Effect of Conserved Intersubunit Amino Acid Substitutions on Hfq Protein Structure and Stability
V. N. Murina1*, B. S. Melnik1, V. V. Filimonov1, M. Ühlein2, M. S. Weiss2, U. Müller2, and A. D. Nikulin1
1Institute of Protein Research, Russian Academy of Sciences, ul. Institutskaya 4, 142290 Pushchino, Moscow Region, Russia; E-mail: thyrada@rambler.ru2Institute for Soft Matter and Functional Materials, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Strasse 15, Berlin 12489, Germany
* To whom correspondence should be addressed.
Received January 21, 2014
Hfq is a thermostable RNA-binding bacterial protein that forms a uniquely shaped homohexamer. Based on sequence and structural similarity, Hfq belongs to the like-Sm (LSm) protein family. In spite of a rather high degree of homology between archaeal and eukaryotic LSm proteins, their quaternary structure is different, usually consisting of five to eight monomers. In this work, the importance of conserved intersubunit hydrogen bonds for the Hfq spatial organization was tested. The structures and stabilities for the Gln8Ala, Asn28Ala, Asp40Ala, and Tyr55Ala Hfq mutants were determined. All these proteins have the same hexamer organization, but their stability is different. Elimination of a single intersubunit hydrogen bond due to Gln8Ala, Asp40Ala, and Tyr55Ala substitutions results in decreased stability of the Hfq hexamer. Tyr55Ala Hfq as well as the earlier studied His57Ala Hfq has reduced protein thermostability, which seems to correspond to an opening of the protein hydrophobic core.
KEY WORDS: Hfq, crystal structure, tertiary structure, protein thermostabilityDOI: 10.1134/S0006297914050113
Hfq is a central mediator of sRNA-based gene regulation in bacteria (for recent reviews see [1-4]) and structurally belongs to the Sm/like-Sm (LSm) protein family with specific ring-like quaternary structures [5]. LSm proteins are built of a five-strand β-barrel capped by an N-terminal α-helix and a variable C-terminal tail. The core part of the proteins contains two conserved sequence motifs called Sm1 (strands β1, β2, and β3) and Sm2 (strands β4 and β5) [6, 7]. The Sm1 motif exhibits homology among all bacterial, archaeal, and eukaryotic proteins [8-11], while the Sm2 motif shows some degree of divergence between bacterial Hfq and eukaryotic/archaeal LSm proteins [7]. In spite of the sequence variations, the overall structure of all LSm proteins is the same, and their ring packing is defined by the contacts between the β4 and β5 strands of two neighboring protein monomers. As a result, there appears within the hexamer a continuous β-sheet with common hydrophobic core passing through the whole ring. Bacterial proteins Hfq form homohexamers exclusively [12], while archaeal Sm-like and eukaryotic Sm proteins exist as homo- and heteroheptamers, respectively [5]. Moreover, the Sm-like protein from cyanophages has C5 symmetry [13], and the LSm3 protein from Saccharomyces cerevisiae has C8 symmetry [14]. The reason for the difference in the packing of the structurally similar LSm proteins is still unknown, and all hypotheses suggested so far were later refuted [7, 14].
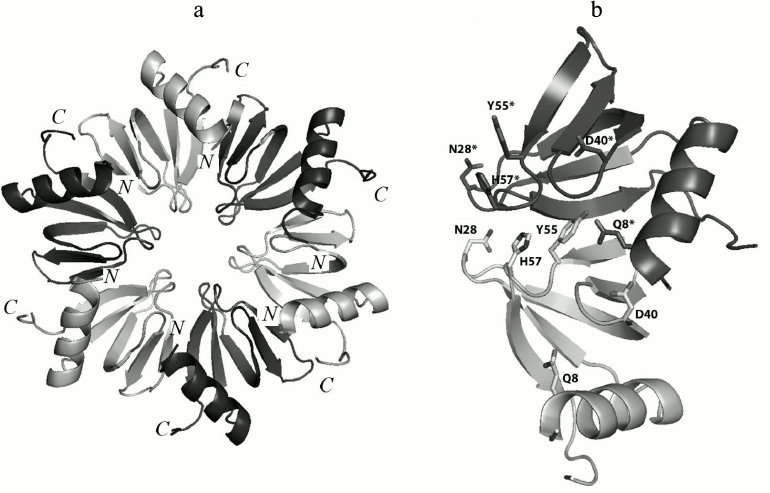
Previously, it has been suggested that inter-monomer hydrogen bonds created by side-chain atoms of conserved amino acid residues, which additionally stabilized the Hfq tertiary structure, play a very important role [15]. There are five conserved amino acids (Gln8, Asn28, Asp40, Tyr55, and His57) organizing inter-monomer hydrogen bonds in Hfq (Fig. 1). Two of them, His57 and Tyr55, belong to the highly-conserved YKHI consensus of the Sm2 motif in bacterial Hfq. It was demonstrated that His57 substitutions reduced the stability of the protein greatly but did not change its spatial structure [16]. In this work, the effects of the Gln8Ala, Asn28Ala, Asp40Ala, and Tyr55Ala substitutions on the structure and stability of Hfq from Pseudomonas aeruginosa (PaeHfq) were studied.
