REVIEW: Primary Radical Ion Pairs in Photosystem II Core Complexes
V. A. Nadtochenko1, I. V. Shelaev1, M. D. Mamedov2, A. Ya. Shkuropatov3, A. Yu. Semenov1,2*, and V. A. Shuvalov2,3*
1Semenov Institute of Chemical Physics, Russian Academy of Sciences, ul. Kosygina 4, 119991 Moscow, Russia2Belozersky Institute of Physical-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: shuvalov@gmail.com; shuvalov@issp.serpukhov.su; semenov@genebee.msu.ru
3Institute of Basic Biological Problems, Russian Academy of Sciences, ul. Institutskaya 2, 142290 Pushchino, Moscow Region, Russia
* To whom correspondence should be addressed.
Received December 3, 2013; Revision received December 15, 2013
Ultrafast absorption spectroscopy with 20-fs resolution was applied to study primary charge separation in spinach photosystem II (PSII) reaction center (RC) and PSII core complex (RC complex with integral antenna) upon excitation at maximum wavelength 700-710 nm at 278 K. It was found that the initial charge separation between P680* and ChlD1 (Chl-670) takes place with a time constant of ~1 ps with the formation of the primary charge-separated state P680* with an admixture of: P680*(1-δ) (P680δ+ChlD1δ–), where δ ~ 0.5. The subsequent electron transfer from P680δ+ChlD1δ– to pheophytin (Pheo) occurs within 13 ps and is accompanied by a relaxation of the absorption band at 670 nm (ChlD1δ–) and bleaching of the PheoD1 bands at 420, 545, and 680 nm with development of the Pheo– band at 460 nm. Further electron transfer to QA occurs within 250 ps in accordance with earlier data. The spectra of P680+ and Pheo– formation include a bleaching band at 670 nm; this indicates that Chl-670 is an intermediate between P680 and Pheo. Stimulated emission kinetics at 685 nm demonstrate the existence of two decaying components with time constants of ~1 and ~13 ps due to the formation of P680δ+ChlD1δ– and P680+PheoD1–, respectively.
KEY WORDS: P680, chlorophyll, pheophytin, photosystem II core complex, primary charge separation, reaction centerDOI: 10.1134/S0006297914030043
Abbreviations: BA, bacteriochlorophyll primary electron acceptor in BRC; BRC, bacterial reaction center; Chl, chlorophyll a; ChlD1 and ChlD2, monomeric chlorophylls located between P680 and pheophytin in D1 and D2 subunits, respectively; D1/D2/cyt b559, PSII RC; P680, special chlorophyll pair in PSII RC; PD1 and PD2, chlorophyll molecules in D1 and D2 subunits and forming P680; Pheo, pheophytin; PheoD1, Pheo located in D1 protein subunit; PSII, photosystem II; PSII core complex, PSII complex with integral antenna; QA, plastoquinone primary electron acceptor; RC, reaction center.
The present work is dedicated to the memory of the great biophysicist
Academician A. A. Krasnovsky and presents an overview of our recent
work on femtosecond (fs) measurements of the primary charge separation
in reaction centers (RCs) of pigment–protein complex of
photosystem II (PSII) under physiological conditions [1-3].
PSII is the light-driven H2O:plastoquinone-oxidoreductase located in thylakoid membranes of cyanobacteria, green algae, and higher plants. PSII is the main source of the oxygen on Earth, and is also involved in the formation of the primary biomass in the biosphere. The electron density map of dimeric PSII core complex from the cyanobacterium Thermosynechococcus elongatus has recently been solved to a resolution of 2.9-1.9 Å [4, 5]. Each monomer of PSII core complex contains RC D1 and D2 proteins, α- and β-subunits of cyt b559, two integral antenna proteins – CP43 and CP47, which carry 13 and 16 chlorophyll a molecules (Chl), respectively, as well as three extrinsic proteins – 33 kDa (PsbO), 17 kDa (PsbV, cyt c550), and 12 kDa (PsbU). Peripheral proteins are required for maintaining the stability and function of the oxygen-evolving complex.
The RC D1/D2 proteins are located approximately symmetrically with respect to the transmembrane region, which is very similar to the arrangement of the L/M subunits in bacterial RC (BRC) [6, 7]. Four Chls (special pair chlorophyll molecules PD1 and PD2, denoted as P680, and two accessory chlorophylls ChlD1 and ChlD2, in BRC denoted as BA,B), two pheophytins (PheoD1 and PheoD2, in BRC denoted as HA,B), and two plastoquinones (QA and QB) are arranged in two symmetrical branches A and B. As in the BRC, electron transfer in PSII is known [8-11] to proceed only along the D1 branch forming P680+Pheo– and then P680+QA–.
