REVIEW: Photosystem II: Its Function, Structure, and Implications for Artificial Photosynthesis
James Barber
Department of Life Sciences, Sir Ernst Chain Building, South Kensington Campus, Imperial College London, London, SW7 2AZ, UK; E-mail: j.barber@imperial.ac.uk
Received October 17, 2013
Somewhere in the region of 3 billion years ago an enzyme emerged which would dramatically change the chemical composition of our planet and set in motion an unprecedented explosion in biological activity. This enzyme used solar energy to power the thermodynamically and chemically demanding reaction of water splitting. In so doing it provided biology with an unlimited supply of hydrogen equivalents needed to convert carbon dioxide into the organic molecules of life. The enzyme, which facilitates this reaction and therefore underpins virtually all life on our planet, is known as Photosystem II (PSII). It is a multisubunit enzyme embedded in the lipid environment of the thylakoid membranes of plants, algae, and cyanobacteria. Over the past 10 years, crystal structures of a 700 kDa cyanobacterial dimeric PSII complex have been reported with ever increasing improvement in resolution with the latest being at 1.9 Å. Thus the organizational and structural details of its many subunits and cofactors are now well understood. The water splitting site was revealed as a cluster of four Mn ions and a Ca ion surrounded by amino acid side chains, of which seven provide ligands to the metals. The metal cluster is organized as a cubane-like structure composed of three Mn ions and the Ca2+ linked by oxo-bonds with the fourth Mn attached to the cubane via one of its bridging oxygens together with another oxo bridge to a Mn ion of the cubane. The overall structure of the catalytic site is providing a framework on which to develop a mechanistic scheme for the water splitting process and gives a blue print and confidence for the development of catalysts for mimicking the reaction in an artificial photo-electrochemical system to generate solar fuels.
KEY WORDS: photosynthesis, photosystem II, structure, water splitting, artificial photosynthesis, manganese cluster, oxygen evolving complexDOI: 10.1134/S0006297914030031
Abbreviations: β-Car, β-carotene; Chl, chlorophyll; DGDG, digalactosyldiacylglycerol; MGDG, monogalactosyldiacylglycerol; OEC, oxygen evolving complex; PG, phosphatidylglycerol; Pheo, pheophytin; PSI(II), photosystem I(II); RC, reaction centre; SQDG, sulfoquinovosyldiacylglycerol.
Prior to the evolution of oxygenic photosynthesis, biology had been
dependent on hydrogen/electron donors such as H2S,
NH3, organic acids, and Fe2+, which were in
limited supply compared with the “oceans” of water with
which planet Earth is blessed. The by-product of the water splitting
reaction is molecular oxygen. The release of this gas also had dramatic
consequences for the development of life, since it created an
oxygen-rich atmosphere and at the same time allowed the ozone layer to
form. With oxygen available, the efficiency of cellular bioenergetics
increased dramatically since for a given amount of substrate, aerobic
respiration provides in the region of ~20 times more energy than
anaerobic respiration. Moreover, new biosynthetic pathways involving
oxygenation reactions were now possible. It was almost certainly these
improvements in metabolism that paved the way for the subsequent
evolution of eukaryotic cells and multicellular organisms. The
establishment of the ozone layer provided a shield against harmful UV
radiation allowing organisms to prosper and diversify on an enormous
scale as testified by the fossil records and by the extent and variety
of living organisms on our planet today. The enzyme, which uses light
energy to split water and form dioxygen, is known as Photosystem II
(PSII) and is truly the engine of life [1].
PHOTOSYSTEM II
PSII is a multisubunit protein complex located in the thylakoid membranes of all types of plants, algae, and cyanobacteria [1]. At its heart is the reaction centre (RC) core where light energy is converted to electrochemical potential energy and where the water splitting reaction occurs. The PSII core consists of two homologous proteins known as D1 and D2 and two further closely related chlorophyll (Chl)-binding proteins (CP), called CP43 and CP47. The former pair has five (A to E) and the latter pair has six (I to VI) transmembrane helices. There are also a number of other low molecular weight subunits, which are rather featureless except for two (PsbE and PsbF) that bind the high potential heme of cytochrome b559 (Cyt b559). Finally the PSII core complex has several extrinsic proteins attached to its lumenal surface, which form a protein shield over the catalytic site of water splitting. Although one of these proteins, PsbO, is ubiquitous to all oxygenic photosynthetic organisms, the others vary between different types of organisms. Plants and green algae bind the PsbP and PsbQ proteins while the PsbU and PsbV proteins are only found in red algae and cyanobacteria [2]. Other extrinsic proteins are transiently bound during the life cycle of PSII. This core complex is serviced by an outer light harvesting system which, in the case of plants and green algae, is composed of intrinsic lhcb proteins which bind Chl a and Chl b. In red algae and cyanobacteria, this outer antenna is replaced by phycobiliproteins, which make up the phycobilisome bound extrinsically to the stromal surface of the PSII core.
The way PSII uses light energy to generate an electrochemical potential in the form of a charge transfer state is essentially the same as for other types of photosynthetic systems. Chlorophyll molecules and other pigments (e.g. β-carotene) absorb visible light and transfer the excitations to a RC where charge separation occurs. The striking difference is that in the case of PSII, the oxidizing “hole” left by charge separation is highly oxidative, so much so that it can drive water splitting. All the redox active cofactors involved in the energy conversion process are bound to the D1 and D2 proteins and the following sequence of reactions occurs: a special form of Chl a P (P680) acts as an exciton trap and is converted to a strong reducing agent after excitation to P680*. P680* reduces a pheophytin a molecule (Pheo) in a few picoseconds to form the radical pair state P680·+Pheo·–. Within a few hundred picoseconds, Pheo·– reduces a firmly bound plastoquinone molecule QA to produce P680·+PheoQA–. P680·+, with its very high redox potential (>1 V), oxidizes a tyrosine residue (TyrZ) to form TyrZ+P680PheoQA– on a nanosecond time scale. This is followed by the deprotonation of TyrZ+ to form a neutral radical TyrZ·. In the millisecond time domain QA– reduces a second plastoquinone QB to form Tyr·P680PheoQAQB– and at about the same time Tyr· extracts an electron from a cluster of four manganese atoms that bind the two substrate water molecules. A second photochemical turnover reduces QB– to QB2– which is then protonated to plastoquinol and released from the QB-binding site of PSII into the lipid bilayer where it is subsequently oxidized by photosystem I (PSI) via the cytochrome b6f complex. Two further photochemical turnovers provide a cluster of four manganese ions and a calcium ion (Mn4Ca2+) with a total of four oxidizing equivalents, which are used to oxidize two water molecules to form dioxygen. Each oxidation state generated within the oxygen-evolving complex (OEC) is represented as an intermediate of the S-state cycle [3, 4], of which there are five (S0 to S4). In addition to these reactions, side reactions can occur under some conditions including the oxidation of Cyt b559, a β-carotene (β-Car) molecule, and a Chl a molecule (ChlZ). These side reactions occur on the tens of millisecond time scale and therefore do not compete with the electron transfer pathway leading to water oxidation. Indeed, they probably only occur when the rate of water oxidation becomes limited and thus provide a protective mechanism against the detrimental reactions resulting from the very high redox potential of the long lived P680 radical cation (P680·+).
Clearly, the reactions of PSII cannot be fully understood in molecular terms without a detailed knowledge of its structure. There are many techniques that give structural information at different degrees of complexity, but none match the overall level of detail gained from X-ray crystallography. PSII is a membrane protein complex making it more difficult to crystallize. Nevertheless, progress during the past 10 years has been substantial and is the focus of this paper. The structural information obtained has improved our understanding of the many reactions of PSII and most importantly, of the water splitting reaction itself, knowledge which may have far reaching implications for the development of new technologies for production of fuel.