Fig. 1. a) Ribbon representation of the Hfq hexamer. The protein monomers are in different colors. b) Amino acid residues involved in hydrogen bond formation between two adjacent monomers in PaeHfq. The figures were produced by PyMOL.
MATERIALS AND METHODS
Site-directed mutagenesis, gene expression, and recombinant protein purification. QuikChange Site-Directed Mutagenesis Kit (Stratagene, USA) was used to prepare mutant forms of PaeHfq. PCR was performed with pET22b(+)/Hfq plasmid and oligonucleotides that contained the desired mutations. The resulted plasmids were verified by sequencing.
Gene expression and protein purification of all mutants (except Tyr55Ala PaeHfq) were performed as described earlier [15]. The Tyr55Ala substitution reduced the thermostability of the protein, and the cell lysate heating step was omitted. For this reason, the previously described purification scheme had to be changed. The cell lysate in 0.5 M ammonium sulfate, 0.5 M NaCl, 50 mM Tris-HCl (pH 8.0) was initially loaded on a butyl-Sepharose column and eluted with 50 mM NaCl, 50 mM Tris-HCl (pH 8.0). The final step of purification was performed using a DEAE-Sepharose anion exchange column eluted with a NaCl gradient from 50 mM to 1 M in 50 mM Tris-HCl (pH 8.0). The protein samples were characterized by 15% SDS-PAGE and by 15% PAGE under non-denaturing conditions (pH 4.5).
Circular dichroism (CD) measurement. CD measurements were performed on a Jasco J600 spectropolarimeter (Japan) equipped with a Julabo F25 computer-controlled thermostat. All spectra and melting experiments were performed using a cell with a 0.1 mm pathlength. The melting experiments were performed by monitoring the ellipticity changing at 220 nm in 0.3 M NaCl, 50 mM Tris-HCl (pH 8.0) containing 1 M guanidine hydrochloride (GuHCl).
Differential scanning calorimetry (DSC). The DSC experiments were carried out using a SCAL-1 microcalorimeter with a cell volume of 0.32 ml at heating rate of 1 K/min within the temperature range between 10 and 120°C, which was achieved by applying the extra pressure of 2.5 atm [17]. The samples were preliminarily equilibrated to 25 mM glycine (pH 2.0 and 3.0) or acetate (pH 4.0) buffers by dialysis. Protein concentrations in DSC experiments ranged from 1 to 2 mg/ml. The partial heat capacity and heat effects were calculated per mole of the monomer assuming 0.73 ml/mg for partial specific volume as described elsewhere [18].
Crystallization of Hfq mutant proteins. Thermal shift assay. Protein crystals were obtained by both hanging-drop and sitting-drop vapor-diffusion methods at 22°C. Thermal shift assay on an iCycler iQ Real Time PCR (Bio-Rad, USA) was used to optimize protein stabilization conditions and additives for crystallization [19, 20]. The in situ crystal screening facility at BL14.1 of BESSY (Berlin, Germany) was applied to test the results of crystallization trials [21]. Details on the protein crystallization conditions are summarized in Table 1.
Table 1. Crystallization conditions
Protein electrophoresis under semi-native conditions. Semi-native gel electrophoresis in 15% polyacrylamide gel containing 0.1% SDS was done as it was described earlier for Hfq from E. coli [22]. The 20-μg aliquots of each protein in the presence of 0.1% SDS were loaded onto 15% polyacrylamide gel passing the heating step. SDS was added to the samples just before loading, which prevented the dissociation of protein multimers prior to the electrophoresis.