It should be noted that PD1 and PD2 are located close to ChlD1 and ChlD2; the distance between the central atoms of PD1 and ChlD1 Mg, and between PD2 and ChlD2, are 10.2-10.4 Å, respectively [12, 13]. The head groups of PD1 and PD2 are in direct van der Waals contact; the Mg–Mg distance is 8.2 Å [12] or 7.6 Å [13]. Although dimeric “special pair” of PD1 and PD2 with parallel orientation of the macrocycles has weaker coupling than that in the BRC special pair [14], the interaction within P680 is stronger than between P680 and ChlD1 molecules.
According to the X-ray structure of PSII crystals, the porphyrin ring planes of PD1 and ChlD1, PD2 and ChlD2, ChlD1 and PheoD1, and ChlD2 and PheoD2 are not parallel. Thus, one can assume that the formation of excimer or exciplex between parallel macrocycles [15] can be observed only for P680, but not for ChlD1, PheoD1 and the other molecules. Recent spectral measurements in photosystem I (PSI) complexes induced by 20-fs pulses at 720 nm have shown [16] that the excimer is initially formed within P700, which has parallel orientation of Chl macrocycles, while primary charge separation occurs in aggregate consisting of six molecules of RC Chls forming the primary donor P700 and the primary electron acceptor A0 [16].
In the study of PSII RC, a key issue is to determine the frequency of the spectral transitions in each of the four chlorophyll molecules and two molecules of pheophytin, as well as to identify the primary charge separated state. According to recent measurements in isolated PSII RCs using 20-fs laser pulses with a maximum at 700 nm (278 K) [1], the light-induced charge separation is initiated within the four excitonically coupled Chl molecules that form the photoactive core of the PSII complex. Primary charge separation with formation of P680+ChlD1– (with ChlD1 absorbing at 670 nm) is similar to the formation of the radical pair P870+BA– in BRC [17, 18]. The formation of P680+ChlD1– is observed within ~1 ps, but the subsequent transfer of an electron to Pheo occurs within ~14 ps.
Although studies concerning the very early light-induced steps in PSII complexes were initiated over three decades ago, there is still considerable debate about the nature of the primary electron donor under physiological conditions, i.e. it is unclear what the initial electron transfer is: whether it starts from excited special pair P680, from accessory ChlD1, or both cofactors are involved in primary charge separation. It should be noted that in all cases transfer of an electron from Pheo– to QA occurs within ~200 ps [19, 20].
Based on some recent publications, the accessory ChlD1 is the primary electron donor in PSII RC; as such, the primary charge separation is due to ChlD1+Pheo– formation both at cryogenic temperatures and under physiological conditions [21-32]. However, as mentioned before, the results obtained in PSII complexes excited by 20-fs pulses with a maximum wavelength 700-710 nm indicate that P680+ChlD1– is the primary ion-radical pair [1, 2]. On the basis of experimental studies performed in isolated PSII RC by kinetic absorption spectroscopy under various excitation conditions at 77 K [33] and the quantitative modeling of the kinetics of absorption changes [34], the possibility of the existence of alternative pathways for charge separation in PSII, i.e. where P680 or ChlD1 play the role of primary electron donor, was considered [33, 34]. Analysis of the kinetics obtained for PSII RC by 2D spectroscopy showed that involvement of the two electron transfer pathways allowed to obtain better agreement with experiment [35].
Recently we showed [1] that the spectroscopic data derived from PSII RC at low temperature (6 K) [36] indicate that the Qy absorption band near 670 nm can be attributed to ChlD1/D2, which is characterized by a positive polarization in the spectra of circular and linear dichroism. Thus, the spectral form of Chl-670 corresponds to ChlD1 [1]. In a recent paper we have presented new experimental data for spinach PSII core complexes using 20-fs photolysis (pump–probe) at 710 nm [2]. These data support our previous results [1] derived from isolated PSII RCs and provide evidence that at physiological temperatures and conditions used by us (20-fs excitation with a maximum at 700 nm, 278 K), the primary electron donor and acceptor are P680* and ChlD1, respectively.
It was previously shown that illumination of oxygen-evolving PSII core complexes at 1.7 K, as well as spinach leaves at 293 K, resulted in the reduction of QA, the action spectrum of which has a band in the 710-730 nm region [37, 38]. Therefore, for the excitation of the sample it was possible to use femtosecond pulses with a peak wavelength at 710 nm.
KINETICS OF PRIMARY REACTIONS OF ELECTRON TRANSPORT IN PSII
RC
Differential spectra of absorbance changes (ΔA) at 278 K derived from isolated PSII RCs in the range of 400-710 nm at various delays (from 0.1 to 28.5 ps) were determined upon excitation of samples with 20-fs laser with maximum wavelength at 700 nm [1]. The main changes were related to the bleaching of the Soret and QY bands of Chl and Pheo molecules at ~430 and 682 nm, respectively, including stimulated emission from these molecules in the red region of spectra. In agreement with previous measurements [9] indicating that both Pheo molecules contribute to the most long wavelength absorption bands of absorption in RC, the bleaching of the Qx band of Pheo at 545 nm is observed at early delay times (0.1-28 ps). The amplitude of the 545-nm bleaching is almost constant within this time period. This observation suggests that the excited state of the RC includes partially Pheo*D1,D2 that is eventually converted to the charge-separated state P680+PheoD1– with similar bleaching at 545 nm.
