Selective Inhibitor of Histone Deacetylase 6 (Tubastatin A) Suppresses Proliferation of Hepatitis C Virus Replicon in Culture of Human Hepatocytes
M. V. Kozlov1*, A. A. Kleymenova1, K. A. Konduktorov1, A. Z. Malikova2, and S. N. Kochetkov1
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; fax: (495) 135-1405; E-mail: kozlovmv@hotmail.com2Kazan Federal University, ul. Kremlyovskaya 18, 420008 Kazan, Republic of Tatarstan, Russia; fax: (843) 292-4448; E-mail: rasssl44@gmail.com
* To whom correspondence should be addressed.
Received March 12, 2014; Revision received April 2, 2014
Acetylation of α-tubulin was studied in cultures of human hepatocytes under the influence of selective inhibitors of histone deacetylases HDAC6 and SIRT-2 – tubastatin A and 2-(3-phenethoxyphenylamino)benzamide, respectively. It was found that in hepatocyte cell line HepG2 acetylated α-tubulin is accumulated preferentially on inhibition of HDAC6 but not of SIRT-2. Under the same conditions, no acetylation of α-tubulin was observed in hepatocyte cell line Huh7. However, the inhibition of HDAC6 with tubastatin A led to hyperacetylation of α-tubulin and simultaneously to decrease in viral RNA concentration in hepatocyte cell line Huh7-luc/neo, which supports propagation of the full genome replicon of hepatitis C virus. The correlation between these two processes points to HDAC6 as a promising cellular target for therapy of hepatitis C.
KEY WORDS: human hepatocytes, acetylation of α-tubulin, HDAC6 and SIRT-2, hepatitis C virus repliconDOI: 10.1134/S0006297914070050
Abbreviations: HCV, hepatitis C virus; HDAC6, Zn2+-dependent histone deacetylase 6; MT, microtubules; SIRT-2, NAD+-dependent histone deacetylase; αTAT1, α-tubulin acetyltransferase of mammals.
Acetylation/deacetylation of α-tubulin is a mechanism of cellular
control of stability of tubulin microtubules (MT) [1]. In mammals α-tubulin acetyltransferase
(αTAT1) seems to be the major and possibly the only
acetyltransferase of α-tubulin, which is highly specific to the
ω-amino group of the Lys40 residue of α-tubulin [2]. Deacetylation of α-tubulin is catalyzed by
two cytoplasmic enzymes: Zn2+-dependent histone deacetylase
HDAC6 (class IIb) and NAD+-dependent histone deacetylase
SIRT-2 (class III) [3-6].
Stable MTs are known to contain a high percentage of acetylated
α-tubulin, whereas dynamic MTs where tubulin subunits are
actively exchanged are nearly non-acetylated [1].
For tubulin microfilaments, the acetylation ↔ deacetylation
equilibrium controls mitosis and positioning of organelles, and it
determines vesicular transport efficiency and the ability of cells to
contract [1, 7].
In 2009, the paper was published that significantly changed ideas concerning the role of HDAC6 in the regulation of MT stability [8]. It was shown that in mouse melanoma cells treated with a selective HDAC6 inhibitor, tubacin, the growth rate of MTs was delayed first of all because of physical contact of catalytically inactive HDAC6 with the apical zone of the MT growth. It was also shown that suppression of Hdac6 gene expression by RNA interference approaches did not influence the growth rate of MTs, although it induced hyperacetylation of α-tubulin. These results are in good agreement with the observation that transgenic mice with Hdac6 gene knockout retained viability despite a high level of acetylation of α-tubulin in many organs [9].
Assessment of the relative contributions of HDAC6 and SIRT-2 to deacetylation of α-tubulin is very important for studies on neurodegenerative diseases caused by oxidative stress [10]. Lateral amyotrophic sclerosis (Charcot’s disease) is characterized by disorders in mitochondrial transport to neuronal axons because of decreased stability of MTs. It was shown in the work of Taes et al. [11] that in a mouse strain deficient in the superoxide dismutase 1 gene, knockout of the Hdac6 gene significantly decelerated the development of the disease and induced hyperacetylation of α-tubulin in nervous system cells. However, knockout of the Sirt2 gene influenced neither the disease development nor the level of α-tubulin acetylation.
