REVIEW: Interaction of Molecular Oxygen with the Donor Side of Photosystem II after Destruction of the Water-Oxidizing Complex
D. V. Yanykin*, A. A. Khorobrykh, O. M. Zastrizhnaya, and V. V. Klimov
Institute of Basic Biological Problems, Russian Academy of Sciences, ul. Institutskaya 2, 142290 Pushchino, Moscow Region, Russia; fax: (4967) 330-532; E-mail: ya-d-ozh@rambler.ru* To whom correspondence should be addressed.
Received November 22, 2013; Revision received December 16, 2013
Photosystem II (PSII) is a pigment–protein complex of thylakoid membrane of higher plants, algae, and cyanobacteria where light energy is used for oxidation of water and reduction of plastoquinone. Light-dependent reactions (generation of excited states of pigments, electron transfer, water oxidation) taking place in PSII can lead to the formation of reactive oxygen species. In this review attention is focused on the problem of interaction of molecular oxygen with the donor site of PSII, where after the removal of manganese from the water-oxidizing complex illumination induces formation of long-lived states (P680+• and TyrZ•) capable of oxidizing surrounding organic molecules to form radicals.
KEY WORDS: photosystem II, reactive oxygen species, manganese, hydroperoxidesDOI: 10.1134/S0006297914030055
Abbreviations: apo-WOC-PSII, PSII membrane fragments deprived of the WOC; Chl, chlorophyll; D1, PSII reaction center polypeptide; DCBQ, 2,6-dichloro-1,4-benzoquinone; DPC, diphenylcarbazide; E0, standard oxidation-reduction potential; ETC, electron transport chain; ΔF, photoinduced changes of chlorophyll fluorescence yield related to the photoreduction of the primary quinone acceptor QA; HO•, hydroxyl radical; H2O2, hydrogen peroxide; HP-OOH, hydrophilic hydroperoxides; LP-OOH, lipophilic hydroperoxides; MCPBA, m-chloroperbenzoic acid; Mn4CaO5 cluster, the catalytic inorganic core of the PSII WOC; (Mn3+)2(di-μ-oxo)-complex, an intermediate both in the photoassembly and disassembly of the WOC; 1O2, singlet oxygen; O2−•, superoxide anion radical; P680, primary electron donor of PSII; Pheo, pheophytin, the primary electron acceptor of PSII; PSII, photosystem II; RC, reaction center; ROOH, organic peroxides; ROS, reactive oxygen species; SOD, superoxide dismutases; Spy-HP, 2-(4-diphenylphosphanylphenyl)-9-(1-hexylheptyl)anthra[2,1,9-def,6,5,10-d′e′f′]diisoquinoline-1,3,8,10-tetraone; TBHP, tert-butylhydroperoxide; TyrZ, redox active tyrosine residue of D1 protein; WOC, water-oxidizing complex.
MOLECULAR ORGANIZATION OF PHOTOSYSTEM II AND POSSIBLE
INTERACTIONS OF OXYGEN WITH THE ELECTRON TRANSPORT CHAIN OF PHOTOSYSTEM
II
Photosystem II (PSII) is a pigment–protein complex located in the thylakoid membrane of oxygenic photosynthetic organisms. The light-dependent reactions of electron transport in PSII lead to water oxidation with the formation of molecular oxygen and protons as well as to the reduction of plastoquinone to plastoquinol. Crystallographic investigations of cyanobacterial PSII from Thermosynechococcus elongatus [1] and Thermosynechococcus vulcanus [2] showed that the complex of PSII contains at least 20 protein subunits, 35 chlorophyll (Chl) molecules, two pheophytin (Pheo) molecules, 11-12 molecules of carotenoids, more than 20 integral lipid molecules, two molecules of plastoquinone, two heme irons, one non-heme iron, one bicarbonate ion, 3-4 calcium atoms (one of which is in the Mn4Ca cluster), and four manganese atoms.
PSII can be divided into three basic functional blocks: (1) light harvesting complex that absorbs photons and transports the excitation energy to the reaction center of PSII, (2) the photochemical reaction center (RC) in which the primary photochemical reaction of charge separation occurs, (3) the water-oxidizing complex (WOC) where water oxidation and oxygen evolution occur.
In the photochemical RC, light energy absorbed by chlorophyll is transformed into the energy of separated charges, and the strongest biological oxidant, P680+•, the oxidized primary electron donor of PSII (with redox potential of 1.1-1.27 V [3-7]) is formed. Until the early 1980s the primary photochemical reaction in PSII was regarded as charge separation between the excited P680 and the primary electron acceptor QA – a bound plastoquinone molecule with redox potential of –130 mV. However, in works published in the late 1970s and early 1980s, it was found that one more electron carrier operates between P680 and QA – pheophytin with redox potential of –610 mV – and that the primary photoreaction consists of the photoformation of the radical ion pair [P680+•Pheo−•] [3, 8-11]. In circumstances where QA is in the reduced state, the lifetime of [P680+•Pheo−•] is 2-4 ns [12, 13].