The kinetics of ΔA at 665 nm includes a fast (completed within 2.5 ps) bleaching with subsequent relaxation with the time constant of 13.3 ps. As will be shown below, these absorbance changes are related to the reduction of the primary electron acceptor (Chl-670, identified as ChlD1) and its subsequent oxidation by further electron transfer to PheoD1.
Significant changes of ΔA spectra in the range 410-470 nm are observed and give evidence in favor of fs and ps formation of the radical anion bands of Chl– and/or Pheo–, which absorb near 450 nm [39]. To reveal the dynamics of this process, the ΔA spectrum of the RC excited state measured at the earliest delay (0.1-0.15 fs) was subtracted from the spectra taken at later delays; as a result, double differential spectra ΔΔA were obtained. It has been suggested that the bleaching of the Soret band is similar for (Chl/Pheo)* and (Chl/Pheo)–. Therefore, the difference ΔΔA should be mainly related to radical anion (Chl/Pheo)– formation. The appearance in the ΔΔA spectra for PSII RC of the band at 445 nm related to radical anion formation [39] occurs at time delays shorter than 1 ps, followed by a further increase in absorption with a characteristic time of 14 ps [1].
PRIMARY AND SECONDARY ION-RADICAL PAIR FORMATION IN PSII CORE
COMPLEXES
As shown earlier, the oxidation of the primary electron donor P680 (684 nm) at low temperature (77 K) was observed in isolated PSII RC complexes in the presence of external electron acceptor SiMo [2] (Fig. 1a). At the same time, the oxidation of Chl-674 (possibly along with P680) with a bleaching at 674 nm upon formation of the state ChlD1+QA– in PSII core complex was observed (Fig. 1d). Note that earlier a bleaching at 674 nm was attributed to PD1 form [23, 30-32, 40-42]. However, analysis of the results obtained upon excitation of isolated RCs and PSII core complexes with 20-fs laser pulses shows that Chl-670 with bleaching and relaxation within ~1 and ~14 ps, respectively, is evidently an intermediary electron acceptor between P680 and Pheo at 278 K and does not seem to be a part of P680 (PD1 or PD2) [1-3].
Fig. 1. a) Spectrum of irreversible difference absorbance changes ΔA (light-minus-dark) in PSII RC in the presence of SiMo (0.1 mM) excited by red light (λ > 600 nm) at 77 K. b) Spectrum of reversible ΔA (time constant is a few seconds) measured with a phosphoroscopic set-up in PSII RC in the presence of DCBQ (1 mM) and SiMo (0.1 mM) with the same excitation as in (a) but at 90 K. c) Spectrum of reversible ΔA (time constant of a few msec) measured with the phosphoroscope in PSII RC in the presence of DPQ (1 mM) and SiMo (0.1 mM) at 90 K. d) Spectrum of reversible ΔA (time constant of a few msec) in PSII core complexes in the presence of SiMo (0.1 mM) at 90 K.
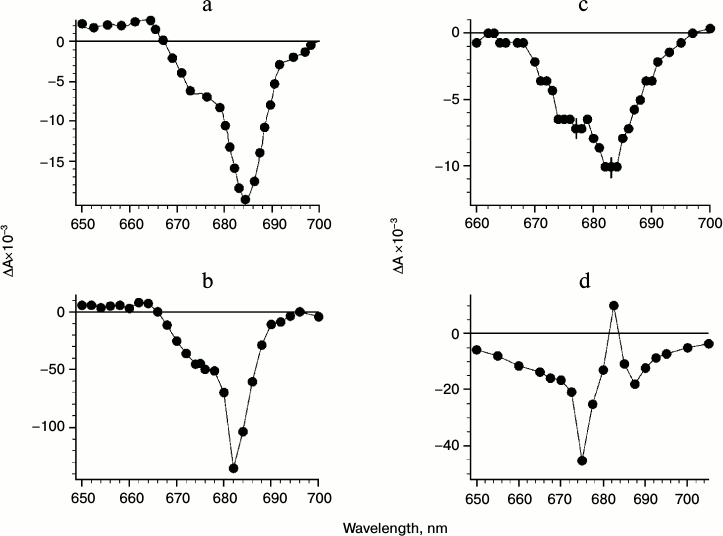
In favor of the participation of ChlD1 (Chl-670) in electron transfer between P680 and Pheo indicate data concerning redox potential changes of P680/P680+ and ChlD1/ChlD1+ induced by the formation of QA– at low temperature in PSII core complexes [1-3]. Assuming fully “frozen” atomic polarizability of protein and solution medium (corresponding to dielectric permittivities (ε) for protein 2.5 and for medium 1.84) at 100 K, we obtained a 44 meV negative shift of midpoint redox potential (Em) of ChlD1/ChlD1+ with respect to that of P680/P680+ induced by the field of QA–. This shift is due to the steric position of both donors with respect to QA– and does not exceed 8-11 meV at room temperature (corresponding to ε for protein 4-6 and ε for medium 81). Taking into account an energy difference of 27 meV between quanta at 674 and 684 nm and close position of LUMO orbitals of P680 and ChlD1, the redox potential of ChlD1/ChlD1+ at low temperature in PSII core complex can become more negative than that of P680/P680+ (17 meV). Note that such effect is observed upon formation of the ChlD1+QA– state with subsequent recombination in the millisecond time domain in PSII core complex. As a result, in PSII core complex the ChlD1 might function as terminal electron donor at low temperature with bleaching at 674 nm along with the P680 bleaching at 684 nm.
