The Role of Cytochrome b5 Structural Domains in Interaction with Cytochromes P450
G. V. Sergeev*, A. A. Gilep, and S. A. Usanov
Institute of Bioorganic Chemistry, Academy of Sciences of Belarus, ul. Akademika Kuprevicha 5/2, 220141 Minsk, Belarus; fax: 375 (172) 63-7274; E-mail: gvserg@iboch.bas-net.by* To whom correspondence should be addressed.
Received November 8, 2013; Revision received December 27, 2013
To understand the role of the structural elements of cytochrome b5 in its interaction with cytochrome P450 and the catalysis performed by this heme protein, we carried out comparative structural and functional analysis of the two major mammalian forms of membrane-bound cytochrome b5 – microsomal and mitochondrial, designed chimeric forms of the heme proteins in which the hydrophilic domain of one heme protein is replaced by the hydrophilic domain of another one, and investigated the effect of the highly purified native and chimeric heme proteins on the enzymatic activity of recombinant cytochromes P4503A4 and P45017A1 (CYP3A4 and CYP17A1). We show that the presence of a hydrophobic domain in the structure of cytochrome b5 is necessary for its effective interaction with its redox partners, while the nature of the hydrophobic domain has no significant effect on the ability of cytochrome b5 to stimulate the activity of cytochrome P450-catalyzed reactions. Thus, the functional properties of cytochrome b5 are mainly determined by the structure of the heme-binding domain.
KEY WORDS: microsomal cytochrome b5, outer mitochondrial membrane cytochrome b5, cytochrome P450, CYP3A4, CYP17A1DOI: 10.1134/S0006297914050046
Two membrane-bound forms of cytochromes b5 encoded by different genes have been found in mammalian cells, and they differ both in cellular localization and functional role. An interesting feature of the microsomal (Mc) isoform of cytochrome b5 is that by an alternative splicing two types of cytochrome b5 are synthesized – “soluble” (98 amino acids) and the membrane-bound (134 amino acids) cytochrome b5 [1].
The outer mitochondrial membrane cytochrome b5 (Om b5) consists of 146 amino acid residues [2]. Comparison of full-length sequences of rat isoforms of Mc b5 and Om b5 indicates that these heme proteins, despite their similarity, have only 46% sequence identity.
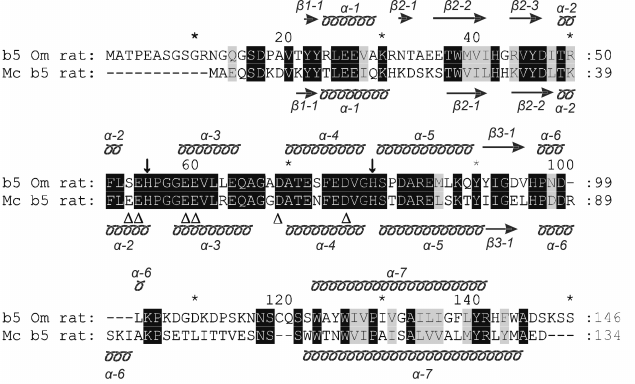
Both b5 isoforms include a hydrophilic N-terminal heme-containing domain and a hydrophobic C-terminal domain responsible for the interaction with the membrane. The N-terminal domain comprising about 100 amino acids is responsible for binding of ferriprotoporphyrin IX. This part of the protein turns to the cytosol and participates in the transport of electrons in redox reactions. The first 20 amino acid residues of the hydrophobic C-terminal domain interact with the lipid bilayer, anchoring the protein in the membrane. It is believed that the part of the C-terminal domain of Mc b5 containing approximately seven terminal amino acid residues is located on the surface of the endoplasmic reticulum. The corresponding C-terminal part of Om b5 containing about 10 residues is probably directed toward the intramembrane space of mitochondria. Moreover, it has been shown that the C-terminal 10 amino acid residues of b5 contain the necessary information about membrane localization of the isoforms [2]: Om b5 in the C-terminal sequence contains positively charged amino acid residues arginine 137 and lysine 144, which are necessary to import the protein to the outer mitochondrial membrane (Fig. 1) [3].
Fig. 1. Alignment of amino acid sequences of rat Mc b5 (P00173, UniProtKB/Swiss-Prot database) and rat Om b5 (P04166, UniProtKB/Swiss-Prot database). The schematic arrangement of secondary structures of the proteins shown above (for Om b5, PDB ID: 4HIL) and bottom (for Mc b5, PDB ID: 1AW3) relative to the compared amino acid sequences. Transmembrane regions of the proteins are hypothetically presented by α-7 helices; ↓, heme-bound histidines residues; Δ, negatively charged amino acid residues of Mc b5 involved in the interaction with P450 during monooxygenase reactions.
The EPR and optical spectral properties of the hydrophilic domains of the rat Om b5 and rat Mc b5 differ only slightly. However, the rat Om b5 redox potential (–107 mV) is about 100 mV more negative than that of rat Mc b5 (0 ± 10 mV) [4]. Om b5 is also characterized by high stability with respect to thermal and chemical denaturation. This is most likely due to the presence of two hydrophobic clusters around the heme pocket in Om b5 [4].
Mc b5 is an important component of the electron transport chain of the endoplasmic reticulum membranes. It receives electrons mainly from NADH through a flavoprotein – NADH-cytochrome b5 reductase, although it may also receive electrons from NADPH-cytochrome P450 reductase [5].
Special attention is devoted to study of the structure and function of b5 domains because they not only determine the physicochemical properties of the heme protein, but they also affect the interaction with its redox partners. The full-length membrane-bound form of Mc b5 supplies electrons for the desaturation and elongation of fatty acids [6] and synthesis of some lipids, including plasmalogen [7] and cholesterol [8]. The “water-soluble” form of Mc b5 reduces hemoglobin in red blood cells [9] and myoglobin in myocytes [10].