The WOC consists of a catalytic inorganic core, the Mn4CaO5-cluster where three manganese atoms, one calcium atom and four oxygen atoms form a cubane-like structure in which they are located at the corners. The fourth manganese atom and one oxygen atom are located outside the cubane. The calcium atom with atoms of manganese as well as the manganese atoms among themselves are linked by μ-oxo bridges. P680+• oxidizes the WOC to form a series of intermediate S-states (S0, S1, S2, S3, and S4), the transition from S4 to S0 is accompanied by oxidation of two water molecules and the formation of O2.
O2 is not only formed in PSII, but it also interacts with the components of the PSII electron transport chain with formation of reactive oxygen species (ROS) – singlet oxygen (1O2), superoxide anion radical (O2−•), hydrogen peroxide (H2O2), and hydroxyl radical (HO•) [14].
There are several types of reactions leading to photoformation of ROS in PSII. Type I is the univalent reduction of O2 to O2−• on the acceptor side, where the generated reduction potential is low enough for the reaction (reaction (1) and figure).
O2 + ē → O2−• (1)
Possible redox interactions of O2 with components of the electron transport in PSII membranes deprived of the WOC (apo-WOC-PSII). On the acceptor side of PSII, molecular oxygen can react with the reduced electron carriers to form superoxide anion radical O2−• (reaction (1)). Oxygen can react with triplet chlorophyll (3P680) with the formation of singlet oxygen 1O2 in circumstances when the system lacks active electron acceptors (that facilitates charge recombination in the primary ion-radical pair of [P680+•Pheo−•] with the formation of 3P680*). On the donor side of apo-WOC-PSII, O2 can react with organic radicals (R•) produced by photooxidation of organic molecules (RH) by the long-lived states of P680+• and TyrZ• (reaction (2)), and peroxyl radicals (ROO•) are formed (reaction (3)). Designations: P680, the primary electron donor of PSII; TyrZ, redox active tyrosine residue of D1 protein; Pheo, pheophytin, the primary electron acceptor of PSII; Mn4CaO5 cluster, catalytic inorganic core of the PSII WOC; QA and QB, the primary and secondary quinone electron acceptors; 3Car, the triplet-state of a carotenoid molecule
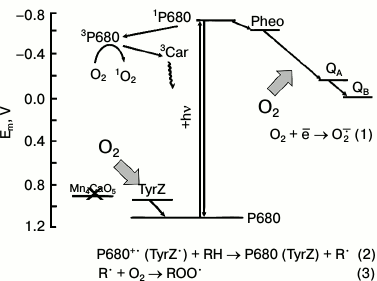
Possible sites of O2−• formation in PSII are the reduced forms of the primary electron acceptor Pheo−• [15] and of the primary and secondary acceptor quinones QA− and QB− [16, 17], and the plastosemiquinone [18-20]. There is also evidence that cytochrome b559 can reduce O2 [21, 22]. Spontaneous or enzyme-catalyzed dismutation of O2−• results in the production of H2O2. H2O2 photoformation in PSII was shown by a luminol-peroxidase assay [21, 23]. The reduction of H2O2 by low-valency transition metals (Fenton reaction) results in the generation of the hydroxyl radical (HO•) having high reactivity.
Type II is photoformation of singlet oxygen (1O2), which is mainly generated through the interaction of the triplet-state chlorophyll (3Chl*) with O2. The formation of 3Chl* can occur in the reaction center of PSII when the system lacks active electron acceptors (which facilitates charge recombination in the primary ion-radical pair of [P680+•Pheo−•] with the formation of 3P680* [24]) as well as in the light-harvesting complexes [25].
Type III is the photoformation of ROS on the donor side. It has been shown that partial impairment of the WOC leads to photoformation of H2O2 on the donor side [17, 23, 26, 27]. The mechanism of the photoformation of H2O2 on the donor side was assumed to consist in two-electron oxidation of two molecules of H2O, although the exact mechanism is yet to be elucidated. H2O2 may be further oxidized to form O2−• or reductively cleaved to form HO•.
OXYGEN PHOTOCONSUMPTION AND FORMATION OF ORGANIC PEROXIDES ON THE
DONOR SIDE OF PSII
When the WOC is destroyed the lifetimes of the oxidized primary (P680) and secondary (TyrZ) electron donors of PSII are significantly increased, and hence their chances of oxidizing the surrounding molecules such as chlorophylls, carotenoids, and amino acids will increase [28-33]. In recent works, we have shown that in addition to the well-known mechanisms of the interaction of O2 with PSII and ROS generation, there is an unknown mechanism of oxygen photoconsumption on the donor side of PSII resulting in the formation of organic peroxides [34-37].
We found an increase in of O2 photoconsumption in PSII preparations at high pH and after manganese removal from the WOC [34, 35]. Based on the study of the effect of diuron, catalase, and exogenous electron donors and acceptors of PSII, we suggested that the oxygen photoconsumption could be caused by two processes: the reduction of O2 to O2−• and the interaction of O2 with organic radicals formed as a result of oxidative activity of P680+• or TyrZ•.
Further, we will consider and discuss our data on the oxygen consumption under continuous and pulsed illumination of PSII preparations after destruction of the WOC by removal of manganese.
