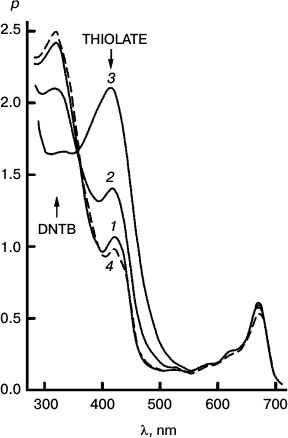
Fig. 4. The fastest recorded sensitized reduction of DTNB by DMOH sensitized by Mg Rhodo with a little 2H Rhodo. Spectra plotted were recorded before irradiation (0) and after 20 (1), 40 (2), 60 (3), 100 (4), 200 (5), and 510 s (6) exposure to 670 nm light. The dashed trace (7) was recorded after 5 days in the dark. Some of the thiolate produced is in the form of a narrow-band 450 nm species, which may be a sort of J-aggregate held together by cations absorbed on the surface of the particles. The 0-100 s difference spectrum (short-dashed trace) represents loss of H2 Rhodo at 678 nm.
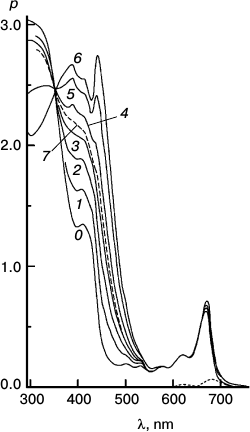
Fig. 5. Reaction sensitized by particles that contain some “tetrameric” Mg Rhodo and a lower concentration of DMOH. Spectral traces recorded before irradiation (0) and after 500 (1), 1000 (2), and 2000 s (3) irradiation at 670 nm. For comparison, the spectrum of an extract of the particles into l-propanol is shown (dashed trace). The difference spectrum corresponding to pigment loss during the first 500 s (short-dashed trace) peaks at 680 nm; there is little loss of pigment after that. In the dark, bands of “tetramer” intensify (not shown). Note the isosbestic point in each near 360 nm.
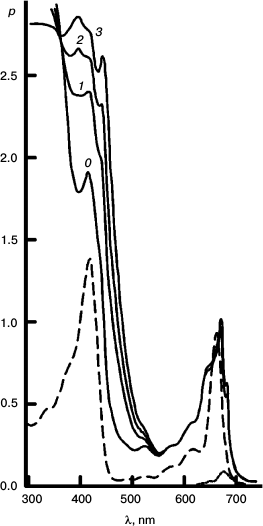
The imidazoline was used as surfactant in all three sets of traces.
Figures 6-9 deal with difference spectra in the red region, accompanying reduction of DTNB by MPBH sensitized by chlorophyll and pheophytin. Spectral traces are shown for three (of five) reductions of DTNB by MPBH, sensitized by a particle preparation with AEMA and cetylpyridinium chloride, in which the fraction of pheophytin ranged from 0 to 1. Difference spectra are plotted so that a peak corresponds to loss of absorbance. Figures 10 and 11 present an idea of mechanisms of studied photosensitized reactions and preliminary evidence of “special pairs” of pigment molecules that facilitate the reaction, as in photosynthesis.
Fig. 6. Reaction photosensitized by Chl only. Spectra plotted before (0) and after 500 (1), 1030 (2), 2505 (3), and 4020 s (4) of irradiation at 670 nm. The difference spectrum shows pigment loss during the first 500 s; there is no appreciable loss after that. It is the slowest reaction, with only a tiny loss of sensitizer initially.
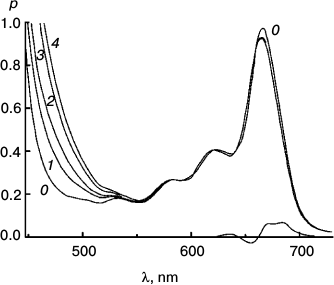
Fig. 7. Reaction photosensitized by Phe prepared in situ by treatment with trifluoroacetic acid. Spectra plotted before (0) and after 100 (1), 200 (2), 500 (3), and 1060 s (4) of irradiation at 670 nm. Difference spectra are plotted for 0-100 s (solid trace) and 500-1060 s (dashed trace) intervals. It was the fastest reaction, but very destructive of sensitizer.