Both the soluble and membrane-bound forms of Mc b5 can accept electrons from NADH-cytochrome b5 reductase. On the other hand, stearoyl-CoA desaturase interacts only with the membrane form of Mc b5 as an electron donor [11].
It was shown that Mc b5 through the carboxyl groups of the hydrophilic domain surrounding the heme pocket interacts with several redox partners such as cytochrome c, hemoglobin, myoglobin, NADH-cytochrome b5 reductase, CYP2B4, metmyoglobin, and others [12].
The primary function of the hydrophobic domain of cytochrome b5 is the interaction of the protein with the membrane, and the charge of the last ten amino acid residues, as mentioned earlier, determines the localization of the protein in the membrane of endoplasmic reticulum or the outer mitochondrial membrane [13-15].
The role of the hydrophobic domain and C-terminal amino acids of b5 in regulating the activity of cytochrome P450 (P450) is not fully elucidated. It has been shown that the hydrophilic form of Mc b5 cannot be reduced by NADPH-cytochrome P450 reductase [16]. Therefore, the presence of full-length cytochrome b5 in the study of the activity of cytochrome P450 in vitro is required. From this perspective, the approach is justified to create a chimeric cytochrome b5 in which the hydrophilic domain of Mc b5 is replaced by a similar part of the Om b5 domain and vice versa. At the same time, there remains a full-size structure of cytochrome b5 and an opportunity to assess the contribution of the b5 domain in the regulation of the activity of cytochrome P450.
In the present study we constructed chimeric forms of Mc and Om cytochromes b5 and analyzed the catalytic activity of recombinant cytochrome CYP3A4 (a major enzyme in the metabolism of lipophilic xenobiotics) and CYP17A1 (a key enzyme in the biosynthesis of androgens, estrogens, and glucocorticoids) in the presence of several forms of cytochrome b5: full length rat Mc b5 and rat Om b5, full length human Om cytochrome b5 (human Om b5), chimeric constructs in which the hydrophilic domain of one isoform is replaced by a hydrophilic domain of the other isoform, and two mutant forms of rat Om b5 (deletion in the N-terminal region).
MATERIALS AND METHODS
Chemicals. We used the magnesium chloride, ampicillin, δ-aminolevulinic acid, isopropyl-β-D-thiogalactopyranoside, LB-broth, TB-broth, Tris base, EDTA, FMN, 2′-AMP, 2,5-ADP-Sepharose, histidine, testosterone, 3-[(3-cholamidopropyl)dimethylammonium]-1-propanesulfonate (CHAPS), NADPH, sodium isocitrate, isocitrate dehydrogenase, methylene chloride, 17α-hydroxyprogesterone, and 17α-hydroxypregnenolone from Sigma (USA), phenylmethylsulfonyl fluoride, sodium chloride, glycerol, sodium cholate, sodium deoxycholate, imidazole, Triton X-100, dithiothreitol, and Tween 20 from Acros Organics (Germany), Emulgen 913 from Kao Atlas (Japan), glycine from AppliChem (Germany), Ni-NTA agarose from Qiagen (USA), and hydroxyapatite from Bio-Rad (USA).
Plasmids and bacterial strains. For cloning polymerase chain reaction (PCR) products, reagent kit pCRTMII (Invitrogen, USA) or pGEM-T-vector (Promega, USA) was used. Expression vectors pCWori+ and pT7 were kindly provided by Dr. R. W. Estabrook (UT Southwestern Medical Center, USA). For expression of cytochromes P450 and b5 the following plasmids were used: pCWori+CYP17Horse [17], pCWori+CYP17Human [17], pCWori+CYP17gpig [17], pCWori+P4503A4 [12], pCWori+Mc b5 rat [18], pCWori+b5 Om rat [19], pT7 b5 om2 [19], pT7 b5 om3 [19], pT7 b5 Om human [20] and pOR262P450red [21].
Construction of expression plasmids. To create chimeric constructs, we used plasmid pCWori containing full-length cDNA rat Mc b5 and rat Om b5. Each of the chimeras consists of a hydrophilic N-terminal portion of one of the proteins and the C-terminal hydrophobic region of the other one. For this purpose, in the sequence of both cytochromes b5, an XhoI restriction site was introduced by site-directed mutagenesis. For introducing XhoI sites in cDNA heme protein sequences, the following oligonucleotides were used:
McB5_XhoI_D: CTGTCGAGTCTAACTCGAGTTGGTGGACC;
McB5_XhoI_R: GGTCCACCAACTCGAGTTAGACTCGACAG;
B5OM_XhoIPR_F: CAATTCATGCCAAAGGAGCTGGGCATATTGG;
B5OM_XhoIPR_R: CCAATATGCCCAGCTCCTTTGGCATGAATTG;
B5OM_XhoI_D: CAATTCATGCAACTCGAGCTGGGCATATTGG;
B5OM_XhoI_R: CCAATATGCCCAGCTCGAGTTGCATGAATTG.
Nucleotides introduced by site-directed mutagenesis are underlined. Plasmids pCWori bearing in their structure sequences of microsomal or mitochondrial cytochrome b5 after the introduction of an XhoI restriction enzyme recognition site were processed using restriction endonucleases XhoI and PstI. The treatment with these restriction enzymes produced two fragments: ~1500 bp (restriction site XhoI at the 5′-end, the restriction site PstI at the 3′-end; a given fragment contains the sequence encoding the C-terminal domain of cytochrome b5) and a ~4000 bp (restriction site PstI at the 5′-end, restriction site XhoI at the 3′-end; a given fragment contains the sequence encoding the N-terminal domain of cytochrome b5). The following procedures were performed with resulting DNA samples: 1) ligation of the fragment containing the C-terminal region of Mc b5 with a fragment containing the N-terminal Om b5 domain, and 2) ligation of the fragment containing the C-terminal Om b5 region with a fragment containing the N-terminal Mc b5 region.