CRYSTAL STRUCTURE OF PSII
Proteins. The first X-ray-derived structural model of PSII used a preparation isolated from the cyanobacterium Thermosynechococcus elongatus and was elucidated by Zouni et al. [5]. This model was limited to a resolution of 3.8 Å, and the Cα backbones of many of the subunits were not fully traced. Although no side chain positioning was deduced, their model confirmed the dimeric organization of the isolated complex [6] and the relative positioning of the major subunits and their transmembrane helices within each monomer derived earlier from electron crystallography [6-10]. It also provided information on the positioning of cofactors involved in excitation transfer and charge separation. Most importantly, the analysis of the diffraction data gave the first direct structural hints of the Mn-cluster of the OEC. Two years later Kamiya and Shen [11] reported a 3.7 Å crystal structure of PSII isolated from Thermosynechococcus vulcanus, a cyanobacterium closely related to T. elongatus and provided additional information to that revealed by Zouni et al. [5]. The tracing of the Cα backbones was more complete and there was some effort made to assign a few amino acids, particularly those of the D1- and D2-proteins, as well as to some regions of the chlorophyll-binding proteins CP43 and CP47. However, it was not until the work of Ferreira et al. [12] that all the protein subunits of PSII were assigned and their complete structures revealed. The PSII complex crystallized was isolated from T. elongatus and its X-ray diffraction-derived structure was refined to a resolution of 3.5 Å. Over 5000 amino acids (close to 100% of this huge complex) were mapped within the dimeric complex, thus revealing for the first time the protein environments of all the major cofactors as well as assigning the location of 18 of the 19 subunits, which made up each monomer. As a consequence, a host of outstanding questions were answered; not least, direct evidence that the phenolic group of TyrZ (D1Tyr161) is H-bonded to nearby D1His190, a necessary requirement for this redox-active tyrosine to function in electron transfer between P680 and the Mn4Ca2+-cluster. Also the finding that glutamate 354 of CP43 (CP43Glu354) was a ligand to the Mn4Ca2+-cluster highlighted another striking discovery among many others.
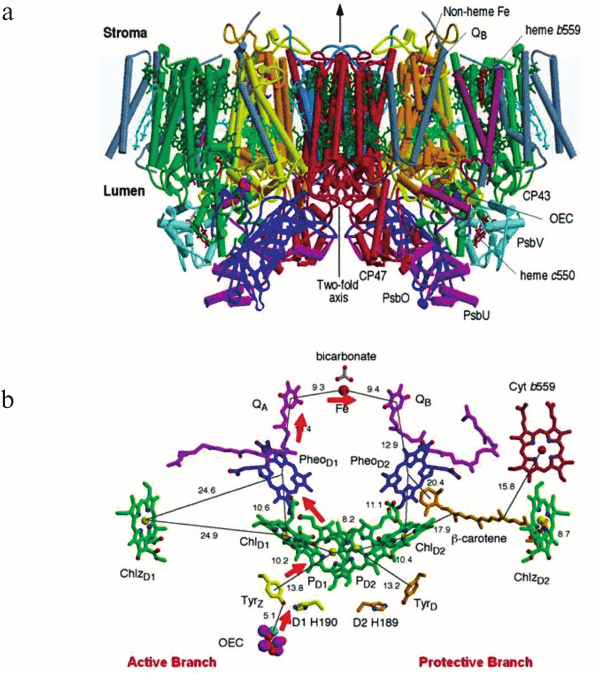
The side view of the PSII dimeric core complex of T. elongatus shown in Fig. 1a (see color insert) is taken from the crystal structure of Ferreira et al. [12]. The PSII dimer has dimensions of 105 Å depth (45 Å in spanning the membrane), 205 Å length, and 110 Å width and has a calculated total molecular mass of about 700 kDa. There were in total 35 transmembrane helices per monomer depicted as cylinders in Fig. 1a. Details of the 19 different subunits are listed in the table with one, Ycf12 (originally suggested to be possibly PsbN), being included based on more recent biochemical analysis of PSII [13] and confirmed by the 1.9 Å crystal structure [14]. Note that the other, non-highlighted proteins in the table are exclusive to the higher plant PSII core complex.
Fig. 1. a) Side view of the structure of PSII, the water splitting enzyme of photosynthesis. This structure was determined by X-ray crystallography [12]. The complex is embedded in the thylakoid membrane spanning between the lumenal and stromal surfaces. It is composed of two monomers related to each other by a 2-fold axis. Each monomer contains 19 different protein subunits with 16 being located within the membrane matrix. In total there are 35 transmembrane α-helices per monomer (depicted by cylinders). The D1- and D2-proteins that compose the RC are shown in yellow and orange, respectively. The Mn3Ca-catalytic centre of the OEC is located on the lumenal side of the complex and is stabilized and shielded by three extrinsic proteins, PsbO, PsbU, and PsbV. b) Organization of the electron transfer cofactors that make up the RC of PSII as revealed by X-ray crystallography [12]. Excitation of the RC via the Chls shown in green leads to the reduction of Pheo resulting in the formation of the radical pair P680•+Pheo•–. The radical cation of P680 is localized on PD1 while the radical anion is located on PheoD1. The electron on PheoD1•– is rapidly donated to a firmly bound plastoquinone QA (shown in purple) and then transferred to a second plastoquinone QB (also shown in purple). This electron transfer is aided by the presence of a non-heme iron located mid-way between them. When the QB plastoquinone has been doubly reduced and protonated the resultant plastoquinol (PQH2) diffuses from the QB-binding site into the lipid matrix of the membrane, P680•+ is reduced by a redox active tyrosine (TyrZ), which then extracts electrons from the Mn4Ca-cluster that constitutes the oxygen-evolving complex (OEC). These electron transfer processes occur mainly on the D1-side of the RC as shown by the red arrows. Some of the symmetrically related cofactors located on the D2-side (PheoD2) are non-functional. Other cofactors on the D2-side (heme of cytochrome b559 shown in red, the β-carotene molecule shown in brown and ChlZD2 in green) are functionally active and seem to play a role in protecting PSII against photoinduced damage.
Location of genes coding for PSII proteins and PSII proteins’
characteristics
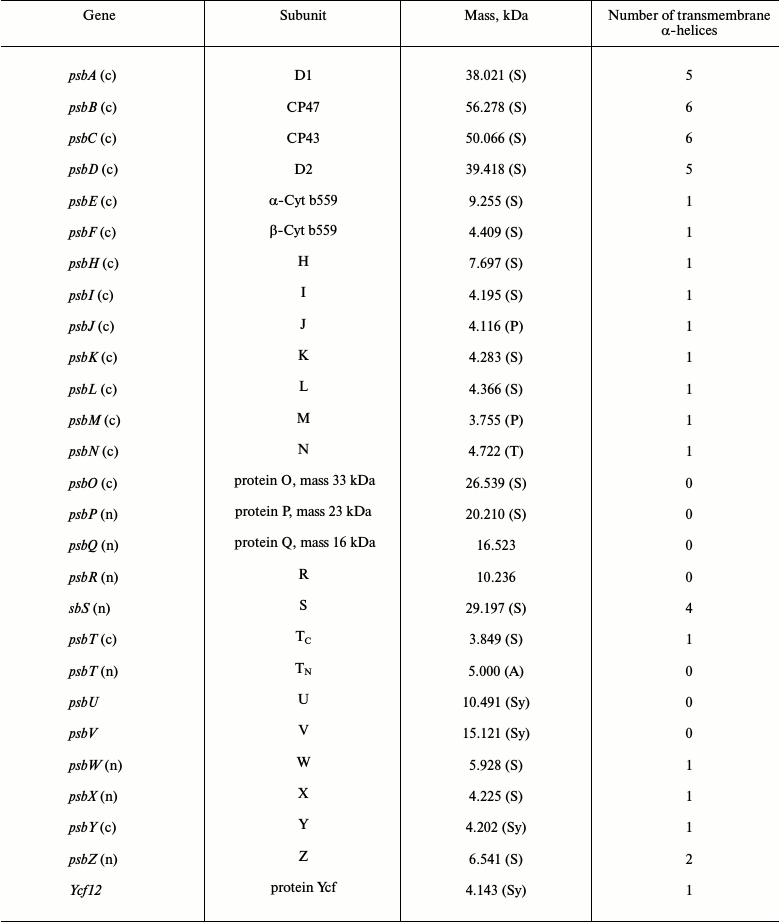
Note: PSII proteins are products of the psbA to psbZ plus
ycf12 genes that occur in oxygenic organisms. In eukaryotic
organisms the psb genes are located in either the chloroplast
(c) or nuclear (n) genes. The molecular masses of the mature
PsbA-PsbZ/Ycf12 proteins are calculated from the protein sequences
reported in the SwissProt database using the MacBioSpec program (Sciex
Corp., Thornhill, ON, Canada) for spinach (S), pea (P), tobacco (T),
Arabidopsis (A), and Synechocystis PCC6803 (Sy).
The three extrinsic proteins PsbO, PsbU, and PsbV form a cap over the catalytic site where oxygen evolution occurs, preventing access by reductants other than water, while some of the low molecular weight subunits are located on the peripheral of the CP43/D1/D2/CP47 cluster where they probably help to stabilize the binding of Chl and β-Car molecules contained within the complex. The exceptions to this are the PsbE and PsbF proteins, which provide histidine ligands for the heme of Cyt b559, and the PsbL, PsbM, and PsbT proteins located at the monomer–monomer interface where they possibly play a role in stabilizing the dimeric nature of the PSII complex. All the small subunits have a single transmembrane helix except for the PsbZ, which has two (see the table). Subsequent X-ray derived models of cyanobacterial PSII by Loll et al. [15], Guskov et al. [16], and Umena et al. [14] agreed with the assignments of the small subunits made by Ferreira et al. [12] with the 1.9 Å confirming the location of Ycf12 adjacent to PsbK and PsbZ. The 2.9 Å crystal structure of Guskov et al. [16] contained an additional transmembrane helix assigned to PsbY, but this peripheral subunit was absent in the structures of Ferreira et al. [12] and Umena et al. [14].