Photoconsumption of O2 after damage or removal of the water-oxidizing complex of PSII. Continuous illumination of functionally active PSII preparations in the presence of exogenous electron acceptors (100 μM 2,6-dichloro-p-benzoquinone (DCBQ) and 1 mM potassium ferricyanide (K3[Fe(CN)6]) resulted in oxygen evolution at the rate of 500 μmol O2/h per mg Chl. Under illumination of the PSII preparations in the absence of exogenous electron acceptors, insignificant oxygen photoconsumption (less than 2 μmol O2/h per mg Chl) was observed [34, 35]. At high pH (8.5-9.0) or upon the removal of manganese from the WOC, a 6-fold increase in the rate of oxygen photoconsumption (up to 10-12 μmol O2/h per mg Chl) was observed. The light-induced O2 uptake was totally suppressed by diuron, the inhibitor of electron transport in PSII.
The study of O2 consumption under illumination of PSII preparations with destroyed WOC (apo-WOC-PSII) by a series of saturating microsecond light flashes is of considerable interest because the flash results in a single charge separation in photochemical RCs and the appearance of only one positive and one negative redox equivalent. It is known that the illumination of the native PSII preparations by a series of saturating microsecond light flashes in the presence of exogenous electron acceptor leads to O2 evolution with a typical oscillation period of four and maximum of O2 yield observed upon the third flash [38, 39]. In our experiments, we observed similar oscillations on illumination of native PSII preparations by a series of microsecond light flashes. It should be noted that the first flash resulted in neither evolution nor consumption of O2 in these PSII preparations. After the removal of Mn from the WOC, only O2 photoconsumption was observed with a maximum on the first flash. Importantly, the amplitude of the signal related to consumption of O2 on the first light flash in apo-WOC-PSII was comparable with the yield of O2 observed in native PSII preparations on the third light flash in the presence of exogenous electron acceptors. If we assume that the yield of O2 evolution in the native PSII preparations on the third saturating flash is one molecule per RC, then the amount of O2 consumed on the first flash in the Mn-depleted PSII will be of 0.8-0.9 molecule of O2 per RC. It is an approximate estimation as the amplitude of the O2 yield on the third light flash in functionally active PSII preparations as well as the amplitude of the O2 uptake on the first light flash in apo-WOC-PSII depends on several factors (type of preparation, the redox state of electron carriers, etc.). Nevertheless, these data indicate that photoconsumption of O2 in apo-WOC-PSII has a very high efficiency comparable with that of O2 evolution in native PSII preparations.
Effect of diuron and exogenous electron donors and acceptors on photoconsumption of O2. As in the case of continuous illumination, O2 uptake induced by microsecond light flashes was almost completely inhibited by diuron, indicating the need for electron transfer in PSII for the oxygen photoconsumption in apo-WOC-PSII [35].
Investigation of dependence of the O2 photoconsumption in apo-WOC-PSII on the concentration of artificial electron acceptors showed that the rate of O2 uptake in the presence of saturating concentrations of artificial electron acceptors (DCBQ and K3[Fe(CN)6]) remains rather high (about 70 and 65% of the initial level for continuous and pulsed illumination, respectively) [35]. The data indicate that approximately 65-70% of the O2 photoconsumed in apo-WOC-PSII may be attributed to the donor side of PSII.
The study of effect of exogenous electron donors on the O2 photoconsumption in apo-WOC-PSII confirms this suggestion [35]. Exogenous electron donors such as ferrocyanide (K4[Fe(CN)6]) and DPC as well as Mn2+ (which is used for the formation of a functionally active Mn4CaO5-cluster during the procedure of photoactivation [40-44]) were used in this investigation. It was shown that the rate of O2 uptake under continuous illumination of apo-WOC-PSII decreased as the concentration of exogenous electron donors grew and the rate was 29, 43, and 35% of control upon the addition of saturating concentrations of K4[Fe(CN)6], DPC, and MnCl2, respectively. The addition of K4[Fe(CN)6] at saturating concentrations resulted in a 80-85% suppression of the flash-induced O2 uptake. A similar effect was observed upon the addition of another exogenous electron donor, DPC, i.e. at a saturating concentration of DPC (100-200 μM) the inhibition of the flash-induced O2 uptake reached from 65 to 70%.
It is known that the removal of manganese from PSII membranes leads to a drastic decrease in photoinduced changes of chlorophyll fluorescence yield (ΔF) of PSII related to the photoreduction of the primary electron acceptor QA, since these preparations are incapable of QA– photoaccumulation in the absence of electron donors. Upon the addition of exogenous electron donors, the photoinduced ΔF is largely restored as a result of increase in electron flow to P680+•, which promotes accumulation of QA– [45]. Comparison of the effect of exogenous electron donor (for example, MnCl2) on the rate of light-induced O2 uptake and the restoration of photoinduced ΔF showed that the restoration of ΔF is accompanied by a decrease in the rate of O2 photoconsumption [35]. Thus, the recovery of electron transport in PSII by exogenous electron donors leads to a decrease in O2 photoconsumption.