Structure determination and refinement. The X-ray diffraction data from the crystals were collected at X8 Proteum (Bruker-AXS, The Netherlands) in the Institute of Protein Research, Russian Academy of Sciences (Pushchino, Russia) or at BESSY HZB beam line 14.1 (Berlin, Germany) [21]. The data were processed by Proteum (Bruker-AXS) software or by the XDS program [23], correspondingly, and scaled in XPREP (Bruker-AXS) or SCALA (CCP4 [24]). Analysis of the diffraction data revealed twinning in all datasets. The protein structures were solved by molecular replacement using PHASER [25] with wild-type PaeHfq as the initial model (PDB 1U1S [15]). In most cases a hexamer was used for the molecular replacement, the exception being Asp40Ala PaeHfq where a monomer was used as the initial model. The structures were refined using the PHENIX package [26]. In the first stage rigid-body refinement was applied, followed by simulated-annealing protocol and conventional residual refinement in combination with manual inspection in Coot [27, 28]. Water molecules were introduced into the models using the “water pick” function of Coot. The highest peaks in the resulting (mFo-DFc)-electron density map were assigned to ions. Detailed statistics of the crystallographic data and refinement are shown in Table 2.
Table 2. Data collection and refinement
statistics
* Statistics for the highest resolution shell are shown in
parentheses.
RESULTS AND DISCUSSION
Crystal structure of Gln8Ala, Asn28Ala, Asp40Ala, and Tyr55Ala PaeHfq. The introduced amino acid substitutions resulted in the modification of the local protein surface, thus influencing the potential protein contacts in the crystal cells. The Gln8Ala, Asn28Ala, and Tyr55Ala substitutions had less effect on the protein crystallization, and the corresponding mutant proteins were crystallized under conditions similar to that of the wild-type protein (Table 1). However Asn28Ala and Tyr55Ala PaeHfq produced small and twinned crystals that resulted in poor low-resolution crystallographic data. Asp40Ala PaeHfq did not form crystals in PEG/MMEPEG screens and was crystallized at a high concentration of ammonium sulfate only. In all cases the crystal packing of the PaeHfq mutant protein was different from that of the wild-type one. Nevertheless, the spatial structure and hexamer organization of the PaeHfq did not change in spite of the different number of protein molecules in the asymmetric unit (Table 1).
Amino acid Gln8 is located in the N-terminal α-helix and forms two inaccessible to solvent hydrogen bonds with the NZ atom of Lys56 of the same monomer and the hydroxyl group of Tyr55 from the adjacent monomer (Fig. 2a). The Gln8–Lys56 contact promotes connecting of the α-helix to the rest of the protein, while the Gln8–Tyr55 contact provides stabilization of the intermonomer interactions. The Gln8Ala substitution eliminated these hydrogen bonds, thereby loosening the protein structure (Fig. 2b). Nevertheless, the analysis of the Gln8Ala PaeHfq structure revealed the missing hydrogen bonds were compensated by water molecules, and the protein structure did not change.
Fig. 2. Detailed views of the protein sites at the selected residues. The (2mFo-DFc)-electron density map superimposed on the refined structures is contoured at the 1.0 σ level. Hydrogen bonds are shown as dotted lines. The asterisk indicated residues from the neighboring monomer. a, b) Region at the Gln8 and Tyr55 residues in the wild-type protein and in the Gln8Ala PaeHfq, correspondingly. c, d) Region at the Asp40 residue in the wild-type and in the Asp40Ala PaeHfq. e) Uridine found at the Hfq U-binding site in Asp40Ala PaeHfq. f) Region at the Asn28 residue in wild-type PaeHfq.
