Experimental Models of Arthritis in Which Pathogenesis Is Dependent on TNF Expression
M. S. Drutskaya1*, G. A. Efimov1, R. V. Zvartsev1, A. A. Chashchina1,2, D. M. Chudakov3, S. V. Tillib2,4, A. A. Kruglov5,6, and S. A. Nedospasov1,2,5,6
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; fax: +7 (499) 135-1405; E-mail: marinadru@gmail.com2Biological Faculty, Lomonosov Moscow State University, Leninsky Gory 1/12, 119234 Moscow, Russia; fax: +7 (495) 939-4309; E-mail: info@mail.bio.msu.ru
3Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: +7 (495) 335-0812; E-mail: office@ibch.ru
4Institute of Gene Biology, Russian Academy of Sciences, ul. Vavilova 34/5, 119334 Moscow, Russia; fax: +7 (499) 135-4105; E-mail: info@genebiology.ru
5Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Leninsky Gory 1/40, 119234 Moscow, Russia; fax: +7 (495) 939-0338; E-mail: fxb@genebee.msu.su
6Lobachevsky State University of Nizhnii Novgorod, pr. Gagarina 23, 603950 Nizhnii Novgorod, Russia; fax: +7 (831) 462-3085; E-mail: unn@unn.ru
* To whom correspondence should be addressed.
Received July 9, 2014
Rheumatoid arthritis (RA) is an autoimmune inflammatory disease characterized by joint damage as well as systemic manifestations. The exact cause of RA is not known. Both genetic and environmental factors are believed to contribute to the development of this disease. Increased expression of tumor necrosis factor (TNF) has been implicated in the pathogenesis of RA. Currently, the use of anti-TNF drugs is one of the most effective strategies for the treatment of RA, although therapeutic response is not observed in all patients. Furthermore, due to non-redundant protective functions of TNF, systemic anti-TNF therapy is often associated with unwanted side effects such as increased frequency of infectious diseases. Development of experimental models of arthritis in mice is necessary for studies on the mechanisms of pathogenesis of this disease and can be useful for comparative evaluation of various anti-TNF drugs. Here we provide an overview of the field and present our own data with two experimental models of autoimmune arthritis – collagen-induced arthritis and antibody-induced arthritis in C57Bl/6 and BALB/c mice, as well as in tnf-humanized mice generated on C57Bl/6 background. We show that TNF-deficient mice are resistant to the development of collagen-induced arthritis, and the use of anti-TNF therapy significantly reduces the disease symptoms. We also generated and evaluated a fluorescent detector of TNF overexpression in vivo. Overall, we have developed an experimental platform for studying the mechanisms of action of existing and newly developed anti-TNF drugs for the treatment of rheumatoid arthritis.
KEY WORDS: rheumatoid arthritis, tumor necrosis factor, anti-cytokine therapy, autoimmune diseasesDOI: 10.1134/S0006297914120086
The increased expression level of tumor necrosis factor (TNF) is associated with many chronic inflammatory states including autoimmune diseases such as the rheumatoid arthritis (RA), Krone’s disease, ulcerative colitis, psoriasis, and ankylosing spondylitis (Bechterew’s disease). Detection of high level of various proinflammatory cytokines, including TNF, in biopsies and cell cultures derived from RA patients suggests their key role in sustaining inflammation in affected joints. This was further confirmed by the fact that significant amelioration of the disease state is observed both in patients and in relevant animal models receiving anti-TNF therapeutics. Studies using genetically modified mice with a transgene encoding human TNF (hTNF) locus (in addition to the endogenous murine tnf gene) has become one of the most informative models that allow study of the role of TNF. In this model, the transgene containing the human tnf gene sequence that was modified in the segment involved in the repression of translation 3′-UTR (untranslated region) led to a high and uncontrollable expression of hTNF. As a result, these mice developed erosive arthritis with a histological picture similar to that observed in patients with RA [1]. Furthermore, mice bearing the same hTNF transgene under the control of T cell-specific “locus control region” of the human cd2 gene, characterized by hTNF overexpression in T-cells [2], were also created. Increased TNF expression only in T-lymphocytes was sufficient to induce the development of arthritis, though this result does not necessarily mean that T-lymphocytes are the source of pathogenic TNF in arthritis. It is interesting that injection of antibodies that blocked IL-1 receptor signaling completely prevented the development of arthritis in these mice [3]. However, IL-1-deficient mice bearing hTNF transgene still developed arthritis, though a reduction in erosion of bone tissue in comparison with control group was observed. Thus, these results indicate that IL-1 is an important effector mediator in this arthritis model and is regulated by TNF, as proposed earlier for RA (Fig. 1). A similar erosive form of arthritis developed in genetically modified mice with a deletion of ARE (3′-AU-rich element) in the 3′-UTR of the tnf gene. These mice were characterized by increased TNF expression; however, crossing of these mice to RAG-1-deficient background did not rescue them from arthritis development, which suggests that lymphocytes with TNF overexpression are not required for disease initiation [4, 5]. It was shown that mice with complete TNF ablation are resistant to collagen-induced arthritis, whereas mice with a cell type-specific inactivation of TNF in macrophages and neutrophils or in T-cells develop the disease (Kruglov et al., manuscript in preparation).