Figure 1a shows differential (light minus dark) absorption spectra derived from isolated PSII RC in the presence of silicomolybdate (SiMo) as an external electron acceptor at 77 K. The figure shows that under irreversible charge separation the P680 may function as an electron donor (bleaching at 684 nm according to Shuvalov et al. [43]). A bleaching and slight blue shift of the band around 670 nm, probably due to ChlD1 oxidation, is observed.
Figure 1 (b and c) shows PSII RC spectra in the presence of SiMo at a higher concentration of dichlorobenzoquinone (DCBQ) (1 mM) (Fig. 1b) or decylplastoquinone (DPQ) (Fig. 1c).
When DCBQ is added before freezing, the bleaching becomes mostly reversible (recombination time in the range of seconds), and the spectrum of ΔA demonstrates the bleaching centered at 684 nm with a shoulder at 674 nm (Fig. 1b). Upon addition of DPQ the recombination time of @ChlD1+QA– is decreased to several milliseconds. This leads to the decrease in the amplitude of absorbance changes because the phosphoroscopic setup has limited resolution in the millisecond time domain (Fig. 1c). In this case the more pronounced bleaching at 674 nm is observed due to the closer position of DPQ to the putative QA site in the RC complex (Fig. 1c). The spectrum in Fig. 1c depicts two bleachings at 684 and 674 nm with the amplitude at 684 nm much smaller than that in Fig. 1b. The spectrum of reversible absorption changes (relaxation time of several milliseconds) in PSII core complexes in the presence of SiMo at 90 K is shown in Fig. 1d. The spectrum shows that reversible millisecond recombination for the @(P680-ChlD1)+QA– state is accompanied by bleaching at 674 nm and blue shift (and bleaching) of the 684 nm band. Similar data were described in [23, 30-32, 40-42] using an assumption about the oxidation of PD1 and reduction of QA. However, we assume that the resulting spectrum can be attributed to the state (P680-ChlD1)+QA– [1-3] in which the positive charge is distributed between P680 and ChlD1.
Figures 2-4 present data of femtosecond experiments with spinach PSII core complexes. To minimize the excitation of antenna Chls, 20-fs pulses centered at 710 nm and energy of 50 nJ were used.
Differential spectra of absorption changes (ΔA) at 278 K in the spectral range 400-725 nm at various delay times (between 0.3 and 455 ps) reveal some important features. These features are clearly evident when considering subtraction of absorption changes measured at 0.15 ps from ΔA registered at later delay times. Figure 2a shows kinetics of absorption changes induced by femtosecond pulses at the 545 nm band reflecting the formation of excited state (unresolved fast time component <20 fs) and the Pheo anion-radical (characteristic time τ = 13 ps). The inset (Fig. 2b) shows the differential spectrum at 33 ps delay caused by bleaching of the QX band of PheoD1. Figure 2c shows the kinetics of absorption changes at 670 nm due to the formation and disappearance of the radical anion ChlD1–. It is seen that the formation of ChlD1– occurs within 0.9 ps, and the kinetics of its decay (14 ps) coincides with the kinetics of Pheo– formation (Fig. 2a).
Fig. 2. a) Kinetics of absorbance changes ΔA at 545 nm in PSII core complex excited by 20-fs pulses at 710 nm. b) The spectrum of ΔA in the 525-565 nm region measured at 33 ps delay and representing the bleaching of the Pheo absorbance band at 545 nm. c) ΔA kinetic at 670 nm; conditions as in Fig. 2a.