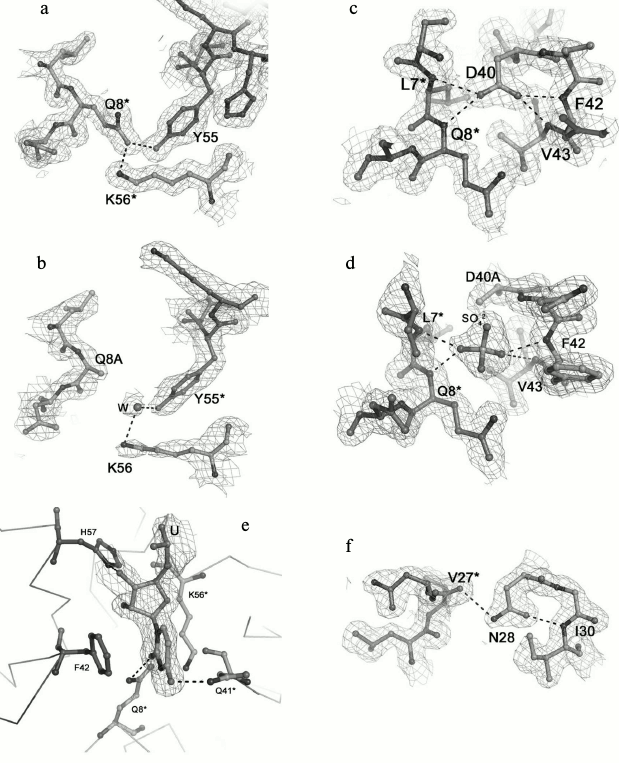
Residue Tyr55 along with the previously studied His57 [16] constitutes part of the highly conserved amino acid consensus in the bacterial Sm2 motif. As mentioned above, it forms an inaccessible to solvent hydrogen bond with residue Gln8 (Fig. 2a). Additionally, the side chain of the tyrosine shields the protein hydrophobic core from the solvent. The Tyr55Ala substitution influenced crystallization appreciably, and protein crystals were obtained only in the presence of Cu2+. Nevertheless, the substitution neither changed the overall structure of the protein nor the conformation of the loop containing Tyr55.
Amino acid Asp40 is located at the C-terminal end of strand β2 (Fig. 2c). This is very crucial for the protein structure since the asparagine side-chain oxygen atoms contacted with four different main-chain nitrogen atoms: Phe42 and Val43 of the same monomer and Leu7 and Asn8 of the adjacent monomer. The lack of the side chain atoms was compensated by appearing of a sulfate ion at this site (Fig. 2d). It was clearly visible in the experimental electron density map. The sulfate was positioned exactly in the place of the asparagine side-chain and formed hydrogen bonds with the residues specified above. Interestingly, Asp40Ala PaeHfq was crystallized with random RNA captured from the cells (Fig. 2e). It was described as a single uridine at the Tyr55-His57 amino acid residues of PaeHfq, since there was only one protein monomer in the asymmetric unit. Earlier it was shown that mutation Asp40Ala increases U-rich RNA-binding ability of Hfq protein [29]. It seems that this mutation caused a natural RNA fragment to fall into a trap.
Amino acid residue Asn28 is located in the loop connecting the β1 and β2 strands of the protein (Fig. 2f). Together with the adjacent Gly29, it is involved in organization of an energetically favorable β-turn between the strands owing to a hydrogen bond between the side-chain oxygen of Asn28 and the main-chain nitrogen of Gly29. On the other hand, the Asn28 side-chain nitrogen connects to the main-chain oxygen atom of Val27 belonging to a neighboring monomer and thus stabilizes the protein hexamer. Although the Asn28Ala substitution abolished both structurally important hydrogen bonds, the mutant PaeHfq was organized as the hexamer in the crystal.
Thus, we conclude that all performed disruptions of a single intermonomer hydrogen bond were incapable of changing the tertiary or quaternary structure of PaeHfq. None of the substitutions introduced any hindrance to form hexamer or to crystallize the protein, though it made the crystallization process more complicated.