All the crystal structures of PSII confirmed that the transmembrane helices of the D1 and D2 proteins are arranged in an almost identical way to those of the L and M subunits of the purple bacterial RC as anticipated by homology [17, 18]. The six transmembrane helices of CP43 and CP47 are arranged in three pairs around a pseudo-3-fold axis as first shown by electron crystallography [7, 8].
These two Chl-binding proteins are located on each side of the D1 and D2 subunits in such a way that their transmembrane helices are related by the same pseudo-2-fold axis that relates the five transmembrane helices of the D1/D2 heterodimer, an organization very similar to that of the RC core of PSI [19, 20]. They are both characterized by having a very large extrinsic loop joining the lumenal ends of helices V and VI.
Cofactors. Chlorophylls and carotenoids. The Ferreira et al. structural analysis [12] identified per monomer 36 Chls and tentatively assigned seven carotenoids, assumed to be all trans-β-Car. It was concluded that CP43 and CP47 bound 14 and 16 Chl a molecules respectively and in the majority of cases these light harvesting Chls were ligated by either α- or β-linkages to conserved histidine side chains and related by the same pseudo-3-fold symmetry axis that relate their transmembrane helices. The Chls were arranged in layers towards the lumenal and stromal surfaces with one Chl in each case located mid-way between the layers. Remarkably the majority of the Chls bound to CP43 and CP47 have counterparts bound in the N-terminal domains of the PsaA and PsaB RC proteins of PSI [20]. The more recent crystal structures of cyanobacterial PSII at 2.9 and 1.9 Å resolution have extended and corrected Ferreira et al. [12] assignment of seven β-Car molecules to 11 all trans-β-Car and concluded that CP43 binds 13 Chls rather than 14. The higher resolution crystal structures also confirmed that the D1/D2 heterodimer bound six symmetrically related Chls as detailed earlier [5, 11, 12] but extended the assignment of one β-Car located within the D2 protein by Ferreira et al. [12] to a second on the D1 side of the heterodimer.
A very important contribution of the most recent PSII crystal structure [14] is that it was at sufficient resolution to identify the location of 2795 water molecules in the dimeric complex. Most of these water molecules form two layers at the stromal and lumenal surfaces. However, a few molecules are located in the membrane spanning region of the complex with seven of them acting as ligands to the Mg of Chls that are not ligated by amino acids (histidines in all cases except one, where CP43Asn39 is a ligand). Water ligands were found for the two accessory Chls (ChlD1 and ChlD2) of the D1/D2 heterodimer as originally suggested by Ferreira et al. [12] while the others are for three Chls of CP47 and two Chls of CP43. Umena et al. [14] noted that most of the vinyl groups of the CP43 and CP47 Chls are positioned in, or near, the same plane of the tetrapyrrole ring, which would be expected to facilitate energy migration between adjacent chlorophylls. While the role of the Chls and β-Car bound to CP43 and CP47 is to capture photons and transfer excitation to the RC, β-Car has additional function, namely to quench harmful Chl triplet states and any singlet oxygen that might be generated. For Chl triplet quenching, they must be located close to the porphyrin head group of Chl, and this is borne out in the crystal structures. The only exceptions are the two β-Car molecules bound to the D1/D2 heterodimer where their role seems to be as electron transfer agents and possibly as singlet oxygen quenchers.
Redox active cofactors. The D1 and D2 proteins bind the cofactors that bring about light induced charge separation leading to the oxidation of water and reduction of the terminal electron/proton plastoquinone acceptor, QB. Together they bind six Chl a, two Pheo, two plastoquinones (PQ), two β-Car, and a non-heme Fe. The Ferreira et al. crystal structure [12], and also those reported by others [5, 11], clearly showed that these cofactors are arranged around the same pseudo-2-fold axis which relates the transmembrane helices of D1 and D2, CP43, and CP47 (see Fig. 1b). The axis passes through the non-heme Fe and through the middle of a cluster of four Chl a molecules called PD1, PD2, ChlD1, and ChlD2 where the suffix denotes binding to the D1 and D2 proteins. Similarly, the two pheophytins are referred to as PheoD1 and PheoD2. The QA and QB are positioned equally on each side of the non-heme Fe and are bound to sites located within the D2 and D1 proteins, respectively. The remaining two Chls (ChlzD1 and ChlzD2) are also symmetrically related, being ligated to histidines located in the B-transmembrane helices of the D1 and D2 proteins.
Photons are captured by the light harvesting pigments and transferred, as excitation energy, via the Chls of CP43 and CP47 to the PD1/PD2/ChlD1/ChlD2-cluster. Although the PD1 and PD2 are located adjacent to each other, excitonic interaction between their tetrapyrrole head groups is weak and they do not form a “special pair” as found in other types of reaction centres. Moreover, the recent high resolution structure [14] has shown that PD1 and PD2 are not perfectly symmetry related, since the vinyl group of PD1 is approximately in the plane of the membrane with its terminal C-atom close to the Mg of PD2 while the PD2 vinyl group is out of the chlorin plane and is not located close to the Mn of PD1. The monomeric character of PD1 and PD2 makes them approximately isoenergetic with each other and also with the two accessory Chls, ChlD1 and ChlD2. Indeed, according to the 1.9 Å structure [14] the π–π and CH–π stacking ranges from 3.3 to 3.5 Å with the shortest distance being between PD1 and ChlD1. Therefore, it seems that excitation energy arriving at this cluster is delocalized over all four Chls to form the P* state [21]. The initial electron transfer probably occurs from ChlD1, since it is the closest to PheoD1, to form the charge transfer state ChlD1·+Pheo·–D1 [22]. This radical pair state is short lived with migration of the “hole” to PD1 to form PD1·+Pheo·–, where PD1·+ corresponds to the long lived P680 radical cation (P680·+) [23].
The electron on PheoD1 then proceeds down the thermodynamic gradient to the terminal PQ electron acceptor bound to the QB site within the D1 protein. As mentioned earlier, this electron transfer occurs in a few milliseconds and is aided by a fast intermediate electron transfer from Pheo·– to the PQ bound in the QA site of the D2 protein and by the non-heme Fe located midway between QA and QB. On receiving two electrons, the PQ is protonated to plastoquinol PQH2 and leaves the PSII complex by detachment from the QB site. A channel leading from the QB site has been identified [15, 24]. This quinone/quinol diffusion channel exits between the subunits of Cyt b559 and PsbJ and is coated with the phytol chains of several Chls and acyl chains of lipids as well as the close proximity of two carotenoids, thus imposing a high degree of lipophilicity. A second potential quinone channel has been postulated based on the assignment of a third PQ molecule in the 2.9 Å PSII structure refined by Guskov et al. [16]. However, this additional PQ molecule was not identified in the 1.9 Å structure of Umena et al. [14].
The reduction of PD1·+ (P680·+) is brought about by removing an electron from the OEC thus driving a manganese ion of the Mn4-Ca2+-cluster into a higher oxidation state. This electron transfer process is aided by a redox active TyrZ (D1Tyr161). This tyrosine has a symmetrically related counterpart within the D2 protein, TyrD (Fig. 2; see colour insert), which can also be oxidized by PD1·+ but is not directly involved in the water splitting reaction. It may, however, because of its long lifetime help to direct primary charge separation to the D1 side of the RC by electrostatic biasing [25]. This symmetrical relationship ends here since the Mn4Ca-cluster is only located on the D1 side while the heme of Cyt b559 is located on the D2 side. Therefore the D1-side of the RC functions directly in energy conversion and water splitting while the D2 side is, in part, involved in protection against photoinduced damage.