Upon the joint addition of artificial electron acceptors and donors, O2 uptake induced by both continuous and pulsed illumination of apo-WOC-PSII was completely inhibited [35]. These results indicate that the photoconsumption of O2 is a result of the redox interaction of O2 with components of PSII. Although the effect of exogenous electron donors and acceptors on the photoconsumption of O2 was somewhat different under pulsed and continuous illumination of apo-WOC-PSII, we can note the following: O2 photoconsumption is the result of reactions occurring on both the acceptor and the donor side of PSII, with predominant (about 70%) photoconsumption of O2 on the donor side of PSII.
It is known that when Mn4CaO5-cluster is totally destroyed the supply of electrons from water to the PSII RC is stopped. In this case P680+• and TyrZ• will live a relatively long time. This can lead to oxidation of the surrounding molecules and photoinhibition of PSII via the donor-side induced mechanism of photoinhibition. The suppression of the O2 photoconsumption in apo-WOC-PSII by exogenous electron donors indicates the relation between the O2 photoconsumption and the formation of the long-lived state of P680+•. The results show that the increase in concentration of exogenous electron donors, on one hand, leads to increase in ΔF restoration and, on the other hand, decrease in the rate of the light-induced O2 uptake in apo-WOC-PSII. One interpretation of these data is that about 70% of oxygen photoconsumption in apo-WOC-PSII occurs on the donor side of PSII.
On the basis of these data, it was assumed that the light-induced O2 uptake in apo-WOC-PSII could be caused by two processes: reduction of O2 to O2−• by reduced electron carriers on the acceptor side of PSII, and the interaction of O2 with organic radicals formed as a result of oxidative activity of P680+• or TyrZ• (reactions (2)-(4)) (the figure):
P680+• (or TyrZ•) + RH → P680 (TyrZ) + R•, (2)
R• + O2 → ROO•, (3)
ROO• + RH → ROOH + R•, (4)
where RH, R•, ROO•, and ROOH represent an organic molecule, its radical, its peroxyl radical, and its peroxide, respectively.
The loss of electron donation to P680+• leads to the photooxidation of the surrounding molecules with the formation of organic radicals R˙. It is known that in solutions organic radicals can interact with O2 to form corresponding peroxyl radicals ROO˙ [46, 47], which are then transformed into hydroperoxides ROOH.
Unusual effect of Mn2+ on O2 consumption under pulsed illumination of apo-WOC-PSII. When studying light-induced O2 consumption, it was found that in contrast to other exogenous electron donors, Mn2+ at micromolar concentrations activated the flash-induced O2 consumption in apo-WOC-PSII [35, 36]. The activating effect of Mn2+ was maximal at 5-100 µM Mn2+ (which corresponds to 2-40 Mn atoms per PSII RC), and upon further increase in Mn2+concentration the effect was lowered, while at higher concentrations (>200-400 µM Mn2+) Mn2+(like other exogenous electron donors to PSII) induced inhibition of the flash-induced O2 consumption [35, 36]. Depending on the sample, the Mn2+ concentration required for the maximum activation of the flash-induced O2 uptake varied from 10 to 100 µM.
The results showed that the activation of the flash-induced O2 uptake was observed at low concentrations of Mn2+, when electron transport on the donor side of PSII was not completely restored. It is possible that manganese is involved in light dependent reactions resulting in the additional photoconsumption of O2. Possible reactions involving Mn2+ in the activation of the flash-induced O2 uptake were proposed:
a) Mn2+ binding in apo-WOC-PSII may lead to structural changes in PSII, which, in turn, may result in the activation of O2 consumption both on the acceptor and the donor sides of PSII;
b) the activation of flash-induced O2 consumption in apo-WOC-PSII upon the addition of catalytic Mn2+ concentration may be due to the oxidation of organic molecules via their interaction with Mn3+ formed as a result of the photooxidation of the added Mn2+ by PSII;
c) Mn2+-induced activation of flash-induced O2 uptake may be due to redox interaction of Mn2+ with ROS (hydroperoxides, O2−•, H2O2) formed upon the illumination of PSII. The reactions can produce radicals, which may cause additional O2 consumption;
d) Mn2+-induced activation of flash-induced O2 uptake in apo-WOC-PSII may reflect the involvement of O2 or its reactive forms in the photoassembly of the inorganic Mn-containing core of the WOC, in particular, in the formation of (Mn3+)2(di-μ-oxo) complex believed to be a key intermediate for the assembly of the Mn cluster.
Mn-depleted PSII preparations are known to be very sensitive to the inhibitory action of light. It is known that the illumination of Mn-depleted PSII preparations results in the irreversible loss of the ability of PSII to be reactivated by Mn2+ [33]. As shown in our work [35], pre-illumination of apo-WOC-PSII for 5 min by continuous light (λ > 650 nm, 900 µmol photons/(m2·s)) resulted in the suppression of photoinduced ΔF reactivation by exogenous Mn2+. Besides, the pre-illumination also inhibited flash-induced O2 consumption, led to a considerable loss of the effect of added Mn2+ on the flash-induced O2 consumption, and suppressed both effects of Mn2+: activation at low Mn2+ concentrations and inhibition at higher Mn2+ concentrations. The results showed that only the donor side of PSII capable of “functional” redox interaction with Mn2+ can be involved in these effects. It is important to note that the photoinhibition procedure caused certain damage to the donor side of PSII, while the photoinduced charge separation in PSII RCs remains active.