It is known that the hepatitis C virus (HCV) uses the host cell cytoskeleton during different stages of its life cycle including cell infection, replication, and translocation of the replicative complex from the perinuclear space to the outer membrane [12-15]. All of these processes are maintained due to interaction of viral proteins Core, NS3, and NS5A with actin and tubulin microfilaments of the cell, and their effectiveness directly depends on dynamic features of the cytoskeleton. Thus, the viral replication in hepatocytes of Huh7 line could be inhibited with any of three inhibitors of the MT polymerization: vinblastin, colchicine, or nocodazole [13].
In 2013, Japanese researchers published the first data on suppression of HCV replication in OR6 line hepatocytes with nonselective inhibitors of histone deacetylases, suberoylanilide hydroxamic acid (SAHA) and trichostatin A (TSA) [16]. They suggested that the antiviral effect of SAHA was due to changes in the expression of some genes as a result of hyperacetylation of histone H3. In our work also published in 2013, we supposed that HDAC6 could be an enzyme responsible for the antiviral effect of benzohydroxamic acids [17]. In this work, the first data are presented on the influence of selective inhibitors HDAC6 and SIRT-2 on acetylation of α-tubulin and the HCV replicon propagation in human hepatocytes.
MATERIALS AND METHODS
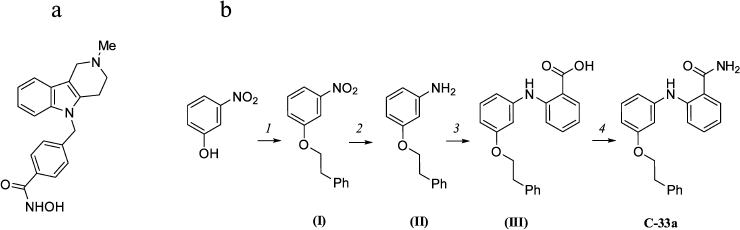
Synthesis of inhibitors of HDAC6 and SIRT-2. The selective inhibitor of HDAC6 tubastatin A (Fig. 1a) was prepared as described earlier in [18]. The selective inhibitor of SIRT-2 C-33a [19] was prepared in four stages (Fig. 1b). The structure of these compounds was confirmed by 1H NMR (400 MHz, DMSO-d6) and 13C NMR spectroscopy (101 MHz, DMSO-d6).
Fig. 1. a) Structure of tubastatin A. b) Scheme of C-33a synthesis: 1) PhCH2CH2OH/PPh3/DIAD/THF; 2) SnCl2·2H2O/EtOH/H2O/HCl; 3) 2-Cl-PhCO2H/K2CO3/Cu/Cu2O/CH3OCH2CH2OH; 4) CDI/NH4TSA/DMF.
1-Nitro-3-phenetoxybenzol (I). 1H NMR – δ 7.79 (dd, J = 8.1, 1.9 Hz, 1H, CH), 7.69 (t, J = 2.2 Hz, 1H, CH), 7.55 (t, J = 8.2 Hz, 1H, CH), 7.40 (dd, J = 8.3, 2.4 Hz, 1H, CH), 7.28-7.36 (m, 4H, CH), 7.15-7.28 (m, 1H, CH), 4.33 (t, J = 6.8 Hz, 2H, CH2), 3.07 (t, J = 6.8 Hz, 2H, CH2); 13C NMR – δ 158.93, 148.73, 137.99, 130.62, 128.90, 128.27, 126.30, 121.85, 115.42, 108.71, 68.90, 34.63.
3-Phenetoxyaniline (II). 1H NMR – δ 7.31 (d, J = 4.3 Hz, 4H, CH), 7.22 (dt, J = 8.8, 4.4 Hz, 1H, CH), 6.89 (t, J = 6.4 Hz, 1H, CH), 6.15 (s, 2H, CH), 6.07 (d, J = 7.7 Hz, 1H, CH), 4.92 (bs, 2H, NH2), 4.07 (t, J = 6.9 Hz, 2H, CH2), 2.99 (t, J = 6.8 Hz, 2H, CH2); 13C NMR – δ 159.27, 149.72, 138.49, 129.39, 128.82, 128.18, 126.11, 106.94, 102.05, 100.13, 67.60, 34.97.