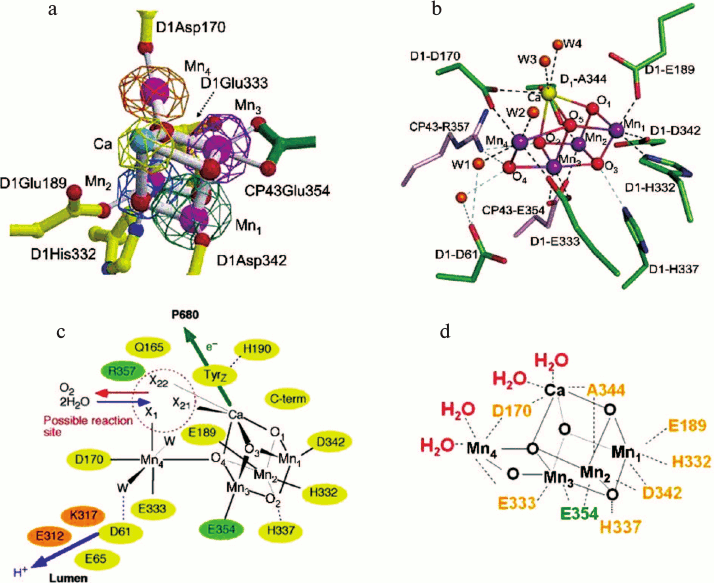
Fig. 2. a) Cubane structural model of the Mn4Ca cluster of the OEC and identification of amino acids within the first coordination sphere deduced from X-ray diffraction analyses at 3.5 Å by Ferreira et al. [12]. Mn ions in magenta, Ca ion in turquoise, bridging oxygens in red, D1 amino acids in yellow, and CP43 amino acid in green. b) Structure of the Mn4CaO5 cluster and its ligand environment as determined at a resolution of 1.9 Å by Umena et al. [14]. Similar colour coding as in Fig. 2a except Ca ion in yellow. c) Schematic view of the OEC published in Ferreira et al. [12]. Residues in D1, D2, and CP43 subunits are shown in yellow, orange, and green, respectively. X11, X21, and X22 were proposed to be substrate water binding positions to Mn4 (X11) and to Ca (X21 and X22), identified from the position of the non-protein ligand and coordination pattern of Mn and Ca ions. Other possible water molecules that were not visible at the 3.5 Å resolution were suggested and indicated as W. Possible hydrogen bonds were also proposed and shown as light-blue dotted lines. d) Diagrammatic representation of the Mn4CaO5 cluster and coordinating ligands determined by Umena et al. [14].
Despite the symmetric arrangement of cofactors on the reducing side, electron transport from PD1 to QB involves only PheoD1 while the other possible electron transfer, via PheoD2, does not occur, a situation which is also found for electron transfer in the RCs of purple photosynthetic bacteria [26]. In fact, the arrangement of the cofactors on the reducing side of PSII is essentially identical to their bacterial counterparts. The only clear exceptions are that one of the ligands for the non-heme Fe of PSII is a bicarbonate ion and not a glutamate as in bacteria and that the QB site is a little larger and in closer contact with the stromal surface than in the bacterial RC. That bicarbonate provides a bidentate ligand for the non-heme Fe has been known for some time [27, 28], and its assignment by Ferreira et at. [12] in their structure has been confirmed [14]. Its function is unclear, but its removal slows the kinetics of QA to QB electron transfer and therefore may represent a feedback mechanism from CO2 fixation to regulate PSII activity [29].
Cyt b559 has a high mid-point potential of +0.4 V and therefore is ideally poised to act as electron donor to PD1·+ (E ~ 1.2 V) and as an electron acceptor from the reduced plastoquinone in the QB site (E ~ 0 V). This cyclic electron transfer does not normally compete with the water oxidation reactions having kinetics in the millisecond timescale and only becomes relevant when the very oxidizing PD1·+ state becomes long-lived and therefore potentially dangerous. Because of the large spatial separation of Cyt b559 and PD1·+, electron flow from the heme to primary donor is aided by the β-Car identified by Ferreira et al. [12], which is positioned strategically between them (see Fig. 1b). The two peripheral ChlzD1 and ChlzD2 also show redox activity and may also act to protect the build-up of long-lived PD1·+ states. Again the β-Car molecules on the D1 and D2 sides would be required to facilitate the long range electron transfer to PD1·+.
Lipids and detergents. The first attempt to assign lipid and detergent molecules in the crystal structure of PSII was made by Loll et al. [15]. They assigned 14 lipids within the monomer: four digalactosyldiacylglycerol (DGDG), six monogalactosyldiacylglycerol (MGDG), three sulfoquinovosyldiacylglycerol (SQDG), one phosphatidylglycerol (PG), and three detergent molecules (β-dodecyl maltoside used for isolating the complex). This assignment was further refined by Guskov et al. [16] raising the total to 25 (seven DGDG, 11 MGDG, five SQDG, two PG) and eight β-dodecyl maltoside molecules. The high lipid content is very unusual for a membrane protein complex and possibly relates to the fact that the D1 protein turns over regularly due to light induced damage [30]. It was noted that the charged lipids (SQDG and PG) are exclusively located at the outer stromal surface while the neutral DGDG and MGDG were located at both surfaces. Assigning specific lipids and detergents using 3.0 to 2.9 Å data is difficult, and it is not surprising that the 1.9 Å crystal structure significantly modified the original assignments to five DGDG, six MGDG, four SQDG, five PG, and more than 15 β-dodecyl maltoside molecules with further refinement required to assign additional densities that are either lipid or detergent.
Chloride. The recognition that chloride is required as a cofactor for photosynthetic oxygen production, particularly for the water splitting reaction of PSII, stems from the pioneering studies of Arnon and Whatley [31], Bove et al. [32], and Izawa et al. [33]. Since then this requirement has been investigated in great depth as detailed in the comprehensive review by van Gorkom and Yocum [34]. When PSII is depleted of Cl ions, the S-state cycle is blocked at S2 [35] and its EPR spectral properties are modified [36, 37]. If Cl ion depletion is carried out on PSII placed in the S3 state, then the subsequent transitions to S0 are blocked and oxygen formation is inhibited [35]. Murray et al. [38] concluded from X-ray crystallographic analyses that there were two Cl-binding sites in the vicinity of the Mn4CaO5 cluster by substitution of Br for Cl ions. These assignments were confirmed by Kawakami et al. [39] and extended by Umena et al. [14]. One of the chloride ions is located close to the amino group of D2-Lys317 and the backbone nitrogen of D1-Glu333, while the other is close to the backbone nitrogens of D1-Asn338 and CP43-Glu354. Both also have two water molecules nearby. Because the side chains of D1-Glu333 and CP43-Glu354 are coordinated to the Mn4CaO5 cluster directly, the two Cl anions may function to maintain the coordination environment of the Mn4CaO5 cluster, thereby allowing the oxygen-evolving reaction to proceed properly [14]. One Cl-binding site close to D2-Lys317 lies at the entrance of hydrogen-bond networks extending towards the lumenal surface and may play a role in facilitating proton efflux from the OEC [38, 40].
Mn4-Ca cluster. The 3.8 Å X-ray diffraction data generated from crystals of PSII isolated from T. elongatus and analysed by Zouni et al. [5] provided hints of the position of the Mn4-cluster within the complex. The electron density assigned to the metal cluster was consistent with four Mn ions based on anomalous diffraction data collected at 1.894 Å and was found to be towards the lumenal surface of PSII on the D1-side of the pseudo-2-fold axis of the RC. The electron density had a “pear-shape” having dimensions of 6.8 × 4.9 × 3.3 Å with its longest axis tilted at about 23° to the membrane plane. Because of the low resolution and the incompleteness of the model, including the absence of side chain positioning, the assignment of the four Mn ions within the density was rather arbitrary. A Mn ion was positioned in the three bulges of the “pear-shaped” density to form an isosceles triangle with a fourth Mn ion placed in the centre of the triangle. The 3 + 1 organization of the four Mn ions gave support to the arrangement previously suggested by Pelequin et al. [41] and earlier by Hasegawa et al. [42], and was also a feature of the crystallographic model of the Mn-cluster derived by Kamiya and Shen [11]. However, Kamiya and Shen modelled the four Mn ions so that they were approximately in the same plane, whereas Zouni et al. [5] had placed the central Mn ion protruding towards the lumenal surface. Importantly, Kamiya and Shen’s map contained electron density connecting to that of the Mn-cluster that was tentatively assigned to side chains of the D1-protein, including Ala344, Asp170, and Glu333 (or His332). They also suggested that, based on weak electron density, D1 His337, D1 Asp189 (or D1 His190), and D1 Tyr73 may also be coordinated to Mn ions of the cluster. Mutagenesis studies already supported the idea that several of these residues could be ligands for the Mn ions [43-45] including those located in C-terminal domain of the D1-protein [46].