Specific effects of manganese and calcium ions on O2 photoconsumption under pulsed illumination of apo-WOC-PSII. To study Mn2+-induced activation of flash-induced O2 uptake in apo-WOC-PSII, the effect of other divalent metal ions (Me2+) on the flash-induced O2 consumption was examined. Two types of metal ions were investigated: (1) non-transition metals with electron configuration ns2, such as Mg and Ca, and (2) transition metals (Co, Fe, V, and Cr), having, like Mn, electronic configuration ns2(n–1)dm [36]. It was shown that the addition of Mg2+ and Ca2+ does not affect the O2 consumption under pulsed illumination of apo-WOC-PSII [36]. This may indicate that the effect of Mn2+ is not associated with structural changes in PSII that could be induced by the metal ions. Most likely, the effect of Mn2+ is associated with the involvement of Mn2+ and/or Mn3+ (as a result of Mn2+ photooxidation) in redox reactions on the donor side of apo-WOC-PSII. The addition of Co2+ (with a high E0 equal to 1.8 V [48]) did not influence the flash-induced O2 consumption in apo-WOC-PSII. Addition of metal ions with a relatively low redox potential: Fe2+, V2+, and Cr2+ (E0 for Fe2+/Fe3+, V2+/V3+, and Cr2+/Cr3+ is equal to 0.771, –0.255, and –0.407 V, respectively [48]) led to a considerable inhibition of the flash-induced O2 consumption [36]. Addition of Fe2+ and V2+, in contrast to Co2+, led to restoration of ΔF in PSII. However, in contrast to Mn2+, which is also able to donate electrons to the apo-WOC-PSII, other ions of transition metals (Co2+, Fe2+, V2+, and Cr2+) did not activate the O2 photoconsumption at any concentration (up to 2.5 mM). These data confirm the unique capability of Mn2+ to activate the flash-induced O2 consumption in apo-WOC-PSII.
Thus, in comparison with other ions of metals used in this work, only Mn2+ ions were able to activate the flash-induced O2 consumption. The absence of a similar effect for other transition metal ions capable of electron donation to apo-WOC-PSII with formation of Me3+ can be explained by: (1) inability of the Me3+ to take part in redox reactions leading to the O2 consumption (for water solutions, E0 for the pairs Mn3+/Mn2+ and Fe2+/Fe3+ is equal 1.51 and 0.771 V, respectively [48]), or (2) redox potential of P680+• (or TyrZ•) is not enough for oxidation of Me2+ to Me3+ which, in turn, could oxidize organic molecules (RH).
As noted above, we suggested that Mn2+-induced activation of the flash-induced O2 consumption in apo-WOC-PSII may reflect the involvement of O2 in the photoassembly of the Mn4O5Ca inorganic core of the WOC of PSII, in particular, in the formation of (Mn3+)2(di-μ-oxo) complex [35]. It is known that Ca2+ is an obligatory cofactor for both proper assembly of Mn-containing cluster and recovery of the oxygen-evolving activity [49]. We considered the effect of Ca2+ on the Mn2+-activated O2 photoconsumption in apo-WOC-PSII. It was found that the effect of Ca2+ depended on concentration of added Mn2+: it was maximum at Mn2+ concentration of 5 µM, lowered at higher Mn2+ concentration, and disappeared at Mn2+ concentration of 100 µM when the effect of Mn2+ itself reached its maximum [36]. It was shown that in the presence of 5 µM MnCl2 the stimulatory effect of Ca2+ grows upon the increase in Ca2+ concentration from 2.5 to 7.5 µM (when the Ca2+/RC ratio is 1-3), while at higher Ca2+ concentrations its effect was lowered so that at Ca2+ concentration of 100 µM its effect disappeared. In the absence of Mn2+, calcium had no effect on the flash-induced O2 consumption in apo-WOC-PSII.
When replacing Ca2+ by Mg2+, no activation of the flash-induced O2 consumption in apo-WOC-PSII was observed either in the absence or in the presence of Mn2+ in the medium, and Mg2+ even caused a decrease in the amplitude of the flash-induced O2 consumption measured in the presence of Mn2+. Subsequent addition of Ca2+ resulted in the elimination of the inhibitory effect of Mg2+, and then activation of the flash-induced O2 consumption took place. In this case, a 14-fold increase in the concentration of Ca2+ (10 and 140 µM Ca2+ in the absence and in the presence of Mg2+, respectively) was required to achieve the maximum activation of Mn2+-induced O2 photoconsumption.
The effect of Ca2+ on the Mn2+-activated O2 photoconsumption in apo-WOC-PSII can also support the idea of the involvement of O2 in the photoassembly of inorganic Mn-containing core of the WOC since this effect was unique for Ca2+ (which is strictly necessary for the restoration of the oxygen-evolving activity of the WOC). But we cannot exclude the possibility that enhancement of O2 photoconsumption reflects the photodestruction of the donor side of PSII in the early stages of the assembly of the enzymatic center of PSII. We suggest that the effect of Ca2+ could be associated with the organization of the binding site(s) for Mn ions in apo-WOC-PSII.