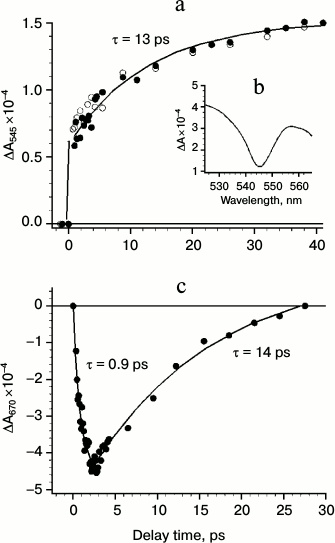
Figure 3 shows kinetics of absorption changes at 460 nm due to the formation (τ ~ 11 ps) and disappearance (τ ~ 250 ps) of PheoD1– anion-radical. Note that the oxidation kinetics of ChlD1– (Fig. 2c, τ ~ 14 ps) agrees quite well with the kinetics of PheoD1 reduction (Fig. 2a, τ ~ 13 ps and Fig. 3, τ ~ 11 ps). This indicates that electron transfer from monomeric ChlD1 to PheoD1 occurs with a characteristic time of 11-14 ps. The kinetics of formation of Pheo– is more than an order of magnitude slower than the oxidation kinetics of P680 and ChlD1– formation (τ = 1 ps). Thus, these data indicate that the primary electron acceptor, at least under the experimental conditions used, is ChlD1, while PheoD1 plays the role of the secondary electron acceptor.
Fig. 3. Kinetics of absorbance changes ΔA at 460 nm in PSII core complexes (conditions as in Fig. 2a). The rise time of the formation of the 460-nm band (Pheo–) is within 11 ± 3 ps, and its relaxation time is 250 ± 50 ps.
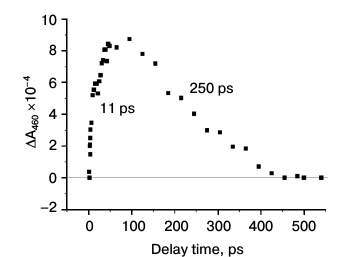
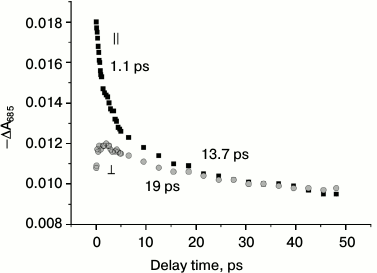
Figure 4 shows that the stimulated emission at wavelength 685 nm measured with a parallel orientation of the electrical vectors of the exciting and probing pulses has at least two decay kinetic phases with lifetimes τ1 ~ 1 ps and τ2 ~ 14 ps (initial anisotropy ~0.25). The τ1 component coincides with the kinetics of formation of P680+ChlD1— [1-3], and the τ2 component coincides with formation of P680+PheoD1— (Figs. 2c and 3). This result shows that the primary ion-radical pair P680+ChlD1— is a quencher of the exited states of the pigments. Thus, under these experimental conditions, the electron transfer reaction to Pheo is not involved in the process of primary charge separation in PSII RC. The decay kinetics of stimulated emission detected with perpendicular orientation of the electric vectors of the exciting and probing pulses demonstrates the appearance of a new component with a significantly lower polarization (anisotropy is 0.08) than that of the first component with lifetime of ~ 1 ps observed in the parallel orientation. The emergence of this new component was accompanied by an absolute increase in amplitude with perpendicular orientation, which is quite unusual for stimulated emission decay. Since about 30% of the stimulated emission decays over a longer time (τ2 ~ 14 ps), we have assumed that the formation of the state P680+ChlD1— can be considered as a mixture of states with charge transfer P680+ChlD1— and excited state P680*. Mixture of states can be represented as P680(1-δ)*(P680δ+ChlD1δ–), where δ ~ 0.5. This mixed state is observed as a stimulated emission at ~685 nm with a low positive polarization, which decays by further electron transfer from ChlD1δ– to Pheo within ~14 ps. Nevertheless, it is possible that the two-component decay kinetics of stimulated emission is due to the existence of two populations of PSII RC decaying with different lifetimes. However, increasing of the perpendicular component with delay time is not consistent with this interpretation.
Fig. 4. Kinetics of absorbance changes ΔA at 685 nm (stimulated emission) in PSII core complexes (conditions as in Fig. 2a). The rise time of the formation of the 685-nm band is 20 fs for both parallel and perpendicular orientation of electric dipole moments for excitation and measuring beams. The decay of the band for parallel polarization has two components with lifetimes of ~1 and ~13.7 ps. For perpendicular polarization the band at 685 nm has two components with approximately the same lifetimes, but the first one increases and the second decays.
As shown in Fig. 3, the band of the Pheo anion-radical at 460 nm completely disappeared at ~450 ps delay, indicating the full electron transfer from Pheo– to QA. It is known that electron transfer to QA must be completed within 450 ps [19]. Thus, the transient spectrum at a delay of 445 ps corresponds to the formation of P680+, because the differential absorption spectrum of QA– does not appear in the visible spectrum.
Figure 5 shows the difference between the transient spectra measured at 23 or 44 ps and the spectrum measured at 455 ps, which is ascribed to the P680+ difference spectrum. Since during this time range the only electron transfer event is the formation of P680+QA–, this ΔΔA spectrum can be assigned to Pheo– formation and has bleaching at 420, 545, 671, and 685 nm and developments at 460 and 660 nm. The spectrum is similar to those previously obtained by accumulation methods [11, 44], and it therefore proves that charge separation between P680* and Pheo is accompanied by the entire transfer of electron density from P680* to Pheo via ChlD1 in the picosecond time domain. This result also confirms that the accumulation method applied previously for the study of PSI [45], BRC [46], and PSII RC [11, 44] demonstrates the photochemical reactions in RCs.