As in the case of the earlier crystal structure [5], the model of Kamiya and Shen did not include a Ca2+ bound close to the Mn-cluster. This was rectified in the crystal structure reported by Ferreira et al. [12]. Anomalous diffraction was collected at a wavelength of 1.89 Å to obtain an electron density profile for the Mn ions and at 2.25 Å to locate Ca2+ (at this wavelength calcium has an anomalous diffraction almost four times stronger than manganese). Again the electron density attributed to the four Mn ions was “pear-shaped” indicative of the 3 + 1 organization and again one Mn was assigned to the small domain and three in the large globular domain, whereas the 2.25 Å wavelength map covers one metal ion in the large domain of the native density. From these data the three Mn ions (Mn1, Mn2, and Mn3) and the Ca2+ located in the large domain were modelled as a cubane organization with bridging oxygens, an organization which was compatible with the native electron density. The fourth Mn ion (Mn4) located in the small domain was modelled so that it was linked to the cubane by one of its bridging oxygens (see Fig. 2a). Based on values determined by EXAFS measurements, the three Mn-di-μ-oxy-Mn bonds of the cubane were spaced at 2.7 Å while the three Mn-di-μ-oxy-Ca2+ bonds at 3.4 Å. The dangler Mn was positioned 3.3 Å from the closest Mn ions of the cubane and about 4 Å from the Ca2+. Although the distances chosen were compatible with distances derived from EXAFS [47], the predicted numbers of each type of bond were not. Nevertheless, density function calculations [48-51] and chemical synthesis of mixed Mn/Ca complexes [52] and more recently by Kanady et al. [53] and Mukherjee et al. [54] suggested that such an organization was thermodynamically stable and chemically feasible despite there being no known similar structure in biology at that time. Moreover, the model provided an important basis for its further refinement by quantum mechanical calculations and for developing detailed mechanisms for the water splitting reaction leading to dioxygen formation [55-62].
Based on this organization of the metal ions, the Mn3CaO4 cubane had four protein side chains as potential ligands: D1Asp342, D1Glu189, D1His332, and CP43Glu354. The identification of the glutamate of CP43 as a Mn-ligand was a surprise and is a residue of a conserved motif Gly-Gly-Glu-Thr-Met-Arg-Phe-Trp-Asp which forms a 310 helix in the large extrinsic loop joining the lumenal ends of transmembrane helices V and VI of this protein. In addition to these four apparent protein ligands, it was noted that the C-terminal residue of the D1 protein, D1Ala344, is located close to Ca2+, and that D1His337 could be hydrogen bonded to one of the bridging oxo-bonds of the cubane. Two side chain densities were available as ligands for the “dangler” Mn(4) outside the cubane cluster, D1Asp170 and D1Glu333, and it was also suggested that D1Asp61 might also function as a second sphere ligand via a bridging water molecule (see Fig. 2c). A non-protein electron density in the vicinity of Mn(4) and Ca2+ was tentatively modelled as a carbonate ion that formed bridging ligands between the two metals. However, the positioning of this density adjacent to the redox active D1Tyr161 (TyrZ) suggested that this could be the site for the binding of substrate water molecules and for the formation of the O–O bond of dioxygen, as diagrammatically shown in Fig. 2c taken from the Ferreira et al. paper [12]. Also located in this potential catalytic site were D1Gln165 and CP43Arg357, which indicated their possible involvement in a hydrogen bonding network and therefore playing a key role in the deprotonation of the substrate water molecules during the catalytic cycle, while D1Asp61 seemed to be strategically located at the mouth of a polar channel that probably functions to facilitate the exit of protons to the lumenal surface. Moreover, as mentioned above, the work of Ferreira et al. [12] also established that D1His190 was at hydrogen bonding distance to D1Tyr161 as required, and predicted, for the oxidation of the latter by the P680·+ to generate the neutral tyrosine radical [63].
The 3.0 Å resolution structure reported by Loll et al. [15] confirmed the subunit and overall amino acid assignment and side chain positioning reported by Ferreira et al. [12] with minor differences within the limitation imposed by the intermediate resolution of both structures. However, as stated above, the improved electron density map allowed Loll et al. [15] to correct and extend the original assignment of carotenoid molecules by Ferreira et al. [12] and to locate several bound lipid molecules. As in the earlier papers from this group [5], the pear-shaped electron density attributed to the metal-cluster of the water splitting site was interpreted as four Mn ions organized in a Y-shape or 3 + 1 arrangement. Based on anomalous diffraction, the Ca2+ was placed in a position similar to that proposed by Ferreira et al. [12], and confirmed by Kargul et al. [64], whereas the positioning of three of the Mn ions were different to those of Ferreira et al. [12] but less so to that in the Zouni et al. model [5]. The fourth Mn ion was assigned to the narrow end of the pear shaped density in a slightly different position to that of Ferreira et al. [12] or Zouni et al. [5]. The same amino acids identified as ligands in the crystal structure of Ferreira et al. [12] were also found by Loll et al. [15], although there were differences in the precise location in some cases. Importantly, they proposed that the carboxylates of the acidic residues formed bridging ligands between adjacent Mn ions and therefore more adequately satisfied the coordination requirements of the metal cluster than in the Ferreira et al. model [12]. However, Loll et al. [15] emphasized that their model for the OEC was unreliable, in part because of the low resolution of the electron density map and in part because of radiation damage. Working with the EXAFS group at Berkeley, and prior to the publication of the Loll et al. crystal structure of 2005 [15], the same group had shown that doses of X-rays typical of those used for diffraction analyses caused the reduction of the Mn-cluster and associated structural changes [65]. This problem of radiation damage occurring at the Mn-cluster had also been highlighted by Grabolle et al. [66]. Because of this, doubts were expressed about the cubane model of Ferreira et al. [12], and several other arrangements of the Mn4Ca-cluster were postulated based on polarized EXAFS studies conducted by the Berkeley group in collaboration with the X-ray crystallography group in Berlin [67]. However, a reinterpretation of the polarized EXAFS data by Sproviero and colleagues at Yale [68] suggested that the cubane model could still be considered as the legitimate organization of the OEC metal cluster.
The validity of the cubane model was confirmed with the most recent 1.9 Å structure from Umena et al. [14] since at this resolution the electron density of individual metals could be resolved and bridging oxygens inferred (Fig. 2b). The resulting model was overall similar to that proposed by Ferreira et al. [12] and refined by Sproviero et al. [49], except an additional oxo bridge was identified linking the Mn outside the Mn3Ca-cubane (Mn4) to a Mn ion of the cubane, which had been previously proposed by Dau et al. [58] and Siegbahn [60].
Four water molecules (W1 to W4) were found to be associated with the Mn4Ca2+O5 cluster, of which W1 and W2 were coordinated to Mn4 and W3 and W4 were coordinated to the Ca2+ (see Fig. 2b). No other water molecules were directly associated with the metal cluster, suggesting that two of the four waters may serve as the substrates for water oxidation in line with the prediction of Ferreira et al. [12]. All of the amino acid residues coordinated to the Mn4Ca2+O5 cluster were the same as those originally identified by Ferreira et al. [12] and confirmed by Loll et al. [15], but their precise arrangement was now revealed (Fig. 2b and shown diagrammatically in Fig. 2d). Of these, D1Glu189 served as a monodentate ligand to Mn1. All of the remaining five carboxylate residues served as bidentate ligands: D1Asp170 as a ligand to Mn4 and Ca, D1Glu333 to Mn3 and Mn4, D1Asp342 to Mn1 and Mn2, D1Ala344 to Mn2 and Ca, and CP43Glu354 to Mn2 and Mn3. In addition, D1His332 was modelled as being coordinated to Mn1. Together with the oxo bridges and waters, these give rise to a saturating ligand environment for the Mn4Ca2+O5 cluster, with each of the four manganese ions having six ligands whereas the calcium has seven ligands.
In addition to the direct ligands of the Mn4Ca2+O5 cluster, Umena et al. [14] confirmed the finding of Ferreira et al. [12] that D1Asp61, D1His337, and CP43Arg357 are located in the second coordination sphere and may have important roles in maintaining the structure of the metal cluster, in agreement with various recent reports showing the importance of these three residues in maintaining the oxygen-evolving activity [45, 69, 70]. Umena et al. [14] noted that one of the guanidinium g-nitrogens of CP43Arg357 is hydrogen-bonded to both O2 and O4 of the Mn4Ca2+O5 cluster, whereas the other is hydrogen-bonded to the carboxylate oxygen of D1Asp170 and to that of D1Ala344. They also noted that the imidazole ε-nitrogen of D1His337 is hydrogen-bonded to a bridging oxo, in agreement with the suggestion of Ferreira et al. [12]. Thus these two residues may function to stabilize the cubane structure of the metal cluster as well as to provide partial positive charges to compensate for the negative charges induced by the oxo bridges and carboxylate ligands of the metal cluster. Ferreira et al. [12] also suggested that D1Asp61 could be H-bonded to a water bridging to Mn4 (Fig. 2c), and Umena et al. [14] did indeed find that the carboxylate oxygen of D1-Asp61 is hydrogen-bonded to W1, and also to O4 indirectly through another water molecule, and they suggested that this residue may also contribute to stabilizing the metal cluster. Furthermore, as postulated by Ferreira et al. [12], D1Asp61 seems to be located at the entrance of a proton exit channel, suggesting that this residue may function in facilitating proton exit from the catalytic site and be aided by the presence of a bound chloride ion [38, 40].