2-(3-Phenetoxyphenylamino)benzoic acid (III). 1H NMR – δ 12.92 (bs, 1H, CO2H), 9.57 (s, 1H, NH), 7.90 (dd, J = 8.0, 1.6 Hz, 1H, CH), 7.39 (ddd, J = 8.6, 7.1, 1.7 Hz, 1H, CH), 7.15-7.36 (m, 7H, CH), 6.76-6.82 (m, 3H, CH), 6.64 (dd, J = 8.2, 1.9 Hz, 1H, CH), 4.19 (t, J = 6.8 Hz, 2H, CH2), 3.02 (t, J = 6.8 Hz, 2H, CH2); 13C NMR – δ 169.78, 159.43, 146.67, 141.84, 138.31, 134.04, 131.77, 130.13, 128.87, 128.21, 126.17, 117.53, 114.26, 113.30, 112.84, 109.24, 107.19, 68.07, 34.88.
2-(3-Phenetoxyphenylamino)benzamide (C-33a). 1H NMR – δ 9.94 (s, 1H, NH), 8.02 (s, 1H, CONH2), 7.70 (d, J = 7.6 Hz, 1H, CH), 7.42 (s, 1H, CONH2), 7.25-7.39 (m, 6H, CH), 7.12-7.25 (m, 2H, CH), 6.80 (t, J = 7.7 Hz, 1H, CH), 6.65-6.75 (m, 2H, CH), 6.55 (d, J = 7.7 Hz, 1H, CH), 4.17 (t, J = 6.6 Hz, 2H, CH2), 3.01 (t, J = 6.4 Hz, 2H, CH2); 13C NMR – δ 169.78, 159.43, 146.67, 141.84, 138.31, 134.04, 131.77, 130.13, 128.87, 128.21, 126.17, 117.53, 114.26, 113.30, 112.84, 109.24, 107.19, 68.07, 34.88.
Cell cultures. Cell lines HepG2 and Huh7 were grown in medium DMEM + F12 (2 : 1) supplemented with 10% FBS (HyClone, USA), 2 mM L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin in the presence of 5% CO2 at 37ºC. The cells were seeded once per three days at the ratio of 1 : 3 or 1 : 5. The line Huh7-luc/neo culture was grown under the same conditions on addition of G418 (330 µg/ml).
Inhibition of HDAC6 and SIRT-2 in hepatocytes. Cells were seeded into a 6-well culture plate. Twenty-four hours later (the monolayer was 40-50%) compounds under test were introduced into the culture medium and the cultures were incubated at 37ºC in the presence of 5% CO2. Three days later (the monolayer in the control sample was 100%) the medium was removed, and the cells were washed with PBS and treated with a lysing reagent (Promega, USA).
Western blot. Proteins of the cell lysates were separated by electrophoresis in 10% polyacrylamide gel with 0.1% SDS and electrotransferred onto a nitrocellulose membrane. The membrane was treated with 5% dry milk (Bio-Rad, USA) in PBST for 60 min at room temperature. Primary antibodies to acetylated lysine diluted 1 : 1000 (ab80178; Abcam, England) and antibodies to tubulin diluted 1 : 10,000 (Sigma, USA) were added, incubated overnight at 4°C, and washed with PBST. Conjugate of horseradish peroxidase with secondary specific antibodies (anti-mouse, anti-rabbit; Santa Cruz, USA) diluted 1 : 10,000 was added and incubated for 50 min at room temperature. Then the membranes were washed with PBST, and the signal was visualized using an ECL-Kit (Pierce-Thermo Scientific, USA) and a High Performance ECL Film (GE Healthcare, England).
Antiviral activity. The Huh7-luc/neo cells were seeded into a 48-well culture plate (the medium did not contain antibiotic G418). After 24 h (the monolayer was 40-50%) the culture medium was supplemented with compounds under testing in different concentrations and incubated at 37ºC in the presence of 5% CO2. Three days later (the monolayer in the control sample was 100%) the medium was removed, the cells were washed with PBS and lysed, and the luciferase activity of the reporter protein was measured using a Luciferase Assay System Kit (Promega) according to the producer’s protocol. Chemiluminescence was measured with a Thermo Luminometer (Labsystems, Israel).