Fig. 5. Difference spectrum of irreversible absorbance changes ΔΔA (light-minus-dark) in PSII core complexes at 278 K excited by 20-fs pulses at 710 nm obtained by subtraction of the spectrum ΔA at 445 ps delay (P680+ spectrum) from spectra ΔA at 23 and 44 ps delays. The bleaching at 545 nm reflects the kinetics of photoreduction of Pheo.
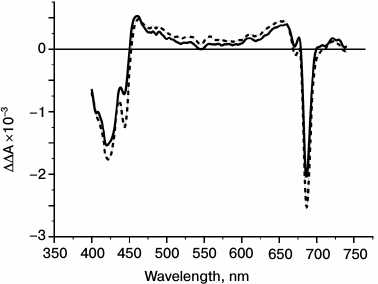
The spectrum of the formation of PheoD1– presented in Fig. 5 also shows an additional bleaching at 670 nm, which was assigned to Chl-670 [1-3]. This feature indicates the close position of the PheoD1 to the Chl-670 molecule, which as we earlier suggested can play a role of the primary electron acceptor ChlD1 functioning between P680* and PheoD1 [1-3].
DISCUSSION
Data presented in this work show that the primary charge separation in RC and in PSII core complexes induced by 20-fs pulses with peak wavelength of 700-710 nm (278 K) is due to the formation of the state P680+ChlD1–. In so doing, the time constant for the formation of P680+ChlD1– is ~0.8 ps, which is revealed from the Chl-670 rise time and delay of stimulated emission at 685 nm. The decay time of the state P680+ChlD1– is ~13 ps measured from Chl-670 decay, from the formation of the Pheo– anion radical band at 460 nm, the bleaching at 545 nm, and from stimulated emission at 685 nm.
The formation of the state P680+PheoD1– in the range of 20-40 ps can be confirmed by the subtraction of the spectrum of state P680+ observed at 445 ps delay from the spectra of P680+PheoD1– measured at 23 and 44 ps (Fig. 5). The resulting spectra are very similar to the spectrum of PheoD1– observed previously by the accumulation method [11, 44].
The bleaching bands and pigment spectral shifts are independent for formations of P680+ and Pheo–, which allows observing the additive sum of its individual features in ΔA spectra at 23 and 44 ps delays. This is in contrast to the situation observed for bacterial RCs where intensive electrochromic shift of the BA band at 800 nm is observed in varying degrees for formation of both P+ and BPheo– ions [46]. The P680+PheoD1– state disappears within 250 ± 50 ps due to electron transfer from PheoD1– to QA. The bleaching (or red shift) of the 670-nm band in the spectrum of the Pheo– formation obtained from fs/ps measurements [1-3] or the accumulation method at room and low temperatures [11, 43, 44] indicates significant interaction and close arrangement of PheoD1 and Chl-670 molecules. The bleaching of the 670-nm band (675 nm at low temperature [2]) in PSII RC and core complexes [1, 2] also indicates the nearby location of the P680 and Chl-670 molecules.
Taking into account the last two observations, we conclude that Chl-670 is located in the vicinity of both P680 and PheoD1. The bleaching of the Chl-670 band in both cases can be due to disappearance of excitonic interaction between Chl-670 and P680 or PheoD1 when two latter electron carriers are oxidized or reduced, respectively. According to the expression for dipole strength (D) of the excitonic band in aggregate [47], the D value for the transition A in aggregate (which is close to transition α in monomer) has a sign depending on the following expression:
DA = B – Cνανβ/(νβ2 – να2),
where να and νβ are frequencies of the transitions in two interacting molecules α and β, D and B are positive, and C is suggested to be a positive constant. If να is frequency for the 670-nm transition in ChlD1 and νβ is for the 680-nm transition in P680 (or PheoD1), then the D value increases for the 670-nm transition and decreases for 680-nm due to excitonic interaction in the aggregate. If interaction is broken by photochemistry in PSII RC, DA is decreased to the value characteristic of the absence of interaction between 670- and 680-nm transitions. Since both oxidation of P680 and reduction of PheoD1 were accompanied by decrease in the 670-nm transition amplitude, we conclude that ChlD1 is located between P680 and PheoD1 and may play the role of the intermediary electron carrier between P680* and PheoD1 as suggested earlier [1-3]. The decay kinetics of the stimulated emission at ~685 nm (Fig. 4) indicates that at least two emitting centers are observed. The first center with maximal positive polarization (~0.25) appears to reflect the emission from the excited states of P680* and/or PheoD1*. This emission decays with a lifetime of ~1 ps due to electron transfer from P680* to ChlD1 with the formation of mixed state P680(1-δ)*(P680δ+ChlD1δ–). This latter state (second center of emission) emits light at 685 nm with smaller positive polarization (~0.08). The decrease in the component with parallel polarization is accompanied by an increase in the same component with perpendicular polarization –ΔA685 (Fig. 4). This evidently shows the formation of a new emitting center at 685 nm, which is probably caused by the formation of the state P680(1-δ)*(P680δ+ChlD1δ–), where δ ~ 0.5. The latter state decays due to further electron transfer to PheoD1 within ~13 ps as observed by fs/ps measurements showing Pheo– formation.