MECHANISM OF OXYGEN FORMATION
Although the geometry of the Mn4Ca-cluster and its ligand field characteristics are now known at a resolution of 1.9 Å, there is uncertainty about which S-state it is in. Because free electrons are generated in the crystal during the collection of X-ray diffraction data, it is likely that the Mn-cluster is reduced to levels lower than the S0-state. From quantum mechanical calculations (DFT, QM/MM) Luber et al. [71] concluded that the Umena et al. model [14] most likely corresponds to a mixture of S0, S–1, and S–2 oxidation states.
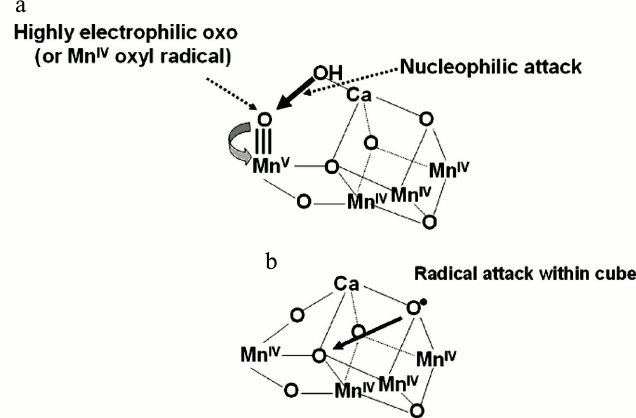
Despite this, the Ferreira et al. cubane model [12] and its subsequent refinements [50, 58, 60] have provided a basis for developing chemical mechanisms for the water oxidation and dioxygen formation. The Mn ion outside the cubane (Mn4) is adjacent to the Ca2+ and their positioning towards the side chains of several key amino acids, including the redox active TyrZ led Ferreira et al. [12] to propose that they provide the “catalytic” surface for binding the two substrate water molecules and their subsequent oxidation (see Fig. 2c). One well-championed mechanism [55, 57, 72-74] would be consistent with a substrate water associated with Mn4 being deprotonated during the S-state cycle and converted to a highly electrophilic oxo (see Fig. 3a). This mechanism is dependent on Mn4 converting to a high oxidation state (possible Mn(V)) during progression to the S4-state just prior to O–O bond formation. Since the other three Mn ions will also progress to high valency states (Mn(IV) (for example S0 (Mn(III), Mn(III), Mn(III), Mn(IV)) to S4 (Mn(V), Mn(IV), Mn(IV), Mn(IV)), then they will act as a further “oxidizing battery” for the Mn(V)-oxo species in the S4-state (see Fig. 3a). In this way the reactive oxo is electron deficient, so much so that it makes an ideal target for a nucleophilic attack by the oxygen of the second substrate water bound within the coordination sphere of the Ca2+ (see Fig. 3a). Deprotonation of the substrate waters would be aided by nearby bases such as CP43 Arg357 and by the weak Lewis acidity of Ca2+. An alternative mechanism proposed by Siegbahn, which is based on in-depth DFT calculations [59-62], suggests that an oxyl radical forms within the Mn3CaO4-cubane and O–O bond formation therefore involves bridging oxo species (see Fig. 3b). This mechanism has recently received some experimental support from Cox et al. [75].
Fig. 3. Two different mechanisms for the final step of the S-state cycle when the dioxygen bond of O2 is formed. a) Mechanism 1. The very high oxidation state of the Mn-cluster, particularly the Mn ion outside the Mn3CaO5-cubane, leads to a highly electron deficient oxo (after deprotonation of water molecules during the S-state cycle). Nucleophilic attack by the hydroxide of the second substrate water within the coordination sphere of Ca2+ leads to O2 formation. b) Mechanism 2. The formation of an oxyl radical on one of the bridging oxygen atoms of the cubane leads to a radical attack of an adjacent oxo-ligand within the Mn3CaO4-cubane. The fifth oxo linking the Mn outside the cubane to a Mn of the cubane is not shown.
ARTIFICIAL PHOTOSYNTHESIS
While some progress has been made in mimicking photosynthesis in artificial systems, researchers have not yet developed components that are both efficient and robust for incorporation into a working system for capturing and storing solar energy in chemical bonds on a large scale as natural photosynthesis does. To date the main focus of research has been to design and synthesize molecular catalysts, which can be linked to a light-driven charge separation system [76]. Dyes have been used for the latter, but inorganic semiconductors offer a more realistic and robust approach for providing the oxidizing and reducing potentials necessary to split water and power reductive chemistry.
Insights gleaned from the recent structural determination of PSII have initiated considerable efforts to identify an artificial catalytic system for water oxidation and hydrogen production using solar energy [77]. The hydrogen produced could be used directly as a source of energy but could also be used, as it is in natural photosynthesis, to reduce carbon dioxide to other types of fuels such as methane, methanol, etc. The challenge is to have a molecular arrangement so that the artificial catalysts efficiently utilize light energy to split water and concomitantly provide reducing potential for hydrogen gas production or CO2 reduction. It has been demonstrated that catalysts based on Mn or on Mn doped with Ca2+ are capable of water splitting and the generation of dioxygen [74, 78-81]. Frei’s group reported that nanostructured manganese oxide clusters supported on mesoporous silica efficiently evolved oxygen in aqueous solution under mild conditions [82].
Complementing the work on Mn has been the earlier discovery that ruthenium-based catalysts, such as the “blue dimer”, can photo-oxidize water to dioxygen [83-85], and the recent spectacular work of Duan et al. [86] has reported a rationally designed Ru-based catalyst with an unprecedentedly turnover rate of 300 per second, which is comparable with that of PSII. But perhaps the most practical catalysts for water splitting so far reported are based on cobalt, which is a relatively abundant element. Kanan and Nocera [87] have described a self-assembling catalyst composed of Co and phosphate or borate ions, which can efficiently produce molecular oxygen from water at neutral pH with a low overpotential akin to that which operates in the OEC of PSII. Dau and his colleagues [88] have revealed important structural details of this Co-based catalyst and found it to have a molecular organization remarkably similar to the Mn3Ca-cubane of PSII. More recently Yin et al. [89] reported a water splitting catalyst comprised of a Co4O4 core stabilized by oxidative resistant polytungstate ligands which, like the Nocera catalyst, is also self-assembling.
Hematite (α-Fe2O3) is a semiconductor that is also capable of photochemically splitting water to molecular oxygen. It has favourable optical band gap (Eg = 2.2 eV), excellent chemical stability in aqueous environments, natural abundance, and low cost [90]. Indeed hematite has been theoretically predicted to achieve a water oxidation efficiency of 16.8% [91]. However, the reported efficiencies of hematite are lower than this predicted value, mainly due to the very short lifetime of photo-generated charge carriers (<10 ps), short hole diffusion length (2-4 nm), slow oxygen evolution reaction kinetics, low flat band potential, and significant reduction in the absorption cross-section at wavelengths approaching the band gap value. Another fundamental limitation of the hematite system is the need for externally applied bias because the conduction band of hematite is lower than the potential required to reduce protons to hydrogen. Nevertheless, it is a system that is receiving considerable attention at the present time [76].
The challenge is to couple these oxygen-producing systems to another catalyst which will use the protons and high energy electrons derived from the water splitting reaction to produce hydrogen gas or reduce carbon dioxide. In the case of the former, considerable progress is being made [92], in part by mimicking the natural hydrogenase enzymes found in a wide variety of microorganisms [93]. Also, a number of inorganic catalysts have been identified with activities that are almost as good as platinum. One such class of catalysts is based on sulfides of Mo and W [94, 95] and another is an alloy composed of nickel, molybdenum, and zinc [96]. Identifying catalysts for the reduction of carbon dioxide, however, is more difficult because using the energy of visible light, multi-electron reactions are required and the emergence of catalysts for generating useful carbon fuels is a considerable challenge [97, 98].
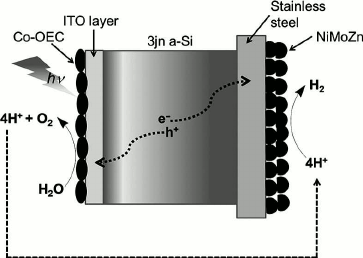
The most successful coupling of catalysts using a semiconductor for light capture and charge separation was reported by Reece and colleagues [96]. They used the semiconductor properties of a triple junction amorphous Si wafer for light capturing and charge separation, the CoPi catalyst for water splitting, and the NiMoZn alloy for the cathodic hydrogen producing catalyst as shown in Fig. 4. This latest discovery is a major step towards the development of efficient, robust, low-cost, and scalable photocatalytic devices for water splitting to generate molecular hydrogen using solar energy.