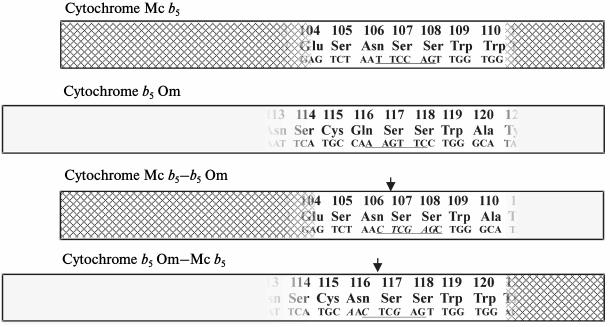
In contrast to microsomal cytochrome b5 whose amino acid sequence was not changed by these procedures, the mitochondrial cytochrome b5 was subject to amino acid replacement 116Gln→116Asn (CAA→aac) (Fig. 2).
Fig. 2. Schematic representation of the full-length sequences of rat cytochromes Mc b5 and Om b5 and constructed on their basis chimeric proteins Mc b5–Om b5 and Om b5–Mc b5. The input region of the site for the restriction enzyme XhoI is underlined. Italics indicate the nucleotide substitutions introduced during site-directed mutagenesis.
The resulting chimeric constructs were analyzed by sequencing and then cloned from pCWori plasmid into pT7 vector using the restriction sites NdeI and SalI. The derived vector constructs pT7 Om b5–Mc b5 and pT7 Mc b5–Om b5 were used to transform E. coli BL21(DE3) cells, and protein expression was analyzed. The level of synthesis of the chimeric protein was from 2 to 8 µmol per liter of culture medium. Strains with the highest expression level were selected for preparative isolation of chimeric cytochromes b5.
Sequencing reactions were performed using a BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA). The sequencing products were analyzed using an Applied Biosystems 3130 Genetic Analyzer.
Expression and isolation of recombinant proteins. Expression and purification of cytochromes b5 used in this work were carried out as described previously [19]. To estimate the expression level of the heme protein, the cell solution was dispensed into two cuvettes with 1 ml each (l = 1 cm), one of which was reduced with a few crystals of sodium dithionite (Sigma, USA), and the other one was oxidized by adding 20 μl of 33% hydrogen peroxide solution, after which the difference spectrum of the A424-413 (ε424-413 = 185 mM–1·cm–1) was recorded.
Expression and isolation of CYP17A1 and CYP3A4 was performed according to [12], and NADPH-cytochrome P450 reductase according to [21].
Titration of CYP3A4 with cytochrome b5. Titration was performed in 50 mM K-phosphate buffer, pH 7.4, containing 0.1% Tween 20 and 20% glycerol. The CYP3A4 concentration was 1.5 µM and the b5 concentration varied depending on the type of protein. Titrations were performed at 22°C in tandem cuvettes. After each addition of b5, the mixture was incubated for 5 min, after which the spectrum was recorded in the range 350-450 nm. The experimental results were plotted as the magnitude of the spectral response versus the concentration of added b5.
The dissociation constant (Kd) was calculated by using the SigmaPlot program:
![]()
where B0 is b5 concentration, P0 is CYP3A4 concentration, dA is the observed value of the absorption, dAmax is maximum change in absorption, and Kd is the dissociation constant for the b5–CYP3A4 complex.
Reconstruction of 17,20-lyase activity of the recombinant CYP17A1. In this work we analyzed the activity of three types of steroid 17α-hydroxylase/17,20-lyases: CYP17A1 of horse (Δ4,5-type, substrate – 17-OH-progesterone), CYP17A1 of guinea pig (Δ4-type, substrate – 17-OH-progesterone), and human CYP17A1 (Δ5-type, substrate – 17-OH-pregnenolone). The reaction was conducted under the following conditions: 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 50 μM substrate, 0.5 μM CYP17A1, and 1 μM NADPH-cytochrome P450 reductase. Concentration of b5 varied in different experiments with ratios of CYP17A1: 0.1 : 1; 0.25 : 1; 0.5 : 1; 1 : 1; 2 : 1 (0.05, 0.125, 0.25, 0.5, and 1 μM, respectively). The enzyme mixture of CYP17A1, NADPH-cytochrome P450 reductase, and b5 was incubated for 5 min at 37ºC and then mixed with buffer. The reaction was started by adding the regeneration mixture containing 0.5 mM NADPH, 8 mM sodium isocitrate, and 0.1 U/ml of isocitrate dehydrogenase. To stop the reaction an aliquot of the reaction mixture (0.5 ml) was mixed with 5 ml of methylene chloride. In the case of Δ5-steroids as substrates, an aliquot of the reaction mixture was placed in boiling water for 15 sec, and then the mixture was cooled to 25ºC and 0.1 units of cholesterol oxidase from Cellulomonas spp. was added. The mixture was incubated at 37ºC for 30 min to convert the Δ5- to Δ4-steroids. Then the reaction mixture was mixed with 5 ml of methylene chloride. The fractions were separated by centrifugation at 3000 rpm for 2 min. The aqueous fraction was removed with an aspirator, and the organic layer was evaporated under a stream of argon. The precipitate was dissolved in 100 μl of 100% methanol and analyzed by HPLC on a HP 1090 Liquid Chromatograph (Hewlett-Packard, USA) instrument (62% methanol, Column Agilent Eclipse Plus C18 5 μm, 4.6 × 150 mm).
Reconstruction of testosterone 6β-hydroxylase activity of recombinant CYP3A4. The reaction was conducted under the following conditions: 50 mM Tris-HCl (pH 7.4), 30 mM MgCl2, 150 μM testosterone, 0.25 μM cytochrome P450 3A4, 0.5 μM NADPH-cytochrome P450 reductase, 40 μg/ml phospholipids, and 125 μg/ml CHAPS. The concentration of cytochrome b5 varied in different experiments and amounted to CYP3A4 ratio: 0.5 : 1; 1 : 1; 2 : 1 (0.125, 0.25, and 0.5 μM, respectively). The products were analyzed as described for recombinant CYP17A1.