Cytotoxicity. The Huh7 cells were seeded into a 96-well culture plate the day previous to addition of the compounds under test. After 24 h (the monolayer was 40-50%) the culture medium was added with the compounds under test in different concentrations and incubated at 37ºC in the presence of 5% CO2. Three days later (the monolayer in the control sample was 100%) the cell viability was determined with an MTT Kit (Sigma-Aldrich, USA) according to the producer’s protocol.
RESULTS AND DISCUSSION
The molecular mechanism of anti-HCV action of derivatives of benzohydroxamic acids that we found is still unknown [17]. According to the literature, benzohydroxamic acid and its para-substituted analogs are polypotent inhibitors of histone deacetylases, but, nevertheless, display a significant selectivity to HDAC6 [20]. This determined the direction and goals of our work, which can be formulated as follows: a) to demonstrate the anti-HCV activity of any powerful inhibitor of HDAC6 from benzohydroxamic derivatives; b) to obtain for this compound concentration dependences of suppression of HCV replication and of accumulation of Ac-α-tubulin; c) to show the correlation between them. For the experimental approach, it was necessary to take into account additionally the SIRT-2 activity, which could also influence the level of MT acetylation.
Choice of inhibitors and their synthesis. In further work we used as HDAC6 inhibitor tubastatin A (Fig. 1a), which a priori corresponded to all necessary requirements: it is a para-substituted benzohydroxamic acid, displays strong and highly selective inhibitory action on HDAC6 in vitro, and induces selective hyperacetylation of α-tubulin in cell culture [21].
To evaluate the contribution of SIRT-2 to the deacetylation of α-tubulin, we chose and synthesized a recently described inhibitor of SIRT-2 – the derivative of 2-aminobenzamide C-33a (Fig. 1b) [19]. It was reported that compound C-33a induced in the cell culture hyperacetylation of α-tubulin but did not influence the acetylation of p53, which was a substrate protein of a relative histone deacetylase of SIRT-1. To prepare this compound, we developed a new scheme of synthesis that allowed us to increase the yield of the product to 11%, as compared to 2.8% in the original work (see Supplement to this paper on the site of Biochemistry (Moscow): http://protein.bio.msu.ru/biokhimiya).
Antiviral activity of tubastatin A and C-33a. The resulting inhibitors of HDAC6 and SIRT-2 were tested for the presence of anti-HCV activity and cytotoxic effect (Fig. 2). Tubastatin A suppressed the HCV replicon with EC50 = 0.3 μM and selectivity of its antiviral effect SI = 37 that was higher than similar parameters of non-substituted benzohydroxamic acid: EC50 = 14 μM and SI = 14 [17]. An unexpected result of testing C-33a was its stimulatory effect on HCV replication (Fig. 2). However, the stimulatory effect of C-33a disappeared at its elevated concentrations, possibly because of an increasing cytotoxic action.
Fig. 2. Antiviral activity and cytotoxicity of tubastatin A (a) and C-33a (b).
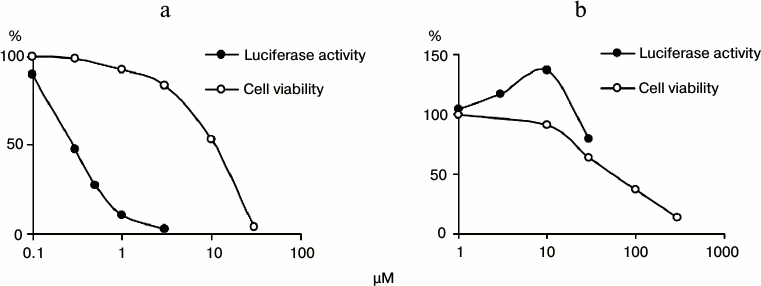
In accordance with the literature, tubastatin A and C-33a caused noticeable acetylation of α-tubulin at concentrations of 2.5 and 10 μM, respectively [19, 21]. The viability of line Huh7 cells at inhibitor concentrations of 3 and 10 μM was, respectively, 84 and 91%, which allowed us to perform the following studies on MT acetylation not taking into account cytotoxicity influence.