Fig. 4. Diagrammatic representation of Nocera’s photocatalytic device for water splitting consisting of a triple n-junction amorphous silicon wafer, Co-based oxygen evolving catalyst (OEC), and NiMoZn H2-evolving catalyst as reported in Reece et al. [96].
The determination of the structure of PSII has provided strong hints of how Nature conducts the remarkable chemistry of water splitting. This new information now provides a blueprint for scientists to seriously consider constructing catalysts that mimic the natural system and provide new technologies to address the energy/CO2 problem that mankind must solve. After all, there is no shortage of water for this reaction, and the energy content of sunlight falling on our planet well exceeds our needs.
This article was written while holding research grants from the European Commission (SOLHYDROMICS project (227192), FP-7-Energy-2008-FET, ArtipHyction project (303435) FCH-JU-2011-1; Eco2CO2 project (309701) FP7-NMP2012) and BBSRC and EPSRC (Grant EP/F00270X/1). It is dedicated to the memory of Academician A. A. Krasnovsky in the celebration of the 100th year of his birth. A. A. Krasnovsky was a pioneer in understanding light driven redox reactions in the context of both natural and artificial photosynthesis with particular emphasize on the generation of oxygen and hydrogen from water.
REFERENCES
1.Barber, J. (2003) Biophys. Quart. Rev.,
36, 71-89.
2.De Las Rivas, J., Balsera, M., and Barber, J.
(2004) Trends Plant Sci., 9, 18-25.
3.Joliot, P., Barbieri, G., and Chabaud, R. (1969)
Photochem. Photobiol., 10, 309-329.
4.Kok, B., Forbush, B., and McGloin, M. (1970)
Photochem. Photobiol., 11, 457-475.
5.Zouni, A., Witt, H. T., Kern, J., Fromme, P.,
Krauss, N., Saenger, W., and Orth, P. (2001) Nature, 409,
739-743.
6.Hankamer, B., Barber, J., and Boekema, E. J. (1997)
Annu. Rev. Plant Phys. Mol. Biol., 48, 641-671.
7.Rhee, K.-H., Morris, E. P., Zheleva, D., Hankamer,
B., Kuhlbrandt, W., and Barber, J. (1997) Nature, 389,
522-526.
8.Rhee, K.-H., Morris, E. P., Barber, J., and
Kuhlbrandt, W. (1998) Nature, 396, 283-286.
9.Hankamer, B., Morris, E. P., Nield, J., Gerle, C.,
and Barber, J. (2001) J. Struct. Biol., 135, 262-269.
10.Hankamer, B., Morris, E. P., Nield, J., Carne,
A., and Barber, J. (2001) FEBS Lett., 504, 142-151.
11.Kamiya, N., and Shen, J. R. (2003) Proc. Natl.
Acad. Sci. USA, 100, 98-103.
12.Ferreira, K. N., Iverson, T. M., Maghlaoui, K.,
Barber, J., and Iwata, S. (2004) Science, 303,
1831-1838.
13.Kashino, Y., Takahashi, T., Inoue-Kashino, N.,
Ban, A., Yohei Ikeda, Y., Satoh, K., and Sugiura, M. (2007) Biochim.
Biophys. Acta, 1767, 1269-1275.
14.Umena, Y., Kawakami, K., Shen, J. R., and Kamiya,
N. (2011) Nature, 473, 55-65.
15.Loll, B., Kern, J., Saenger, W., Zouni, A., and
Biesiadka, J. (2005) Nature, 438, 1040-1044.
16.Guskov, A., Kern, J., Gabdulkhakov, A., Broser,
M., Zouni, A., and Saenger, W. (2009) Nat. Struct. Mol. Biol.,
16, 334-342.
17.Barber, J. (1987) Trends Biochem. Sci.,
12, 321-326.
18.Michel, H., and Deisenhofer, J. (1988)
Biochemistry, 27, 1-7.
19.Schubert, W. D., Klukas, O., Saenger, W., Witt,
H. T., Fromme, P., and Krauss, N. (1998) J. Mol. Biol.,
280, 297-341.
20.Murray, J. W., Duncan, J., and Barber, J. (2006)
Trends Plant Sci., 11, 152-158.
21.Durrant, J. R., Klug, D. R., Kwa, S. L. S., van
Grondelle, R. V., Porter, G., and Klug, D. R. (1995) Proc. Natl.
Acad. Sci. USA, 92, 4798-4802.
22.Holzwarth, A. R., Muller, M. G., Rees, M.,
Nowaczyk, M., Sander, J., and Rogner, M. (2006) Proc. Natl. Acad.
Sci. USA, 103, 6895-6900.
23.Barber, J., and Archer, M. D. (2001) J.
Photochem. Photobiol., A142, 97-106.
24.Murray, J. W., and Barber, J. (2007) J.
Struct. Biol., 159, 228-237.
25.Faller, P., Debus, R. J., Brettel, K., Sugiura,
M., Rutherford, A. W., and Boussac, A. (2001) Proc. Natl. Acad. Sci.
USA, 98, 14368-14373.
26.Rutherford, A. W. (1989) Trends Biochem.
Sci., 14, 227-232.
27.Hienerwadel, R., and Berthomieu, C. (1995)
Biochemistry, 34, 16288-16297.
28.Hasegawa, K., Ono, T. A., Inoue, Y., and
Kusunoki, M. (1999) Chem. Phys. Lett., 300, 9-19.
29.Govindjee and van Rensen, J. J. S. (1993) in
The Photosynthetic Reaction Center (Deisenhofer, J. B., and
Norris, J., eds.) Academic Press, San Diego, pp. 357-389.
30.Barber, J., and Andersson, B. (1992) Trends
Biochem. Sci., 17, 61-66.
31.Arnon, D. I., and Whatley, F. R. (1949)
Science, 110, 554-556.
32.Bove, J. M., Bove, C., Whatley, F. R., and Arnon,
D. I. (1963) Z. Naturforsch., 18b, 683-688.
33.Izawa, S., Heath, R. L., and Hind, G. (1969)
Biochim. Biophys. Acta, 180, 388-389.
34.Van Gorkom, H. J., and Yochum, C. F. (2005) in
Photosystem II. The Light-Driven Water Plastoquinone Oxidoreductase
(Wydrzynski, T. J., and Satoh, K., eds.) Springer, Dordrecht, pp.
307-327.
35.Wincencjusz, H., van Gorkom, H. J., and Yocum, C.
F. (1997) Biochemistry, 36, 3663-3670.
36.Boussac, A., Setif, P., and Rutherford, A. W.
(1992) Biochemistry, 31, 1224-1234.
37.Ono, T.-A., Noguchi, T., Inoue, Y., Kusunoki, M.,
Yamaguchi, H., and Oyanagi, H. (1995) J. Am. Chem. Soc.,
117, 6386-6387.
38.Murray, J. W., Maghlaoui, K., Kargul, J., Ishida,
N., Lai, T.-L., Rutherford, A. W., Sugiura, M., Boussac, A., and
Barber, J. (2008) Energy Environ. Sci., 1, 161-166.
39.Kawakami, K., Umena, Y., Kamiya, N., and Shen,
J.-R. (2009) Proc. Natl. Acad. Sci. USA, 106,
8567-8572.
40.Rivalta, I., Amin, M., Luber, S., Vassiliev, S.,
Pokhrel, R., Umena, Y., Kawakami, K., Shen, J.-R., Kamiya, N., Bruce,
D., Brudvig, G. W., Gunner, M. R., and Batista, V. S. (2011)
Biochemistry, 50, 6312-6315.
41.Peloquin, J. M., Campbell, K. A., Randall, D. W.,
Evanchik, M. A., Pecoraro, V. L., Armstrong, W. H., and Britt, R. D.
(2000) J. Am. Chem. Soc., 122, 10926-10942.
42.Hasegawa, K., Ono, T. A., Inoue, Y., and
Kusunoki, M. (1999) Chem. Phys. Lett., 300, 9-19.
43.Diner, B. A. (2001) Biochim. Biophys.
Acta, 1503, 147-163.
44.Debus, R. J. (2001) Biochim. Biophys.
Acta, 1503, 164-186.
45.Debus, R. J. (2008) Chem. Rev.,
252, 244-258.
46.Diner, B. A., Nixon, P. J., and Farchaus, J. W.
(1991) Curr. Opin. Struct. Biol., 1, 546-554.
47.Yachandra, V. K. (2002) Phil. Trans. Roy. Soc.
Lond. B, 357, 1347-1358.