Experimental evidence for photoformation of organic peroxides on the donor side of apo-WOC-PSII. We verified our suggestion that the light-induced O2 consumption on the donor side of PSII is related to interaction of O2 with radicals (R•) produced by photooxidation of organic molecules (RH) with long-lived states of P680+• and TyrZ•, and organic peroxides (ROOH) are formed as the final product [37]. To identify the hydroperoxides photoproduced in apo-WOC-PSII, a lipophilic fluorescence probe 2-(4-diphenylphosphanylphenyl)-9-(1-hexylheptyl)anthra[2,1,9-def,6,5,10-d′e′f ′]diisoquinoline-1,3,8,10-tetraone (Spy-HP) specific to peroxides was used. Spy-HP reacts with hydroperoxides to form its oxidized product, Spy-HPOx, resulting in a significant increase in fluorescence [50]. To obtain insight into the specificity of the probe for various peroxide species, the interaction of Spy-HP with m-chloroperbenzoic acid (MCPBA), tert-butylhydroperoxide (TBHP), and H2O2 was studied. The data showed that the reaction of lipophilic hydroperoxides LP-OOH (for example, MCPBA) with Spy-HP was completed within 5 min, while the interaction of the fluorescence probe with hydrophilic hydroperoxides HP-OOH (for example, TBHP) and H2O2 was not completed after 180 min [37].
It was shown that illumination of untreated PSII membranes for 3 min led to just a small increase in the fluorescence intensity of Spy-HP, while illumination of apo-WOC-PSII resulted in a significant (8-fold) increase in the fluorescence intensity of the probe [37]. To characterize the photoproduced peroxide species in apo-WOC-PSII, the illuminated Mn-depleted PSII membranes were incubated for various periods of time with Spy-HP, and the fluorescence increase was monitored. Two components, fast and slow, were distinguishable in this dependence: the fast component was the fluorescence increase in the initial 5 min, and the slow one was the subsequent monotonic increase. Based on the finding that the reaction rate of peroxides with Spy-HP depended on their hydrophobicity, the fast component was attributed to the formation of LP-OOH, and the slow one to HP-OOH. To examine whether the O2−• and H2O2 (which are formed upon illumination of PSII membranes) contributed to the R-OOH fluorescence, the experiments were done in the presence of superoxide dismutase (SOD) and catalase. It was found that the addition of these enzymes did not suppress the formation of LP-OOH and HP-OOH. These data indicated that neither O2−• nor H2O2 significantly contributed to the oxidation of Spy-HP. There was a slight stimulation of Spy-HPOx fluorescence by the addition of SOD and catalase. This might have been due to the disappearance of H2O2, which, had it been present, could have acted as a PSII donor. To eliminate the possible influence of O2−• and H2O2, all subsequent experiments were performed in the presence of SOD and catalase.
The effects of diuron, electron donors (DPC and K4[Fe(CN)6]), and electron acceptor (K3[Fe(CN)6]) on the ROOH photoformation in apo-WOC-PSII were examined. A considerable (70-75%) suppression of the formation of LP-OOH and HP-OOH by diuron confirmed that this photoreaction was related to electron flow in PSII. The electron acceptor ferricyanide decreased the photoproduction of both LP-OOH and HP-OOH by 10% [37]. This effect of ferricyanide can be explained if we assume the production of a small amount of ROOH on the acceptor side. Another explanation is that the ferrocyanide (produced as a result of ferricyanide photoreduction) donated electrons and thereby suppressed the ROOH formation on the donor side. The result indicated that the ROOH formed on the acceptor side, if any, constitutes a minor fraction of the total detected ROOH.
When the electron donor DPC was added, the photoproduction of both LP-OOH and HP-OOH was decreased by 80-90%. This was ascribable to the electron donor blocking the photoaccumulation of the cation-radical P680+• or TyrZ•, and thereby suppressing the production of organic peroxides in apo-WOC-PSII. Another electron donor, ferrocyanide, showed a similar degree of suppression.
It was found that the half-lives of LP-OOH and HP-OOH (approximately 12 min) photoproduced in apo-WOC-PSII were not affected by either DPC or K4[Fe(CN)6] [37]. Thus the suppression of ROOH formation in apo-WOC-PSII by DPC and K4[Fe(CN)6] was ascribed to their electron donation to PSII. These results using Spy-HP were thus in good correspondence with the O2 photoconsumption data and support our hypothesis that the photoconsumption of O2 on the PSII donor side leads to the formation of ROOH via radical chain reactions.
Upon addition of FeCl2 (a Fenton reagent that induces the decomposition of peroxides) to illuminated apo-WOC-PSII, the increase in fluorescence induced by the oxidation of Spy-HP was not observed, while it did not quench the fluorescence of Spy-HPOx that had been produced via the oxidation of Spy-HP with 0.5 µM MCPBA. Thus, it was confirmed that the detected oxidants that were formed in apo-WOC-PSII were peroxides.