Acetylation of α-tubulin in hepatocytes. Our next step was to test the Ac-α-tubulin level in hepatocytes of HepG2, Huh7, and Huh7-luc/neo lines normally and in the presence of inhibitors of HDAC6 and SIRT-2. Figure 3 shows that for all three cell lines the normal level of Ac-α-tubulin was lower than the detection level. On incubation of the cells with the inhibitor of SIRT-2, a weak acetylation was observed only in the HepG2 line cells. In the presence of the inhibitor of HDAC6, α-tubulin was hyperacetylated in hepatocytes of HepG2 and Huh7-luc/neo lines but not in hepatocytes of Huh7 line.
Fig. 3. Acetylation of α-tubulin in cultures of human hepatocyte HepG2, Huh7, and Huh7-luc/neo normally and in the presence of tubastatin A or C-33a.
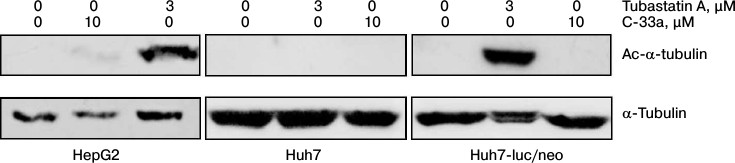
Based on these findings, we concluded that in the cells of all three lines the total activity of HDAC6 and SIRT-2 normally is prevalent above the αTAT1 activity, and as a result the equilibrium acetylation ↔ deacetylation is strongly shifted to the right for α-tubulin; moreover, the contribution of SIRT-2 to deacetylation of α-tubulin is either very insignificant or completely absent. Finally, the high sensitivity of Huh7-luc/neo line cells to inhibition of HDAC6 in comparison with native cells of Huh7 line is probably explained by intensification of both acetylation and deacetylation of MTs in the presence of the HCV replicon.
Correlation between α-tubulin acetylation and inhibition of HCV. To study the dependence between the antiviral activity of tubastatin A and inhibition of HDAC6, Huh7-luc/neo line cells were grown in the presence of the inhibitor (0-3 μM) and analyzed for luciferase activity and content of Ac-α-tubulin as a reporter protein of HDAC6 activity. The results presented in Fig. 4 obviously confirm this correlation. Thus, it is highly probable that the anti-HCV action of tubastatin A is due to suppression of HDAC6 activity. However, it remains unclear whether functioning of the HCV replicon depends on acetylation of MTs or of some other protein substrates of HDAC6.
Fig. 4. Correlation between HDAC6 inhibition and suppression of HCV replication. a) Huh7-luc/neo line cells were grown in the presence of 0, 0.5, 1, and 3 μM tubastatin A and analyzed for Ac-α-tubulin content. The amount of Ac-α-tubulin in the samples detected by Western blotting was evaluated by densitometry. b) To design the correlation dependence, the maximal value of Ac-α-tubulin was conventionally taken as 100%, whereas the other points were calculated according to the densitogram. The resulting values of luciferase activity are presented as the inhibition curve in percent.
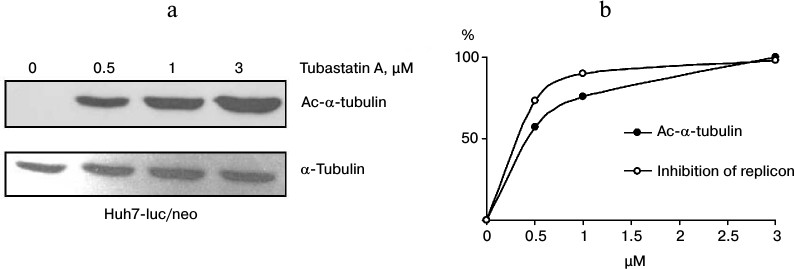
It is interesting that in addition to α-tubulin, HDAC6 also controls the acetylation level and the activity of: a) chaperone Hsp90, which is essential for stabilization of the viral protein NS3, and b) peroxiredoxins Prx1/2 regulating redox potential of the cell, which is an important parameter of HCV adaptation [22-25]. Perhaps due to simultaneous action in all the above-listed lines, HDAC6 can significantly affect the propagation of the virus.