48.Lundberg, M., and Siegbahn, P. E. M. (2004)
Phys. Chem. Chem. Phys., 6, 4772-4780.
49.Sproviero, E. M., Gascon, J. A., McEvoy, J. P.,
Brudvig, G. W., and Batista, V. S. (2006) J. Chem. Theory
Comput., 2, 1119-1134.
50.Sproviero, E. M., Gascon, J. A., McEvoy, J. P.,
Brudvig, G. W., and Batista, V. S. (2007) Curr. Opin. Struct.
Biol., 17, 173-180.
51.Batista, V. S., Sproviero, E. M., Gascon, J. A.,
McEvoy, J. P., and Brudvig, G. W. (2008) Coord. Chem. Rev.,
252, 395-415.
52.Mishra, A., Wernsdorfer, W., Abboud, K. A., and
Christou, G. (2005) Chem. Commun., No. 1, 54-56.
53.Kanady, S., Tsui, E., Day, M., and Agapie, T.
(2011) Science, 333, 733-736.
54.Mukherjee, S., Stull, J. A., Yano, J.,
Stamatatos, T., Pringouri, K., Stich, T. A., Abboud, K. A., Britt, R.
D., Yachandra, V. K., and Christou, G. (2012) Proc. Natl. Acad. Sci.
USA, 109, 2257-2262.
55.McEvoy, J. P., and Brudvig, G. W. (2004) Phys.
Chem. Chem. Phys., 6, 4754-4763.
56.McEvoy, J. P., and Brudvig, G. W. (2006) Chem.
Rev., 106, 4455-4483.
57.Brudvig, G. W. (2008) Philos. Trans. R. Soc.
Lond. B, 363, 1211-1218.
58.Dau, H., Grundmeier, A., Loja, P., and Haumann,
M. (2008) Philos. Trans. R. Soc. Lond. B, 363,
1237-1243.
59.Siegbahn, P. E. M. (2006) Chemistry – A
European Journal, 12, 9217-9237.
60.Siegbahn, P. E. M. (2008) Chemistry,
14, 8290-8302.
61.Siegbahn, P. E. M. (2009) Acc. Chem. Res.,
42, 1871-1880.
62.Siegbahn, P. E. M. (2012) Phys. Chem. Chem.
Phys., 14, 4849-4856.
63.Hoganson, C. W., and Babcock, G. T. (1997)
Science, 277, 1953-1956.
64.Kargul, J., Maghlaoui, K., Murray, J. W., Deak,
Z., Boussac, A., Rutherford, A. W., Vass, I., and Barber, J. (2007)
Biochim. Biophys. Acta, 1767, 404-413.
65.Yano, J., Kern, J., Irrgang, K.-D., Latimer, M.
J., Bergmann, U., Glatzel, P., Pushkar, Y., Biesiadka, J., Loll, B.,
Sauer, K., Messinger, J., Zouni, A., and Yachandra, V. K. (2005)
Proc. Natl. Acad. Sci. USA, 102, 12047-12052.
66.Grabolle, M., Haumann, M., Muller, C., Liebisch,
P., and Dau, H. (2006) J. Biol. Chem., 281,
4580-4588.
67.Yano, J., Kern, J., Sauer, K., Latimer, M. J.,
Pushkar, Y., Biesiadka, J., Loll, B., Saenger, W., Messinger, J.,
Zouni, A., and Yachandra, V. K. (2006) Science, 314,
821-825.
68.Sproviero, E. M., Gascon, J. A., McEvoy, J. P.,
Brudvig, G. W., and Batista, V. S. (2008) J. Am. Chem. Soc.,
130, 3428-3442.
69.Hwang, H. J., Dilbeck, P., Debus, R. J., and
Burnap, R. L. (2007) Biochemistry, 46, 11987-11997.
70.Service, R. J., Hillier, W., and Debus, R. J.
(2010) Biochemistry, 49, 6655-6669.
71.Luber, S., Rivalta, I., Umena, Y., Kawakami, K.,
Shen, J.-R., Kamiya, N., Brudvig, G. W., and Batista, V. S. (2011)
Biochemistry, 50, 6308-6311.
72.Messinger, J., Badger, M., and Wydrynski, T.
(1995) Proc. Natl. Acad. Sci. USA, 92, 3209-3213.
73.Pecoraro, V. L., Baldwin, M. J., Caudle, M. T.,
Hsieh, W.-Y., and Law, N. A. (1998) Pure Appl. Chem., 70,
925-929.
74.Limberg, J., Vrettos, J. S., Liable-Sands, L. M.,
Rheingold, A. L., Crabtree, R. H., and Brudvig, G. W. (1999)
Science, 283, 1524-1527.
75.Cox, N., Rapatskiy, L., Su, J.-H., Pantazis, D.
A., Sugiura, M., Kulik, L., Dorlet, P., Rutherford, A. W., Neese, F.,
Boussac, A., Lubitz, W., and Messinger, J. (2011) J. Am. Chem.
Soc., 133, 3635-3648.
76.Tran, P. D., Wong, L. H., Barber, J., and Loo, J.
S. C. (2012) Energy Environ. Sci., 5, 5902-5918.
77.Eisenberg, E., and Gray, H. B. (2008) Inorg.
Chem., 47, 1697-1699.
78.Tagore, R., Crabtree, R. H., and Brudvig, G. W.
(2008) Inorg. Chem., 47, 1815-1823.
79.Gao, Y., Crabtree, R. H., and Brudvig, G. W.
(2012) Inorg. Chem., 51, 4043-4050.
80.Najafpour, M. M., Ehrenberg, T., Wiechen, M., and
Kurz, P. (2010) Angewandte Chemie, 49, 2233-2237.
81.Zaharieva, I., Najafpour, M. M., Wiechert, M.,
Haumann, M., Kurz, P., and Dau, H. (2011) Energy Environ. Sci.,
4, 2400-2408.
82.Jiao, F., and Frei, H. (2010) Chem. Commun., 46,
2920-2922.
83.Gersten, S. W., Samuels, G. J., and Meyer, T. J.
(1982) J. Am. Chem. Soc., 104, 4029-4030.
84.Liu, F., Concepcion, J. J., Jurss, J. W.,
Cardolaccia, T., Templeton, J. L., and Meyer, T. J. (2008) Inorg.
Chem., 47, 1727-1752.
85.Romero, I., Rodriguez, M., Sens, C., Mola, J.,
Kollipara, M. R., Francas, L., Mas-Marza, E., Escriche, L., and Llobet,
A. (2008) Inorg. Chem., 47, 1824-1834.
86.Duan, L., Bogoglian, F., Mandal, S., Stewart, B.,
Privalot, T., Llobet, A., and Sun, L.-C. (2012) Nature
Chemistry, 4, Pages:418-423.
87.Kanan, M. W., and Nocera, D. G. (2008)
Science, 321, 1072-1075.
88.Risch, M., Khare, V., Zaharieva, I., Gerencser,
L., Chernev, P., and Dau, H. (2009) J. Am. Chem. Soc.,
131, 6936-6937.
89.Yin, Q., Tan, J. M., Besson, C., Geletii, Y. V.,
Musaev, D. G., Kuznetsov, A. E., Luo, Z., Hardcastle, K. I., and Hill,
C. L. (2010) Science, 328, 342-345.
90.Sivula, K., Le Formal, F., and Graetzel, M.
(2011) ChemSusChem., 4, 432-449.
91.Murphy, A. B., Barnes, P. R. F., Randeniya, L.
K., Plumb, I. C., Grey, I. E., Horne, M. D., and Glasscock, J. A.
(2006) Int. J. Hydrogen Energy, 31, 1999-2017.
92.Wang, M., Chen, L., and Sun, L. (2012) Energy
Environ. Sci., 5, 6763-6778.
93.Tran, P. D., Artero, V., and Fontecave, M. (2010)
Energy Environ. Sci., 3, 727-747.
94.Zong, X., Han, J., Ma, G., Yan, H., Wu, G., and
Li, C. (2011) J. Phys. Chem. C, 115, 12202-12208.
95.Merki, D., and Hu, X. (2011) Energy Environ.
Sci., 4, 3878-3888.
96.Reece, S. Y., Hamel, J. A., Sung, K., Jarvi, T.
D., Esswein, A. J., Pijpers, J. J. H., and Nocera, D. G. (2011)
Science, 334, 645-648.
97.Fujita, E. (2000) in McGraw-Hill Yearbook of
Science & Technology (Licker, M. D., ed.) McGraw-Hill Book Co,
New York, pp. 71-74.
98.Arakawa, H., Aresta, M., Armor, J. N., Barteau,
M. A., Beckman, E. J., and Bell, A. T. (2001) Chem. Rev.,
101, 953-996.