Analytical methods. The purity of protein samples of cytochrome b5 were analyzed by gel electrophoresis in a 15% polyacrylamide gel under denaturing conditions [22] on a Mini Protean II instrument (Bio-Rad, USA). The concentration of the purified b5 was determined from the absolute absorption spectrum of the oxidized form of the heme protein (ε413 = 117 mM–1·cm–1) [23].
RESULTS
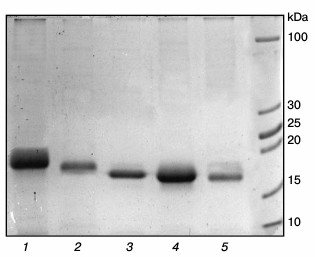
Expression of chimeric cytochromes b5. Purified preparations of chimeric cytochromes b5 are characterized by absorption spectra typical for cytochrome b5 (absorption maxima at 413 nm for the oxidized form and at 424, 527, and 557 nm for the reduced form). Polyacrylamide gel electrophoresis under denaturing conditions showed high purity of the chimeric proteins with molecular weights corresponding to theoretically calculated 17.2 kDa for the chimeric cytochrome Om b5–Mc b5 and 16.6 kDa for the chimeric Mc b5–Om b5 cytochrome (Fig. 3).
Fig. 3. Electrophoresis of preparations of different cytochromes b5 in 15% polyacrylamide gel under denaturing conditions. Lanes: 1) human Om b5; 2) rat Om b5; 3) Om b5–Mc b5; 4) Mc b5–Om b5; 5) rat Mc b5.
Titration of CYP3A4 by various forms of cytochrome b5. Human CYP3A4 can metabolize a wide range of chemical compounds. The CYP3A4 content reaches up to 60% of all cytochromes P450 present in liver cells. CYP3A4 has broad substrate specificity but exhibits a unique stereospecificity in hydroxylation reactions of most substrates. For example, the enzyme catalyzes the 2β-, 6β-, and 15β-hydroxylation of testosterone, 6β- and 16α-hydroxylation of progesterone, and 1- and 4-hydroxylation of midazolam [24].
In the reconstituted system containing NADPH-cytochrome P450 reductase and phospholipids, full-length Mc b5 stimulates reaction of testosterone 6β-hydroxylation catalyzed by CYP3A4, while the hydrophilic form of Mc b5 has no such an effect. At the same time, it was shown that the principal for realization of the stimulating effect of Mc b5 on P450-dependent reactions is the presence of a hydrophobic domain and a heme prosthetic group [12].
The interaction of cytochrome b5 with CYP3A4 results in transition of heme iron of the latter from low- to high-spin state accompanied by change in the absorption spectrum of CYP3A4 – decrease in absorbance at 417 nm and increase at 393 nm. On the basis of the spectral changes, it is possible to calculate the dissociation constant of the CYP3A4–b5 complex (Fig. 4 and Table 1).
Fig. 4. Spectrophotometric titration of CYP3A4 by various forms of cytochrome b5: 1) rat Mc b5; 2) rat Om2 b5; 3) Mc b5–Om b5; 4) human Om b5; 5) Om b5–Mc b5; 6) rat Om b5.
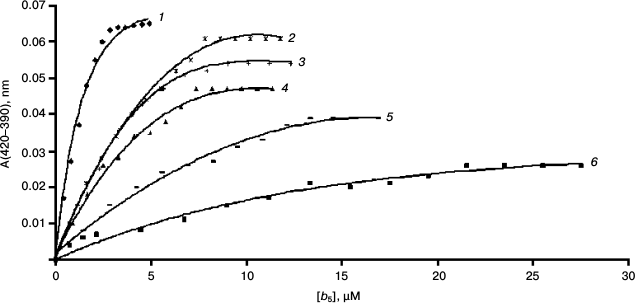
Table 1. Dissociation constants
(Kd) for complex formation of CYP3A4 with various
forms of cytochrome b5
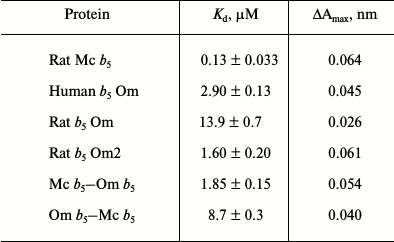
The interaction of Mc b5 with CYP3A4 demonstrates the smallest value of the dissociation constant. Rat Om b5 and human Om b5 less efficiently (100- and 20-fold, respectively) interact with CYP3A4 as compare to rat Mc b5. Truncated rat cytochrome b5 (rat Om2 b5) with deleted first 11 amino acid residues 9-fold more effectively interacts with CYP3A4 than the full length rat Om b5. Differences in the length of the hydrophilic fragment of two isoforms of b5 may be the reasons for various catalytic properties of the proteins. Removal of the N-terminal amino acid residues may reduce the rigidity of the structure of the Om b5 hydrophilic domain, which in turn promotes interaction with the CYP3A4 protein.
The difference in the dissociation constants during titration of CYP3A4 by chimeric cytochromes b5 is up to 5-fold. The chimera Mc b5–Om b5 having hydrophilic domain of microsomal b5 changes the spin state of CYP3A4 most effectively. The Kd of chimera Om b5–Mc b5 is close to the value constant found for the full-size Om b5. Thus, we conclude that the main role in the effective interaction of CYP3A4 with b5 belongs to the hydrophilic domain of the latter, although the presence of a hydrophobic moiety is required for the effective interaction of the heme proteins.