To determine the yields of LP-OOH and HP-OOH in apo-WOC-PSII, the light-intensity dependence of the photoproduction of both types of ROOH was investigated. LP-OOH and HP-OOH showed different light-saturation curves (at 25 and 750 μmol photon/(m2·s), respectively). At the light intensity of 750 μmol photon/(m2·s) for 3 min, the yield of LP-OOH and HP-OOH in apo-WOC was 4 and 200 molecules per RC of PSII, respectively. In this case, the photoproduction rates of LP-OOH and HP-OOH were 0.37 and 18 μmol/h per mg Chl, respectively. The photoproduction rate of ROOH was comparable to the O2 photoconsumption rate in apo-WOC-PSII (11-12 μmol/h per mg Chl) [37].
Thus, the results present experimental evidence for photoproduction of ROOH on the donor side of PSII deprived of components of the WOC.
Physiological relevance of the peroxides formed on the donor side of PSII. At the present time, the physiological role of organic peroxides formed on the donor side of PSII is unknown. It is believed that donor side-induced photoinhibition starts with the inactivation of the WOC. Illumination of apo-WOC-PSII will lead to the enzymic degradation of D1 protein. It is generally accepted that specific proteases are involved in this process. However, there is little information about the mechanism of the degradation of D1 protein. We can propose that the formation of LP-OOH lead to modification of the D1 protein, and this type of modification can be an initial event to trigger the protein degradation in the donor side-induced photoinhibition. Small reactive aldehydes such as malondialdehyde and acrolein, derived from lipid peroxides or HP-OOH, have been shown to occur and to modify several PSII proteins in vivo under heat stress conditions [51]. Although the accumulation levels of LP-OOH and HP-OOH in apo-WOC-PSII are very low and their chemical identity has yet to be elucidated, the formation of these potentially reactive species should be of significant importance in the photoinhibition mechanisms. On the other hand, the process of ROOH formation on the donor side of apo-WOC-PSII may be involved in the formation of the inorganic core of the WOC during photoreactivation. This speculation is based on the fact that the addition of catalytic concentration of Mn2+ as well as ions of Ca2+ leads to increase in the flash-induced O2 consumption in apo-WOC-PSII.
This work was supported by the Russian Foundation for Basic Research (grants 12-04-31166 and 12-04-90044) and the Russian Academy of Sciences Presidium Program “Molecular and Cell Biology”.
REFERENCES
1.Guskov, A., Kern, J., Gabdulkhakov, A., Broser, M.,
Zouni, A., and Saenger, W. (2009) Nat. Struct. Mol. Biol.,
16, 334-342.
2.Umena, Y., Kawakami, K., Shen, J.-R., and Kamiya,
N. (2011) Nature, 473, 55-60.
3.Klimov, V. V., Allakhverdiev, S. I., Demeter, S.,
and Krasnovsky, A. A. (1979) Dokl. AN SSSR, 249,
227-230.
4.Rappaport, F., Guergova-Kuras, M., Nixon, P. J.,
Diner, B. A., and Lavergne, J. (2002) Biochemistry, 41,
8518-8527.
5.Ishikita, H., Loll, B., Biesiadka, J., Saenger, W.,
and Knapp, E.-W. (2005) Biochemistry, 44, 4118-4124.
6.Allakhverdiev, S. I., Tomo, T., Shimada, Y., Kindo,
H., Nagao, R., Klimov, V. V., and Mimuro, M. (2010) Proc. Natl.
Acad. Sci. USA, 107, 3924-3929.
7.Allakhverdiev, S. I., Tsuchiya, T., Watabe, K.,
Kojima, A., Los, D. A., Tomo, T., Klimov, V. V., and Mimuro, M. (2011)
Proc. Natl. Acad. Sci. USA, 108, 8054-8058.
8.Klimov, V. V., Klevanik, A. V., Shuvalov, V. A.,
and Krasnovsky, A. A. (1977) FEBS Lett., 82, 183-186.
9.Klevanik, A. V., Klimov, V. V., Shuvalov, V. A.,
and Krasnovsky, A. A. (1977) Biofizika, 236,
241-244.
10.Klimov, V. V., and Krasnovsky, A. A. (1981)
Photosynthetica, 15, 592-609.
11.Klimov, V. V. (2005) in Discoveries in
Photosynthesis (Govindjee, Beatty, J. T., Gest, H., and Allen, J.
F., eds.) Springer, pp. 275-281.
12.Klimov, V. V., Allakhverdiev, S. I., and
Pashchenko, V. Z. (1978) Dokl. AN SSSR, 242,
1204-1207.
13.Shuvalov, V. A., Klimov, V. V., Dolan, E.,
Parson, W. W., and Ke, B. (1980) FEBS Lett., 118,
279-282.
14.Pospisil, P. (2009) Biochim. Biophys.
Acta, 1787, 1151-1160.
15.Ananyev, G. M., Renger, G., Wacker, U., and
Klimov, V. (1994) Photosynth. Res., 41, 327-338.
16.Bekina, R. M., Lebedeva, A. F., and Shuvalov, V.