Current approaches for treatment of hepatitis C are based on chemotherapy with the use of α-interferon, a nucleoside analog of ribavirin, and inhibitors of viral proteinase (NS3) telaprevir and boceprevir. The last two preparations allow the treatment time to be shortened from 52 to 24 weeks, but their long-term application is associated with a high probability of appearance of resistant forms of the virus in the patient’s body [26]. Obviously, the problem of acquired resistance can be overcome mainly due to an additional influence on the host cell proteins involved in the intracellular replication of the virus. In the present work, we have shown for the first time that histone deacetylase 6 is a promising cellular target for development of new approaches for chemotherapy of hepatitis C.
This work was supported by the Russian Foundation for Basic Research (project No. 14-04-00221).
REFERENCES
1.Singh, B. N., Zhang, G., Hwa, Y. L., Li, J., Sean,
C., Dowdy, S. C., and Jiang, S. W. (2010) Nonhistone protein
acetylation as cancer therapy targets, Expert Rev. Anticancer
Ther., 10, 935-954.
2.Shida, T., Cueva, J. G., Xu, Z., Goodman, M. B.,
and Nachury, M. V. (2010) The major α-tubulin K40
acetyltransferase αTAT1 promotes rapid ciliogenesis and efficient
mechanosensation, PNAS, 107, 21517-21522.
3.Matsuyama, A., Shimazu, T., Sumida, Y., Saito, A.,
Yoshimatsu, Y., Seigneurin-Berny, D., Osada, H., Komatsu, Y., Nishino,
N., Khochbin, S., Horinouchi, S., and Yoshida, M. (2002) In vivo
destabilization of dynamic microtubules by HDAC6-mediated
deacetylation, EMBO J., 21, 6820-6831.
4.Hubbert, C., Guardiola, A., Shao, R., Kawaguchi,
Y., Ito, A., Nixon, A., Yoshida, M., Wang, X. F., and Yao, T. P. (2002)
HDAC6 is a microtubule-associated deacetylase, Nature,
417, 455-458.
5.North, B. J., Marshall, B. L., Borra, M. T., Denu,
J. M., and Verdin, E. (2003) The human Sir2 ortholog, SIRT2, is an
NAD+-dependent tubulin deacetylase, Mol. Cell,
11, 437-444.
6.Nahhas, F., Dryden, S. C., Abrams, J., and Tainsky,
M. A. (2007) Mutations in SIRT2 deacetylase which regulate enzymatic
activity but not its interaction with HDAC6 and tubulin, Mol. Cell.
Biochem., 303, 221-230.
7.Janke, C., and Bulinski, J. C. (2011)
Posttranslational regulation of the microtubule cytoskeleton:
mechanisms and functions, Nat. Rev. Mol. Cell Biol., 12,
773-786.
8.Zilberman, Y., Ballestrem, C., Carramusa, L.,
Mazitschek, R., Khochbin S., and Bershadsky, A. (2009) Regulation of
microtubule dynamics by inhibition of the tubulin deacetylase HDAC6,
J. Cell Sci., 122, 3531-3541.
9.Witt, O., Deubzer, H. E., Milde, T., and Oehme, I.
(2009) HDAC family: what are the cancer relevant targets? Cancer
Lett., 277, 8-21.
10.Simoes-Pires, C., Zwick, V., Nurisso, A.,
Schenker, E., Carrupt, P. A., and Cuendet, M. (2013) HDAC6 as a target
for neurodegenerative diseases: what makes it different from the other
HDACs? Mol. Neurodegener., 8, 7-22.
11.Taes, I., Timmers, M., Hersmus, N.,
Bento-Abreu, A., Van Den Bosch, L., Van Damme, P., Auwerx, J., and
Robberecht, W. (2013) Hdac6 deletion delays disease progression
in the SOD1G93A mouse model of ALS, Hum. Mol.
Genet., 22, 1783-1790.
12.Roohvand, F., Maillard, P., Lavergne, J. P.,
Boulant, S., Walic, M., Andreo, U., Goueslain, L., Helle, F., Mallet,
A., McLauchlan, J., and Budkowska, A. (2009) Initiation of hepatitis C
virus infection requires the dynamic microtubule network, J. Biol.
Chem., 284, 13778-13791.
13.Bost, A. G., Venable, D., Liu, L., and Heinz, B.
A. (2003) Cytoskeletal requirements for hepatitis C virus (HCV) RNA
synthesis in the HCV replicon cell culture system, J. Virol.,
77, 4401-4408.