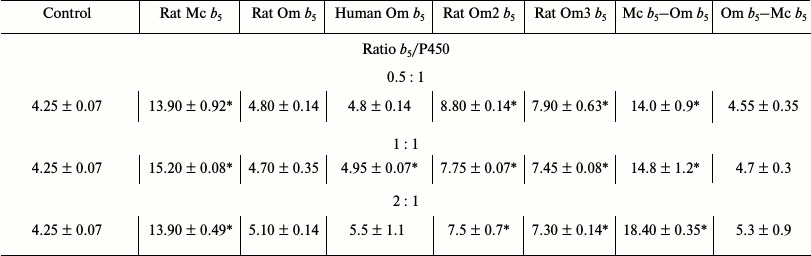
Reconstruction of testosterone 6β-hydroxylase activity of recombinant CYP3A4. Rat Mc b5 stimulates the testosterone oxidation reaction catalyzed by CYP3A4 by 3.0-3.5-fold in all studied ratios (Table 2). At the same time, Om b5 (either from rat or human) stimulates the activity of CYP3A4 only in 1.1-1.3-fold. The ability of rat Om2 b5 and rat Om3 b5 (first 13 amino acid residues deleted) mutants to stimulate the activity of CYP3A4 is increased 2-fold as compared to the full-length heme protein. These data are consistent with the results of titration – changes in the integrity of the hydrophilic domain of Om b5 facilitate its greater plasticity in the interaction with redox partners.
Table 2. Catalytic activity
(min–1) for 6β-hydroxylation of testosterone by
CYP3A4 in the presence of various b5 forms
* Activity values significantly different from control (significance
level of p = 0.05).
The chimera Mc b5–Om b5 increases testosterone 6β-hydroxylase activity of recombinant CYP3A4 by 3-4-fold. Despite the fact that the protein sequence includes a hydrophobic domain of Om b5, the activity level is comparable to the degree of activation by full-length Mc b5. The second chimeric protein, Om b5–Mc b5, stimulates the activity of CYP3A4 similarly to human or rat full-size Om b5 by 1.1-1.2-fold.
Analysis of CYP3A4 activity in the presence of various forms of b5 indicates the existence of correlation – the level of the stimulating effect is determined by the nature of the hydrophilic domain in the structure of the chimera. Based on these data, it is possible with some degree of confidence to say that the origin of the hydrophobic domain is not essential, while it is important that a hydrophobic moiety should present in the structure of the chimera (previously published data support this conclusion and indicate the absence of interaction of truncated microsomal b5 with NADPH-cytochrome P450 reductase [16]).
Reconstruction of 17,20-lyase activity of recombinant CYP17A1 Equus caballus. Steroid 17α-hydroxylase-C17,20-lyase (CYP17) (EC 1.14.99.0) is a key regulatory enzyme of the biosynthetic pathway of androgens. CYP17A1 catalyzes both 17α-hydroxylation and the C17-C20 side-chain cleavage of C21 steroids [25, 26]. Both Mc b5 and Om b5 can stimulate 17,20-lyase activity of CYP17A1 [27].
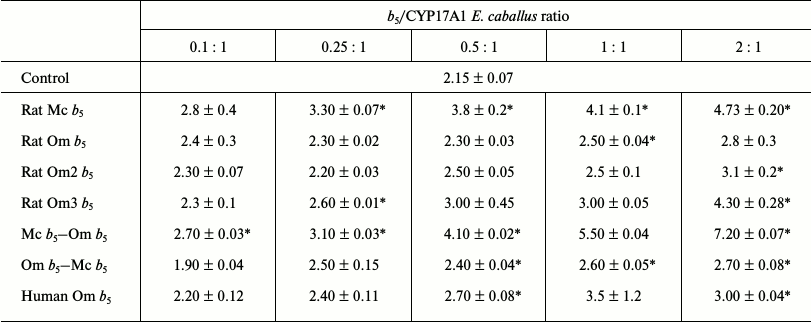
Rat Mc b5 stimulates 17,20-lyase activity (17OH-P4 as substrate) of CYP17A1 E. caballus in all investigated ratios (Table 3). The degree of stimulation increases with increasing CYP17A1/b5 ratio and reaches its maximum at ratio 1 : 2 (in 1.7-2.2-fold), after which further increase in b5 content reduces the stimulatory effect (data not shown).
Table 3. Catalytic activity
(min–1) for 17,20-lyase reactions using 17-OH
progesterone as substrate catalyzed by CYP17A1 Equus caballus in
the presence of various forms of cytochrome b5
* Activity values significantly different from control (significance
level of p = 0.05).
Rat Om b5 slightly stimulated 17,20-lyase activity (17OH-P4) of CYP17A1 E. caballus at all investigated ratios. The maximum stimulating effect was observed at high ratios of CYP17A1/b5 of 1 : 1 and 1 : 2 (by 1.3-fold).
Chimeric cytochrome Om b5–Mc b5 slightly stimulates 17,20-lyase activity (17OH-P4) of CYP17A1 E. caballus at all investigated ratios. The maximum effect was achieved at ratios of CYP17A1/b5 of 1 : 1 and 1 : 2 (by 1.3-fold).
Cytochrome Mc b5–Om b5 at all investigated ratios stimulated 17,20-lyase activity (17OH-P4) of CYP17A1 E. caballus. The stimulatory effect increased with increasing b5/CYP17A1 ratio and reached 3.25-fold.
Rat Om2 b5 at all investigated ratios slightly stimulated 17,20-lyase activity (17OH-P4) of CYP17A1 E. caballus. The maximum difference in the reaction rate relative to the control was observed at high b5/CYP17A1 ratios of 1 : 1 and 2 : 1 and reached 1.1-1.4-fold. Statistically significant stimulatory effect was observed only at 2 : 1 ratio (1.4-fold). A similar pattern was observed for rat cytochrome Om3 b5, but the stimulating effect increased 2-fold.
Human Om b5 stimulated 17,20-lyase activity (17OH-P4) of CYP17A1 E. caballus at all investigated ratios. The stimulation increased with increasing ratio and reached 1.4-1.5-fold.