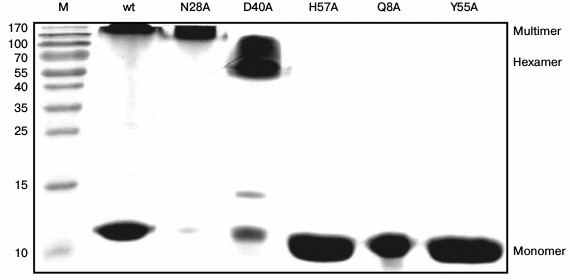
Stability of Gln8Ala, Asn28Ala, Asp40Ala, and Tyr55Ala PaeHfq hexamers. To examine the stability of the wild-type and mutant PaeHfq quaternary structure, PAGE experiments were carried out under semi-native conditions (Fig. 3). The results demonstrated the presence of wild-type PaeHfq multimers besides hexamers in solution, which was in good agreement with data obtained for the wild-type Hfq from E. coli (EcoHfq) [22]. The performed amino acid substitutions in PaeHfq led to disruption of the protein multimers except for the Asn28Ala mutant. The hydrogen bond between the nitrogen of the Asn28 side chain and main-chain carboxyl oxygen of Val27 of an adjacent protein monomer was accessible to solvent in the PaeHfq. Our results demonstrate that the elimination of this accessible to solvent hydrogen bond did not influence the stability of the PaeHfq hexamer, in contrast to elimination of inaccessible ones. Gln8Ala, His57Ala, and Tyr55Ala PaeHfq dissociated to monomers under the semi-native conditions, which is in good agreement with the earlier reported results for the Gln8Ala and Tyr55Ala EcoHfq mutants [22]. Since these substitutions resulted in elimination of a single intersubunit hydrogen bond without any visible changes in the protein structure, it can be assumed that this intervention was the reason for the decrease in the hexamer stability. In contrast, the mutant protein Asp40Ala PaeHfq predominantly existed as hexamers. The Asp40Ala PaeHfq crystal structure demonstrates that this substitution results in the creation of a binding site for a multivalent ion such as sulfate that is able to compensate for the lost hydrogen bonds. It seems that this compensation provides additional stabilization of the Asp40Ala PaeHfq hexamer in comparison with the wild-type protein.
Fig. 3. PAGE under semi-native conditions for the wild-type, Asn28Ala, Asp40Ala, His57Ala, Gln8Ala, and Tyr55Ala PaeHfq proteins. M is a protein molecular mass marker. Positions of monomer, hexamer, and multimeric PaeHfq are marked.
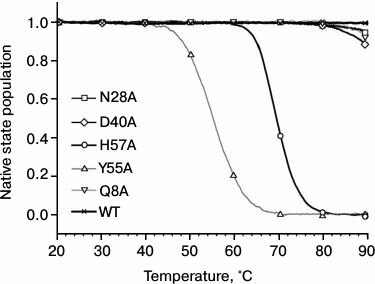
Influence of substitutions on PaeHfq secondary structure stability. To examine possible changes in structure stability, the temperature-induced unfolding of PaeHfq and its mutants was monitored by CD and scanning calorimetry. In accordance with X-ray data, the CD spectra of the mutants were practically coinciding with those of the native hexamer at room temperature. However, the melting profiles of the mutants were remarkably different (Fig. 4). Because of high structure stability, the heating experiments were carried out in the presence of 1 M GuHCl as previously described [16]. It was found that the Gln8Ala, Asn28Ala, and Asp40Ala substitutions change the CD melting curves only slightly, while the Tyr55Ala variant is even less stable than the His57Ala mutant.
Fig. 4. Normalized melting curves of PaeHfq mutant forms. Melting was performed using ellipticity at 220 nm at pH 8.0 in presence of 1 M GuHCl.
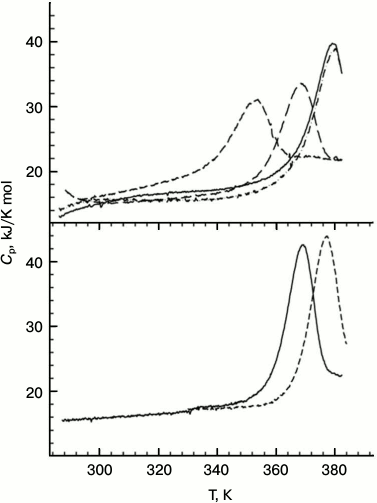
Measurements of the protein stability by microcalorimetry were carried out in the acidic pH range in an attempt to avoid aggregation of the unfolded forms that was observed at neutral pH. In this case, no denaturant was added since the DSC experiments can be run up to 120°C by applying extra pressure to the samples. All the Hfq mutants except for Tyr55Ala have sharp heat capacity peaks at very high temperatures (the transition midpoint for the wild-type protein is about 116°C; Fig. 5) and, in accordance with the CD data, were less stable than wild-type Hfq. Although the main heat capacity peak looked rather sharp and asymmetric, which is typical for a two-state unfolding of multimers [30], more detailed analysis showed that even in the case of the wild-type protein, not speaking of the mutants, the transitions are not fully cooperative. This deviation from a classical two-state transition is more clearly seen in the case of the Gln8Ala mutant, which is only slightly less stable than the wild-type protein, as the position of the main peak does not depend on protein concentration. Thus, a more realistic model for the temperature-induced unfolding of the Hfq hexamers would look like:
N6 ↔ 6I → 6U.