We thank F. E. Gostev for assistance in femtosecond measurements and V. A. Shkuropatova, M. I. Vishnev, and A. A. Zabelin for assistance in preparation of PSII complexes.
Financial support from the Programs “Molecular and Cell Biology”, “The Fundamentals of Technologies of Nanostructures and Nanomaterials”, “Leading Scientific Schools” (SS-307.2012.4), Russian Foundation for Basic Research grants (11-04-00818, 11-04-91330, 12-04-00821, 13-04-40297H, 13-04-40298H, 13-04-40299H) are gratefully acknowledged.
REFERENCES
1.Shelaev, I. V., Gostev, F. E., Nadtochenko, V. A.,
Shkuropatov, A. Ya., Zabelin, A. A., Mamedov, M. D., Semenov, A. Y.,
Sarkisov, O. M., and Shuvalov, V. A. (2008) Photosyn. Res.,
98, 95-103.
2.Shelaev, I. V., Gostev, F. E., Vishnev, M. I.,
Shkuropatov, A. Ya., Ptushenko, V. V., Mamedov, M. D., Sarkisov, O. M.,
Nadtochenko, V. A., Semenov, A. Y., and Shuvalov, V. A. (2011) J.
Photochem. Photobiol. B: Biol., 104, 44-50.
3.Nadtochenko, V. A., Semenov, A. Y., and Shuvalov,
V. A. (2014) Biochim. Biophys. Acta, Feb 7, doi:
10.1016/j.bbabio.2014.01.026 [Epub ahead of print].
4.Guskov, A., Kern, J., Gabdulkhakov, A., Broser, M.,
Zouni, A., and Saenger, W. (2009) Nature, 16,
334-342.
5.Umena, Y., Kawakami, K., Shen, J.-R., and Kamiya,
N. (2011) Nature, 473, 55-60.
6.Michel, H., Epp, O., and Deisenhofer, J. (1986)
EMBO J., 5, 2445-2451.
7.Komiya, H., Yeates, T. O., Rees, D. C., Allen, J.
P., and Feher, G. (1988) Proc. Natl. Acad. Sci. USA, 85,
9012-9016.
8.Shkuropatov, A. Ya., Khatypov, R. A., Volshchukova,
T. S., Shkuropatova, V. A., Ovens, T. G., and Shuvalov, V. A. (1997)
FEBS Lett., 420, 171-174.
9.Shkuropatov, A. Ya., Khatypov, R. A., Shkuropatova,
V. A., Zvereva, M. G., Ovens, T. G., and Shuvalov, V. A. (1999) FEBS
Lett., 450, 163-167.
10.Raszewski, G., Saenger, W., and Renger, T. (2005)
Biophys. J., 88, 986-998.
11.Klimov, V. V., Klevanik, A. V., Shuvalov, V. A.,
and Krasnovsky, A. A. (1977) FEBS Lett., 82, 183-186.
12.Ferreira, K. N., Iverson, T. M., Maghlaoui, K.,
Barber, J., and Iwata, S. (2004) Science, 303,
1831-1838.
13.Loll, B., Kern, J., Saenger, W., Souni, W., and
Biesiadka, J. (2005) Nature, 438, 1040-1044.
14.Ivashin, N., and Larsson, S. J. (2005) J.
Phys. Chem. B, 109, 23051-23060.
15.Beens, H., and Weller, A. (1975) in Organic
Molecular Photophysics (Birks, J. B., ed.) John Willey & Sons,
London, Vol. 11, pp. 159-215.
16.Shelaev, I. V., Gostev, F. E., Mamedov, M. D.,
Sarkisov, O. M., Nadtochenko, V. A., Shuvalov, V. A., and Semenov, A.
Y. (2010) Biochim. Biophys. Acta, 1797,
1410-1420.
17.Yakovlev, A. G., Shkuropatov, A. Ya., and
Shuvalov, A. V. (2002) Biochemistry, 41, 14019-14027.
18.Khatypov, R. A., Khmelnitskiy, A. Yu., Khristin,
A. M., and Shuvalov, V. A. (2010) Dokl. Biochem. Biophys.,
430, 24-28.
19.Nuijs, A. M., van Gorkom, H. J., Plijter, J., and
Duysens, L. N. M. (1986) Biochim. Biophys. Acta, 848,
167-175.