When analyzing the effects of all studied cytochromes b5 on the activity of CYP17A1 E. caballus, the following trends are observed: Mc b5 stimulates the 17,20-lyase activity of CYP17A1. Similar stimulating profile was observed for the chimeric protein Mc b5–Om b5. Rat Om b5 was at least 1.5-fold less active in the stimulation of CYP17A1 E. caballus than rat Mc b5, and the stimulatory effects were observed at the highest ratios. Truncated rat Om2 b5 and the Om b5–Mc b5 chimera exhibited similar to rat Om b5 ability to stimulate the activity of CYP17A1. The other truncated cytochrome b5 (rat Om3 b5) exhibited stimulatory effect comparable to the effect of rat Mc b5. Human Om b5 stimulated the hydroxylation reaction more actively than rat Om b5, but this effect was observed only at high ratios and did not reach the level of Mc b5 (maximum 70% of the stimulatory effect of rat Mc b5).
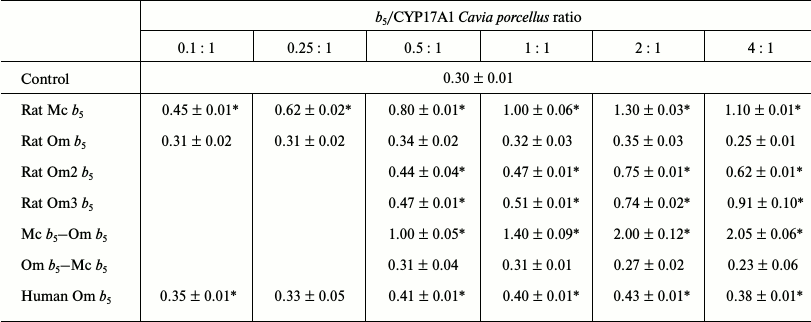
Reconstruction of 17,20-lyase activity of recombinant CYP17A1 Cavia porcellus. Rat Mc b5 activated 17,20-lyase activity of CYP17A1 C. porcellus to the ratio CYP17A1/b5 = 1 : 2, reaching 4.4-fold increase, and subsequent increase in b5 concentration in the reaction mixture decreased the stimulatory effect (Table 4).
Table 4. Catalytic activity
(min–1) for 17,20-lyase reactions using 17-OH
progesterone as substrate catalyzed by CYP17A1 Cavia porcellus
in the presence of various forms of cytochrome
b5
* Activity values significantly different from the control (significance
level of p = 0.05).
Rat Om b5 had no stimulating effect on the 17,20-lyase activity of CYP17A1 C. porcellus at all investigated ratios. Decrease in hydroxylase activity below the control level at ratio CYP17A1/b5 = 1 : 4 is due to the excess of b5.
Human Om b5 had little stimulatory effect on 17,20-lyase activity of CYP17A1 C. porcellus at all investigated ratios. The maximum increase in activity of 1.3-1.4-fold was observed at high ratios of b5 to CYP17A1. It is noteworthy that the degree of activation using human Om b5 is higher than when rat Om b5 is added to the reaction mixture.
Rat Om2 b5 stimulated 17,20-lyase activity of CYP17A1 C. porcellus at ratios CYP17A1/b5 from 1 : 0.5 to 1 : 4 by 1.5-2.5-fold. This stimulatory effect of rat Om2 b5, when compared with effect of full-length rat Om b5 action, may be due to: 1) the composition of the 11 amino acids deleted (charged amino acid residues are present) or the spatial location of a remote part that could prevent activity of rat Om b5, or 2) removal of amino acids, resulting in increased mobility of the hydrophilic b5 domain, which contributes to more effective interaction of the protein with the redox partners (by allosteric effect or by increasing the rate of electron transfer).
Rat Om3 b5 stimulated 17,20-lyase activity of CYP17A1 C. porcellus in 1.6-3.1-fold. The reasons for the stimulating effect may be the factors described above for rat Om2 b5.
The chimera Mc b5–Om b5 activated the 17,20-lyase activity of CYP17A1 C. porcellus at all investigated ratios. The maximum stimulating effect observed at CYP17A1/b5 = 1 : 4 was 6.8-fold.
The chimera Om b5–Mc b5 did not activate the 17,20-lyase activity of CYP17A1 C. porcellus at any investigated ratio.
Thus, there is a clear trend that for the activation of 17,20-lyase reaction of CYP17A1, cytochrome b5 must contain the hydrophilic domain of Mc b5 or a similar analog (rat Om2 b5 and rat Om3 b5). The origin of the hydrophobic domain does not change the effect of b5. Om b5 (human and rat) have similar activation profile, but human Om b5 more actively stimulates hydroxylase than rat Om b5.
Reconstruction of 17,20-lyase activity of recombinant CYP17A1 Homo sapiens. In studying the effect of b5 on 17,20-lyase reaction of CYP17A1 H. sapiens, together with available cytochromes b5, recombinant human Mc b5 was used to evaluate the factor of interaction of the two proteins (CYP17A1 and b5) from one organism.
Rat Mc b5 stimulated the 17,20-lyase reaction of transformation of 17-hydroxypregnenolone to dehydroepiandrosterone catalyzed by CYP17A1 H. sapiens at all investigated ratios. The maximum stimulating effect was 1.8-fold at ratio CYP17A1/b5 = 1 : 2, and a further increase in the ratio lead to a decrease in the stimulation (Table 5).
Table 5. Catalytic activity
(min–1) for 17,20-lyase reactions using 17-OH
pregnenolone as substrate catalyzed by CYP17A1 H. sapiens in the
presence of various forms of cytochrome b5
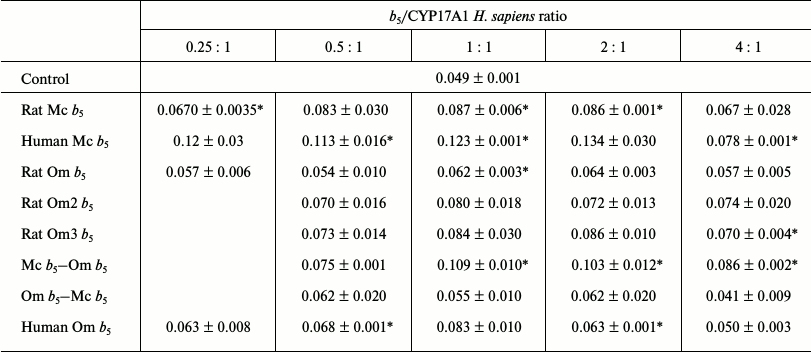
* Activity values significantly different from the control (significance
level of p = 0.05).