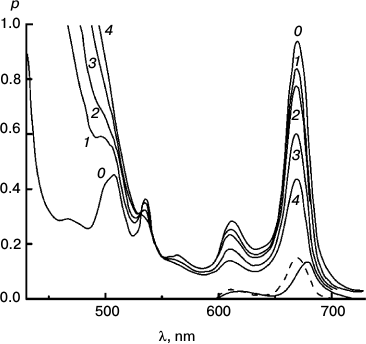
Fig. 8. Reaction with pheophytin added to particles with Chl. Spectra plotted before (0) and after 200 (1), 400 (2), and 2000 s (3) irradiation at 670 nm. Difference spectra are plotted for the 0-200 s (solid trace) and 400-2000 s (dashed trace) intervals. To obtain the (0-0-0) trace, enough of the difference spectrum of Fig. 6 was subtracted from the 0-200 s trace to eliminate the blue shift responsible for the dip at 654 nm. This reaction was intermediate in speed and pigment loss.
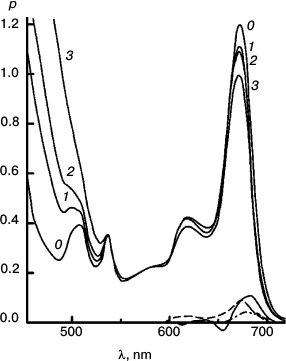
Fig. 9. Semilog plot of the rate of thiolate production (Δρ/Δt) measured at 470 nm in the course of chlorophyll-photosensitized oxidation of DNTB, against time of irradiation at 670 nm using the systems containing different concentrations of DNTB and a reducing agent MPBH. Rates for all reactions are represented as the superposition of two exponential decays. There is usually a brief induction period during which residual oxygen is reduced. The data show biphasic nature of the reactions and the loss of minor spectral components. Details of these experiments are described in ref. [10].
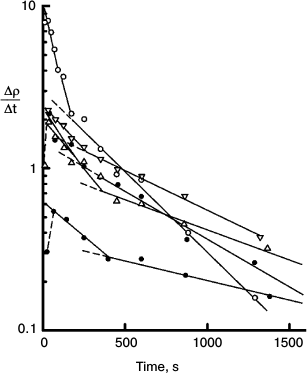
Fig. 10. Estimates (the third column) of the exothermicity of transition of excited singlet states to ion pairs for listed pigments.

Fig. 11. Diagram of the proposed reversible reduction of DNTB by trisubstituted hydrazines taken from ref. [10].
Thus, results of this work demonstrate the possibility of generating reaction center models in an artificial heterogeneous system containing organic polymers. The system finally achieved might, with proper modifications, serve as a means of generating power from solar energy.
I would first of all like to thank my laboratory assistants, postdoctoral fellows, staff, and colleagues at CFKRL and Arizona State University whose advice and assistance have made this work possible. I also wish to thank my mentors and colleagues at Shell Development Company who introduced me to polymer chemistry. I wish to acknowledge the guidance of the late Prof. Melvin Calvin in the earliest stages of my research, and the Organizing Committee and the Editorial board of the journal Biochemistry (Moscow) for giving me the opportunity to present this work at a Symposium and in a journal issue dedicated to the 100th birth anniversary of the late Prof. A. A. Krasnovsky. Much of this work has been supported by the U. S. Department of Energy.
REFERENCES
1.Krasnovsky, A. A. (1960) Ann. Rev. Plant
Physiol., 11, 363-410.
2.Seely, G. R. (1965) J. Phys. Chem.,
69, 2779-2782.
3.Seely, G. R. (1969) J. Phys. Chem.,
73, 117-124.
4.Seely, G. R. (1971) J. Phys. Chem.,
75, 1667-1670.
5.Seely, G. R. (1970) J. Phys. Chem.,
74, 219-227.
6.Seely, G. R. (1976) J. Phys. Chem.,
80, 447-451.
7.Seely, G. R., and Haggy, G. A. (1987) J. Phys.
Chem., 91, 440-447.
8.Seely, G. R., and Haggy, G. A. (1988) J.
Photochem. Photobiol. A: Chemistry, 42, 337-346.
9.Seely, G. R., and Rehms, A. A. (1991) Photochem.
Photobiol., 53, 675-688.
10.Seely, G. R. (1995) Photosynth. Res.,
45, 203-217.