Fig. 5. Temperature dependences of wild-type PaeHfq (lower panel) and the Gln8Ala mutant (upper panel) in the acidic pH range. In the lower panel: pH 2.0 (solid line) and pH 3.0 (dashed line) both at 1.5 mg/ml. In the upper panel: pH 2.0 (short dash), pH 3.0 (long dash), pH 4.0 (dash-dot line, c = 0.7 mg/ml; solid line, c = 1.4 mg/ml).
The degree of cooperativity depends on the population of the intermediate state, i.e. on the relation between thermodynamic parameters of the first and second transitions. It seems that in the case of the wild-type protein the dissociation constant reflecting the strength of the intersubunit contacts is relatively small, while in the mutants the dissociation occurs more easily. The extreme case is represented by the Tyr55Ala mutant with intersubunit interactions weakened so much that its unfolding curve looks like a set of peaks corresponding to the melting of monomers, dimers, and other oligomers.
Unfortunately, the enthalpy change for the dissociation step should be rather small, resulting in a broad heat capacity peak added to the left shoulder of the main unfolding peak. For this reason and due to not very high precision of the instrument baseline, accurate analysis of the melting curves based on scheme above was not possible.
We conclude that the elimination of a single intersubunit inaccessible to solvent hydrogen bond decreased the stability of the PaeHfq hexamer. Most substitutions decrease the protein thermostability only slightly, first of all due to weakening of intersubunit contacts, as was originally planned. Although at room temperature the variants His57Ala and Tyr55Ala restore their hexamer structure as other mutants, their melting profiles are different and demonstrate large decrease in structure stability, most likely due to increased accessibility of the hydrophobic core of the protein.
We are thankful to S. V. Nikonov for critical discussion of the results.
This research was supported by the Russian Foundation for Basic Research (research projects 13-04-00783-a and 14-04-31215-mol_a), by the Molecular and Cell Biology Program of the Russian Academy of Sciences, and by the German-Russian Cooperation for Development and Use of Accelerator-Based Light Sources (05K10CBB).
REFERENCES
1.Wagner, E. G. H. (2013) RNA Biol.,
10, 619-626.
2.Wilusz, C. J., and Wilusz, J. (2013) RNA
Biol., 10, 592-601.
3.Regnier, P., and Hajnsdorf, E. (2013) RNA
Biol., 10, 602-609.
4.Vogel, J., and Luisi, B. F. (2011) Nat. Rev.
Microbiol., 9, 578-589.
5.Wilusz, C. J., and Wilusz, J. (2005) Nat.
Struct. Mol. Biol., 12, 1031-1036.
6.Valentin-Hansen, P., and Eriksen, M. (2004)
Mol. Microbiol., 51, 1525-1533.
7.Sauter, C. (2003) Nucleic Acids Res.,
31, 4091-4098.
8.Kambach, C., Walke, S., Young, R., Avis, J. M., de
la Fortelle, E., Raker, V., Luhrmann, R., Li, J., and Nagai, K.
(1999) Cell, 96, 375-387.
9.Nielsen, J. S., Boggild, A., Andersen, C. B. F.,
Nielsen, G., Boysen, A., Brodersen, D. E., and Valentin-Hansen, P.
(2007) RNA, 13, 2213-2223.
10.Murina, V. N., and Nikulin, A. D. (2011)
Biochemistry (Moscow), 76, 1434-1449.
11.Kilic, T., Sanglier, S., Van Dorsselaer, A., and
Suck, D. (2006) Protein Sci., 15, 2310-2317.
12.Brennan, R. G., and Link, T. M. (2007) Curr.
Opin. Microbiol., 10, 125-133.
13.Das, D., Kozbial, P., Axelrod, H. L., Miller, M.
D., McMullan, D., Krishna, S. S., Abdubek, P., Acosta, C., Astakhova,
T., Burra, P., Carlton, D., Chen, C., Chiu, H.-J., Clayton, T., Deller,
M. C., Duan, L., Elias, Y., Elsliger, M.-A. M., Ernst, D., Farr,
C., Feuerhelm, J., Grzechnik, A., Grzechnik, S. K., Hale, J., Han, G.