Human Mc b5 stimulated 17,20-lyase of CYP17A1 H. sapiens at all ratios. Even at low ratios the stimulatory effect is 2.3-fold and it reaches 2.7-fold with an increase in the ratio.
Rat Om b5 did not stimulate the 17,20-lyase reaction catalyzed by CYP17A1 H. sapiens at any ratio.
Human Om b5 slightly stimulated the 17,20-lyase reaction of CYP17A1 H. sapiens at all ratios. The maximum stimulating effect was observed at ratio 1 : 1 and was 1.7-fold (not statistically significant).
The chimera Mc b5–Om b5 stimulated the 17,20-lyase reaction of CYP17A1 H. sapiens at all ratios. The maximum stimulating effect was observed at ratio 1 : 1 and was 2.2-fold. The stimulation profile was comparable to the profile of the full-length rat Mc b5.
The chimera Om b5–Mc b5 did not stimulate the 17,20-lyase reaction of CYP17A1 H. sapiens at any ratio. The maximum increase in the reaction rate as compared to the control was 1.3-fold (not statistically significant). The stimulation profile was comparable to the profile of full-length rat Om b5.
Rat Om2 b5 stimulated the 17,20-lyase reaction of CYP17A1 H. sapiens. The maximum stimulating effect was observed at ratio 1 : 1 and was 1.6-fold. It should be noted that this truncated form more actively stimulated the hydroxylase activity than the full-length rat Om b5 (however, these differences were not statistically significant).
Rat Om3 b5 stimulated the 17,20-lyase reaction of CYP17A1 H. sapiens. The maximum stimulating effect was observed at ratio 1 : 2 and was 1.7-1.8-fold. The scatter of values in duplicates show statistically insignificant differences with the control (with the exception of ratio 4 : 1), but the profile can be seen that the mutant more actively stimulates CYP17A1 activity than a full length rat Om b5.
DISCUSSION
There are at least three hypotheses to explain the stimulatory effect of cytochrome b5 on cytochrome P450-catalyzed reactions. The first hypothesis presumes that cytochrome b5 is directly involved in electron transfer to P450 during the reaction. According to this hypothesis, reduced cytochrome b5 directly transmits the second electron needed to convert the complex P450Fe(II)O2 to form an “active oxygen”. This hypothesis is supported by experiments showing the transfer of electrons from reduced cytochrome b5 to cytochrome P450 and the ability of cytochrome b5 to reduce the effect of “uncoupling” in the reactions catalyzed by P450 [28].
The second hypothesis suggests complex formation between cytochrome P450 and b5. This complex receives electrons from NADPH-cytochrome P450 reductase. Reduced by two electrons, the complex uses these electrons for oxidation reactions to activate molecular oxygen [29].
A third hypothesis explains the stimulatory effect of cytochrome b5 by presuming that it induces conformational changes in P450 (thus changing the spin state), which modifies its redox potential and in turn promotes interaction of cytochrome P450 with substrate or an electron donor – NADPH-cytochrome P450 reductase. It was shown that CYP3A4-catalyzed oxidation of testosterone and nifedipine is indeed stimulated in this manner. Apo-cytochrome b5 stimulates the transfer of electrons from NADPH-cytochrome P450 reductase to CYP3A4 [30], suggesting that the stimulatory effect of cytochrome b5 is not associated with the presence of heme and is only due to conformational changes. However, later it was shown that the data are simply explained by the formation of the active holo-cytochrome b5 from the apo-cytochrome b5 and heme (from denatured forms of P450 – cytochrome P420) [1]. The mutant form of cytochrome b5, which is not able to bind heme, is not able to stimulate reactions catalyzed by P450. This confirms the important role of the cytochrome b5 prosthetic group in carrying out cytochrome b5 functions.
Study of structural features of Om b5 revealed the presence of two hydrophobic chains of amino acid residues surrounding the heme. These networks probably afford rigidity and increased resistance to chemical and thermal denaturation to hydrophilic domain of Om b5. However, the rigidity of the structure apparently prevents effective interaction of Om b5 with P450 and NADPH-cytochrome P450 reductase. The results of the present study confirm this hypothesis. Full-length Om b5 (of rat and human) is ten times less effective (compared to the Mc b5) in interaction with CYP3A4 and accordingly several times less effective in stimulating the activity of CYP3A4 and CYP17A1. Changes in the structural integrity of the hydrophilic rat Om b5 domain are observed in rat Om2 b5 and Om3 b5 mutants. This leads to more effective interaction of cytochrome b5 with P450. This is reflected in a decrease in Kd (interaction with CYP3A4) and stimulation of the oxidation of testosterone (CYP3A4) and the 17,20-lyase reaction (CYP17A1).
The presence of a hydrophobic domain in b5 structure is essential for effective interaction with its redox partners, but its origin has no significant effect on the ability of b5 to stimulate the activity of P450. This is confirmed by experiments with chimeric cytochrome Mc b5–Om b5. This heme protein stimulates the activity of CYP3A4 and CYP17A1 at level similar to full-length Mc b5. The chimera Om b5–Mc b5 stimulates the P450 reaction similarly to the full-size rat Om b5.
Despite the fact that human Om b5 contains similar to rat Om b5 networks of hydrophobic amino acid residues, these proteins differ in their ability to interact with CYP3A4. The Kd for human Om b5 is five times less than that of rat Om b5. However, the abilities to stimulate the activity of CYP3A4 and CYP17A1 for the studied natural forms of Om b5 are at a comparable level.