14.Lai, C. K., Jeng, K. S., Machida, K., and Lai, M.
M. (2008) Association of hepatitis C virus replication complexes with
microtubules and actin filaments is dependent on the interaction of NS3
and NS5A, J. Virol., 82, 8838-8848.
15.Wolk, B., Buchele, B., Moradpour, D., and Rice,
C. M. (2008) A dynamic view of hepatitis C virus replication complexes,
J. Virol., 82, 10519-10531.
16.Sato, A., Saito, Y., Sugiyama, K., Sakasegawa,
N., Muramatsu, T., Fukuda, S., Yoneya, M., Kimura, M., Ebinuma, H.,
Hibi, T., Ikeda, M., Kato, N., and Saito, H. (2013) Suppressive effect
of the histone deacetylase inhibitor suberoylanilide hydroxamic acid
(SAHA) on hepatitis C virus replication, J. Cell. Biochem.,
114, 1987-1996.
17.Kozlov, M. V., Kleymenova, A. A., Romanova, L.
I., Konduktorov, K. A., Smirnova, O. A., Prasolov, V. S., and
Kochetkov, S. N. (2013) Benzohydroxamic acids as potent and selective
anti-HCV agents, Bioorg. Med. Chem. Lett., 23,
5936-5940.
18.Kozlov, M. V., Kleymenova, A. A., Konduktorov, K.
A., and Kochetkov, S. N. (2013) A new synthesis of a highly selective
inhibitor of histone deacetylase 6 —
N-hydroxy-4-(2-methyl-1,2,3,4-tetrahydropyrido[4,3-b]indol-5-ylmethyl)benzamide –
tubastatin A, Bioorg. Khim., 39, 117-120.
19.Suzuki, T., Khan, M. N., Sawada, H., Imai, E.,
Itoh, Y., Yamatsuta, K., Tokuda, N., Takeuchi, J., Seko, T., Nakagawa,
H., and Miyata, N. (2012) Design, synthesis, and biological activity of
a novel series of human sirtuin-2-selective inhibitors, J. Med.
Chem., 55, 5760-5773.
20.Wagner, F. F., Olson, D. E., Gale, J. P., Kaya,
T., Weiver, M., Aidoud, N., Thomas, M., Davoine, E. L., Lemercier, B.
C., and Holson, E. B. (2013) Potent and selective inhibition of histone
deacetylase 6 (HDAC6) does not require a surface-binding motif, J.
Med. Chem., 56, 1772-1776.
21.Butler, K. V., Kalin, J., Brochier, C., Vistoli,
G., Langley, B., and Kozikowski, A. P. (2010) Rational design and
simple chemistry yield of a superior, neuroprotective HDAC6 inhibitor,
tubastatin A, J. Am. Chem. Soc., 132, 10842-10846.
22.Kovacs, J. J., Murphy, P., Gaillard, S., Zhao,
X., Wu, J. T., Nicchitta, C. V., Yoshida, M., Toft, D. O., Pratt, W.
B., and Yao, T. P. (2005) HDAC6 regulates Hsp90 acetylation and
chaperone-dependent activation of glucocorticoid receptor, Mol.
Cell., 18, 601-607.
23.Ujino, S., Yamaguchi, S., Shimotohno, K., and
Takaku, H. (2009) Heat-shock protein 90 is essential for stabilization
of the hepatitis C virus nonstructural protein NS3, J. Biol.
Chem., 284, 6841-6846.
24.Parmigiani, R. B., Xu, W. S., Venta-Perez, G.,
Erdjument-Bromage, H., Yaneva, M., Tempst, P., and Marks, P. A. (2008)
HDAC6 is a specific deacetylase of peroxiredoxins and is involved in
redox regulation, PNAS, 105, 9633-9638.
25.Waris, G., Turkson, J., Hassanein, T., and
Siddiqui, A. (2005) Hepatitis C virus (HCV) constitutively activates
STAT-3 via oxidative stress: role of STAT-3 in HCV replication, J.
Virol., 79, 1569-1580.
26.Wyles, D. (2012) Beyond telaprevir and
boceprevir: resistance and new agents for hepatitis C virus infection,
Top Antivir. Med., 20, 139-145.
Supplementary Methods (PDF)