A. (1976) Dokl. AN SSSR, 231, 739-742.
17.Ananyev, G., Wydrzynski, T., Renger, G., and
Klimov, V. (1992) Biochim. Biophys. Acta, 1100,
303-311.
18.Khorobrykh, S. A., and Ivanov, B. N. (2002)
Photosynth. Res., 71, 209-219.
19.Khorobrykh, S. A., Mubarakshina, M. M., and
Ivanov, B. N. (2004) Biochim. Biophys. Acta, 1657,
164-167.
20.Mubarakshina, M. M., and Ivanov, B. N. (2010)
Physiol. Plant., 140, 103-110.
21.Kruk, J., and Strzalka, K. (1999) Photosynth.
Res., 62, 273-279.
22.Pospisil, P., Snyrychova, I., Kruk, J., Strzalka,
K., and Naus, J. (2006) Biochem. J., 397, 321-327.
23.Ananyev, G. M., and Klimov, V. V. (1988) Dokl.
AN SSSR, 298, 1007-1011.
24.Krasnovsky, A. A. (1982) Photochem.
Photobiol., 36, 733-741.
25.Pospisil, P. (2012) Biochim. Biophys.
Acta, 1817, 218-231.
26.Ananyev, G. M., and Klimov, V. V. (1989)
Biokhimiya, 54, 1587-1597.
27.Klimov, V., Ananyev, G., Zastrizhnaya, O.,
Wydrzynski, T., and Renger, G. (1993) Photosynth. Res.,
38, 409-416.
28.Jegerschoeld, C., Virgin, I., and Styring, S.
(1990) Biochemistry, 29, 6179-6186.
29.Hanley, J., Deligiannakis, Y., Pascal, A.,
Faller, P., and Rutherford, A. W. (1999) Biochemistry,
38, 8189-8195.
30.Tracewell, C. A., Vrettos, J. S., Bautista, J.
A., Frank, H. A., and Brudvig, G. W. (2001) Arch. Biochem.
Biophys., 385, 61-69.
31.Telfer, A., Frolov, D., Barber, J., Robert, B.,
and Pascal, A. (2003) Biochemistry, 42, 1008-1015.
32.Telfer, A., Barber, J., and Evans, M. (1988)
FEBS Lett., 232, 209-213.
33.Klimov, V. V., Shafiev, M. A., and Allakhverdiev,
S. I. (1990) Photosynth. Res., 23, 59-65.
34.Khorobrykh, S. A., Khorobrykh, A. A., Klimov, V.
V., and Ivanov, B. N. (2002) Biochemistry (Moscow), 67,
683-688.
35.Yanykin, D. V., Khorobrykh, A. A., Khorobrykh, S.
A., and Klimov, V. V. (2010) Biochim. Biophys. Acta,
1797, 516-523.
36.Yanykin, D. V., Khorobrykh, A. A., Khorobrykh, S.
A., Pshybytko, N. L., and Klimov, V. V. (2013) Photosynth. Res.,
117, 367-374.
37.Khorobrykh, S. A., Khorobrykh, A. A., Yanykin, D.
V., Ivanov, B. N., Klimov, V. V., and Mano, J. (2011)
Biochemistry, 50, 10658-10665.
38.Joliot, P., Barbieri, G., and Chabaud, R. (1969)
Photochem. Photobiol., 10, 309-329.
39.Joliot, P., Joliot, A., Bouges, B., and Barbieri,
G. (1971) Photochem. Photobiol., 14, 287-305.
40.Cheniae, G. M., and Martin, I. F. (1971)
Biochim. Biophys. Acta, 253, 167-181.
41.Tamura, N., and Cheniae, G. (1987) Biochim.
Biophys. Acta, 890, 179-194.
42.Ananyev, G. M., and Klimov, V. V. (1988) Dokl.
AN SSSR, 298, 1007-1011.
43.Ananyev, G. M., and Dismukes, G. C. (1996)
Biochemistry, 35, 4102-4109.
44.Hwang, H. J., McLain, A., Debus, R. J., and
Burnap, R. L. (2007) Biochemistry, 46, 13648-13657.
45.Klimov, V. V., Allakhverdiev, S. I., Shuvalov, V.
A., and Krasnovsky, A. A. (1982) FEBS Lett., 148,
307-312.
46.Hawkins, C. L., and Davies, M. J. (2001)
Biochim. Biophys. Acta, 1504, 196-219.
47.Denisov, E. T., and Afanas’ev, I. B. (2005)
Taylor & Francis Group CRC, Press is an imprint of Taylor &
Francis Group.
48.Dobos, D. (1975) Electrochemical Data: a
Handbook for Electrochemists in Industry and Universities,
Elsevier, NY, p. 339.
49.Baranov, S. V., Tyryshkin, A. M., Katz, D.,
Dismukes, G. C., Ananyev, G. M., and Klimov, V. V. (2004)
Biochemistry, 43, 2070-2079.
50.Soh, N., Ariyoshi, T., Fukaminato, T., Nakano,
K., Irie, M., and Imato, T. (2006) Bioorg. Med. Chem. Lett.,
16, 2943-2946.
51.Yamauchi, Y., and Sugimoto, Y. (2010)
Planta, 231, 1077-1088.