W., Jaroszewski, L., Jin, K. K., Johnson, H. A., Klock, H. E.,
Knuth, M. W., Kumar, A., Marciano, D., Morse, A. T., Murphy, K.
D., Nigoghossian, E., Nopakun, A., Okach, L., Oommachen, S., Paulsen,
J., Puckett, C., Reyes, R., Rife, C. L., Sefcovic, N., Sudek, S., Tien,
H., Trame, C., Trout, C. V., van den Bedem, H., Weekes, D., White,
A., Xu, Q., Hodgson, K. O., Wooley, J., Deacon, A. M., Godzik, A.,
Lesley, S. A., and Wilson, I. A. (2009) Proteins, 75,
296-307.
14.Naidoo, N., Harrop, S. J., Sobti, M.,
Haynes, P. A., Szymczyna, B. R., Williamson, J. R., Curmi, P. M. G.,
and Mabbutt, B. C. (2008) J. Mol. Biol., 377,
1357-1371.
15.Nikulin, A., Stolboushkina, E., Perederina, A.,
Vassilieva, I., Blaesi, U., Moll, I., Kachalova, G., Yokoyama, S.,
Vassylyev, D., Garber, M., and Nikonov, S. (2005) Acta
Crystallogr. D. Biol. Crystallogr., 61, 141-146.
16.Moskaleva, O., Melnik, B., Gabdulkhakov, A.,
Garber, M., Nikonov, S., Stolboushkina, E., and Nikulin, A. (2010)
Acta Crystallogr. F, 66, 760-764.
17.Senin, A. A., Potekhin, S. A., Tiktopulo, E. I.,
and Filomonov, V. V. (2000) J. Therm. Anal. Calorim.,
62, 153-160.
18.Privalov, P. L., Plotnikov, V. V., and Filimonov,
V. V. (1975) J. Chem. Thermodynam., 7, 41-47.
19.Phillips, K., and de la Pena, A. H. (2011)
Curr. Protoc. Mol. Biol., Chap. 10, Unit 10.28.
20.Reinhard, L., Mayerhofer, H., Geerlof, A.,
Mueller-Dieckmann, J., and Weiss, M. S. (2013) Acta Crystallogr.
Sect. F. Struct. Biol. Cryst. Commun., 69, 209-214.
21.Mueller, U., Darowski, N., Fuchs, M. R., Forster,
R., Hellmig, M., Paithankar, K. S., Puhringer, S.,
Steffien, M., Zocher, G., and Weiss, M. S. (2012) J.
Synchrotron Radiat., 19, 442-449.
22.Panja, S., and Woodson, S. A. (2012) J. Mol.
Biol., 417, 406-412.
23.Kabsch, W. (2010) Acta Crystallogr. D. Biol.
Crystallogr., 66, 125-132.
24.Winn, M. D., Ballard, C. C., Cowtan, K. D.,
Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E.
B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N.,
Pannu, N. S., Potterton, E. A, Powell, H. R., Read, R. J., Vagin, A.,
and Wilson, K. S. (2011) Acta Crystallogr. D. Biol.
Crystallogr., 67, 235-242.
25.McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P.
D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007) J. Appl.
Crystallogr., 40, 658-674.
26.Adams, P. D., Afonine, P. V., Bunkoczi, G.,
Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L.-W.,
Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W.,
Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S.,
Terwilliger, T. C., and Zwart, P. H. (2010) Acta Crystallogr. D.
Biol. Crystallogr., 66, 213-221.
27.Emsley, P., and Cowtan, K. (2004) Acta
Crystallogr. D. Biol. Crystallogr., 60, 2126-2132.
28.Emsley, P., Lohkamp, B., Scott, W. G., and
Cowtan, K. (2010) Acta Crystallogr. D. Biol. Crystallogr.,
66, 486-501.
29.Mikulecky, P. J., Kaw, M. K., Brescia, C. C.,
Takach, J. C., Sledjeski, D. D., and Feig, A. L. (2004) Nat. Struct.
Mol. Biol., 11, 1206-1214.
30.Boudker, O., Todd, M. J., and Freire, E. (1997)
J. Mol. Biol., 272, 770-779.