Reconstruction of the 17,20-lyase activity of CYP17A1 H. sapiens in the presence of Om b5 (human or rat) revealed how crucial the origin from one species of both cytochromes is: CYP17A1 and Om b5. The experimental results indicate that the replacement of rat Om b5 by human Om b5 has no significant effect on the activation profile of CYP17A1 H. sapiens. Thus, the inability to stimulate the activity of CYP17A1 is not associated with the species origin of Om b5.
The results of this study suggest the importance of the hydrophilic domain in the structure of cytochrome b5 for stimulation of the activity of P450. We first described the interaction of Om b5 with CYP3A4 and its inability to effectively activate CYP3A4 due to rigid structure of the hydrophilic domain. Despite the fact that the results do not answer the question relative to intrinsic mechanism during regulation of the activity of P450, they clearly demonstrate the importance of plasticity of the structure of cytochrome b5 for its effective interaction with its redox partners.
The created chimeric cytochromes b5 are good model systems to study the role of cytochrome b5 domains in redox reactions involving P450.
REFERENCES
1.Giordano, S. J., and Steggles, A. W. (1993)
Biochim. Biophys. Acta, 1172, 95-100.
2.Altuve, A., Silchenko, S., Lee, K. H., Kuczera, K.,
Terzyan, S., Zhang, X., Benson, D. R., and Rivera, M. (2001)
Biochemistry, 40, 9469-9483.
3.Kuroda, R., Ikenoue, T., Honsho, M., Tsujimoto, S.,
Mitoma, J. Y., and Ito, A. (1998) J. Biol. Chem., 273,
31097-31102.
4.Altuve, A., Wang, L., Benson, D. R., and Rivera, M.
(2004) Biochem. Biophys. Res. Commun., 314, 602-609.
5.Enoch, H. G., Fleming, P. J., and Strittmatter, P.
(1979) J. Biol. Chem., 254, 6483-6488.
6.Keyes, S. R., Alfano, J. A., Jansson, I., and
Cinti, D. L. (1979) J. Biol. Chem., 254, 7778-7784.
7.Paltuaf, F., Prough, R. A., Masters, B. S., and
Johnston, J. M. (1974) J. Biol. Chem., 249, 2661-2662.
8.Fukushima, H., Grinstead, G. F., and Gaylor, J. L.
(1981) J. Biol. Chem., 256, 4822-4826.
9.Gacon, G., Lostanlen, D., Labie, D., and Kaplan, J.
C. (1980) Proc. Natl. Acad. Sci. USA, 77, 1917-1921.
10.Livingston, D. J., McLachlan, S. J., La Mar, G.
N., and Brown, W. D. (1985) J. Biol. Chem., 260,
15699-15707.
11.Strittmatter, P., Spatz, L., Corcoran, D.,
Rogers, M. J., Setlow, B., and Redline, R. (1974) Proc. Natl. Acad.
Sci. USA, 71, 4565-4569.
12.Guryev, O. L., Gilep, A. A., Usanov, S. A., and
Estabrook, R. W. (2001) Biochemistry, 40, 5018-5031.
13.Honsho, M., Mitoma, J. Y., and Ito, A. (1998)
J. Biol. Chem., 273, 20860-20866.
14.Borgese, N., Gazzoni, I., Barberi, M., Colombo,
S., and Pedrazzini, E. (2001) Mol. Biol. Cell, 12,
2482-2496.
15.Ito, A. (1980) J. Biochem., 87, 73-80.
16.Enoch, H. G., and Strittmatter, P. (1979) J.
Biol. Chem., 254, 8976-8981.
17.Gilep, A. A., Guryev, O. L., Shet, M., Fisher,
C., Usanov, S. A., and Estabrook, R. (1999) Mol.
Steroidogenesis, 29, 85-86.
18.Chudaev, M. V., Gilep, A. A., and Usanov, S. A.
(2001) Biochemistry (Moscow), 66, 667-681.
19.Sergeev, G. V., Gilep, A. A., Estabrook, R. W.,
and Usanov, S. A. (2006) Biochemistry (Moscow), 71, 790-799.
20.Sergeev, G. V., Gilep, A. A., Usanov, S. A., and
Vasilevskaya, A. V. (2010) Trudy BGU. Mol. Biol., 5,
179-184.
21.Shen, A. L., Porter, T. D., Wilson, T. E., and
Kasper, C. B. (1989) J. Biol. Chem., 264, 7584-7589.
22.Laemmli, U. K. (1970) Nature, 227,
680-685.
23.Chudaev, M. V., and Usanov, S. A. (1997)
Biochemistry (Moscow), 62, 401-411.
24.Harlow, G. R., and Halpert, J. R. (1997) J.
Biol. Chem., 272, 5396-5402.
25.Chung, B. C., Picado-Leonard, J., Haniu, M.,
Bienkowski, M., Hall, P. F., Shively, J. E., and Miller, W. L. (1987)
Proc. Natl. Acad. Sci. USA, 84, 407-411.
26.Grigoryev, D. N., Kato, K., Njar, V. C., Long, B.
J., Ling, Y. Z., Wang, X., Mohler, J., and Brodie, A. M. (1999)
Anal. Biochem., 267, 319-330.
27.Soucy, P., and Luu-The, V. (2002) J. Steroid
Biochem. Mol. Biol., 80, 71-75.
28.Perret, A., and Pompon, D. (1998)
Biochemistry, 37, 11412-11424.
29.Schenkman, J. B., Voznesensky, A. I., and
Jansson, I. (1994) Arch. Biochem. Biophys., 314, 234-241.
30.Yamazaki, H., Johnson, W. W., Ueng, Y. F.,
Shimada, T., and Guengerich, F. P. (1996) J. Biol. Chem., 271,
27438-27444.