20.Eckert, H. J., Wiese, N., Bernarding, J.,
Eichler, H. J., and Renger, G. (1988) FEBS Lett., 240,
153-158.
21.Prokhorenko, V., and Holzwarth, A. R. (2000)
J. Phys. Chem. B., 104, 11563-11578.
22.Renger, G., and Renger, T. (2008) Photosynth.
Res., 98, 53-80.
23.Diner, B. A., Schlodder, E., Nixon, P. J.,
Coleman, W. J., Rappaport, F., Laverge, J., Vermaas, W. F. J., and
Chisholm, D. A. (2001) Biochemistry, 40, 9265-9281.
24.Frese, R. N., Germano, M., de Weerd, F. L., van
Stokkum, I. H. M., Shkuropatov, A. Ya., Shuvalov, V. A., van Gorkom, H.
J., van Grondelle, R., and Dekker, J. P. (2003) Biochemistry,
42, 9205-9213.
25.Germano, M., Gradinaru, C. C., Shkuropatov, A.
Ya., van Stokkum, I. H. M., Shuvalov, V. A., Dekker, J. P., van
Grondelle, R., and van Gorkom, H. J. (2004) Biophys. J.,
86, 1664-1672.
26.Groot, M. L., Pawlowicz, N. P., van Wilderen, L.
J. G. W., Breton, J., van Stokkum, I. H. M., and van Grondelle, R.
(2005) Proc. Natl. Acad. Sci. USA, 102, 13087-13092.
27.Holzwarth, A. R., Muller, M. G., Reus, M.,
Nowaczyk, M., Sander, J., and Rogner, M. (2006) Proc. Natl. Acad.
Sci. USA, 103, 6895-6900.
28.Pawlowicz, N. P., Groot, M.-L., van Stokkum, I.
H. M., Breton, J., and van Grondelle, R. (2007) Biophys. J.,
93, 2732-2742.
29.Di Donato, M., Cohen, R. O., Diner, B. A.,
Breton, J., van Grondelle, R., and Groot, M. L. (2008) Biophys.
J., 94, 4783-4795.
30.Raszewski, G., Diner, B. A., Schlodder, D. E.,
and Renger, T. (2008) Biophys. J., 95, 105-119.
31.Schlodder, E., Renger, T., Raszewski, G.,
Coleman, W. J., Nixon, P. J., Cohen, R. O., and Diner, B. A. (2008)
Biochemistry, 47, 3143-3154.
32.Renger, T., and Schlodder, E. (2010) Chem.
Phys. Chem., 116, 1141-1153.
33.Romero, E., Stokkum, I. H. M., Novoderezhkin, V.
I., Dekker, J. P., and van Grondelle, R. (2010) Biochemistry,
49, 4300-4307.
34.Novoderezhkin, V. I., Romero, E., Dekker, J. P.,
and van Grondelle, R. (2011) Chem. Phys. Chem., 12,
681-688.
35.Gelzinis, A., Valkunas, L., Fuller, F. D.,
Ogilvie, J. P., Mukamel, S., and Abramavicius, D. (2013) New J.
Phys., 15, 075013.
36.Germano, M., Shkuropatov, A. Ya., Permentier, H.,
de Wijn, R., Hoff, A. J., Shuvalov, V. A., and van Gorkom, H. J. (2001)
Biochemistry, 40, 11472-11482.
37.Hughes, J. L., Smith, P., Race, R., and Krausz,
E. (2006) Biochim. Biophys. Acta, 1757, 841-851.
38.Heber, U., and Shuvalov, V. A. (2005)
Photosynth. Res., 84, 85-91.
39.Fujita, I., Davis, M. S., and Fajer, Y. J. (1978)
J. Am. Chem. Soc., 100, 6280-6282.
40.Schlodder, E., Coleman, W. J., Nixon, P. J.,
Cohen, R. O., Renger, T., and Diner, B. A. (2008) Philos. Trans.
Roy. Soc. Lond. Biol. Sci., 363, 1197-1202.
41.Hillmann, B., and Schlodder, E. (1995)
Biochim. Biophys. Acta, 1231, 76-88.
42.Hillmann, B., Brettel, K., van Mieghem, F.,
Kamlowski, A., Rutherford, A. W., and Schlodder, E. (1995)
Biochemistry, 34, 4814-4827.
43.Shuvalov, V. A., Heber, U., and Schreiber, U.
(1989) FEBS Lett., 258, 27-31.
44.Klevanik, A. V., Klimov, V. V., Shuvalov, V. A.,
and Krasnovsky, A. A. (1977) Dokl. AN SSSR, 236,
241-244.
45.Shuvalov, V. A. (1975) Biochim. Biophys.
Acta, 430, 113-121.
46.Shuvalov, V. A., and Klimov, V. V. (1976)
Biochim. Biophys. Acta, 440, 587-599.
47.Tinoco, I., Jr. (1962) Adv. Chem. Phys.,
4, 113-160.