In spite of the fact that anti-TNF therapy is widely used in clinics (see the review by Astrakhantseva et al. in this issue), the mechanism of its action is still not fully understood. It is clear that anti-TNF blockers either bind a soluble form of TNF or interfere with binding of TNF to its receptors. At the same time, interaction of TNF blockers with membrane-bound TNF can induce antibody-dependent or complement-mediated cytotoxicity, reduce expression of other proinflammatory cytokines that are regulated by TNF (such as IL-1 and IL-6), and, in turn, lead to reduced TNF expression as a feedback mechanism [6]. Furthermore, it was demonstrated that expression of chemokines that attract cells to the site of inflammation (RANTES, MCP-1, IL-8) is also reduced as a result of anti-TNF treatment. This effect is also observed due to a decreased expression of adhesion molecules on endothelial cells and a reduction in angiogenesis, both of which are TNF-dependent processes. In RA patients subjected to treatment with infliximab, an increase in the number of circulating regulatory T cells was also observed [7]. In general, anti-TNF therapy efficiently reduces the level of inflammation locally at the affected sites, and it probably also results in systemic effects on differentiating cells, including those in the lymphoid organs. In addition, the mechanism of action of TNF blockers on “pain receptors” was recently described, which undoubtedly contributes to overall success of this type of therapy in patients [8].
It is quite difficult to give an exact assessment of the side effects associated with each anti-TNF therapeutic, mainly because they are often prescribed in combination with other drugs such as methotrexate or even cyclosporin, which is immunosuppressive. The most frequent and well-documented side effect is possible reactivation of latent infections, the most serious being tuberculosis. This undesirable complication from anti-TNF treatment is based on one of the non-redundant physiological functions of TNF – its role in formation and maintenance of the structural integrity of bactericidal granulomas [9, 10]. In this regard, prior to the initiation of anti-TNF therapy all patients have to undergo medical examination to rule out mycobacterial infection. Side effects from anti-TNF therapy are often associated with other granulomatous diseases: histoplasmosis, coccidioidomycosis, and cryptococcosis. Among fungal diseases, such opportunistic infections as mucormycosis and candidiasis have been reported. More often, leishmaniasis and some chronic viral infections (such as the human herpes virus type 3, hepatitis B, and cytomegalovirus) arise or become aggravated [6]. There are also some data suggesting that systemic TNF ablation may possibly increase the risk of malignancy: skin cancer, lymphoma, gastrointestinal cancers, etc. However, addressing this issue is quite complicated since patients with psoriasis or RA are already predisposed to an increased risk of lymphoma regardless of the treatment [6]. Moreover, there is some indication that TNF inhibitors can actually be the cause of some manifestations of autoimmune diseases. Among such diseases are chronic inflammatory demyelinating polyneuropathies with syndromes similar to a systemic lupus erythematosus. The latter characterized by an increase in autoantibodies to nuclear DNA and histones is seen in the blood of patients. The most paradoxical is a report on an increased number of cases of psoriasis in patients undergoing anti-TNF therapy [6].
Thus, TNF plays a key role in maintenance of the protective immune response to intracellular pathogens and has an important homeostatic function, but at the same time is one of the main cytokines associated with the induction of autoimmune diseases. Reproducible experimental animal models are necessary for study of the mechanisms of action of different TNF inhibitors and for understanding the nature of possible side effects. In the current study, we describe two experimental protocols for autoimmune arthritis induction in mice: collagen-induced arthritis and collagen antibody-induced arthritis in C57Bl/6 and BALB/c mice, as well as in newly generated humanized TNF knock-in mice recently created in our laboratory, in which the human tnf gene functionally replaces the mouse gene. The latter mouse model allows evaluation of all clinically approved TNF inhibitors in the context of experimental therapy in mice specific against human rather than murine TNF. The only exception is etanercept, which inhibits both human and murine TNF. TNF-deficient mice are protected against collagen-induced arthritis. Treatment of BALB/c mice with etanercept considerably reduces the symptoms of arthritis induced by arthritogenic antibodies. In vivo imaging of TNF expression in the effected joint was performed using a new fluorescent TNF detector.
MATERIALS AND METHODS
Animals. C57Bl/6 and BALB/c mice as well as TNF-deficient mice [11] and humanized hTNFKI mice generated on C57Bl/6 genetic background were 8-10 weeks of age upon the initiation of the experiments. Only female mice were used for the experiments. The mice were bred and maintained under specific pathogen-free conditions at the Pushchino Animal Facility of the Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences.
Fluorescent TNF detector for in vivo imaging. The construct carrying a gene encoding one-domain camel antibody Vhh41 [12] connected to a gene encoding red fluorescent Katushka protein [13] by a flexible glycine-serine linker was cloned into the expression vector pET28 (Novagen, Germany) in such a way that there is a hexahistidine sequence at the C-end. The resulting genetic construct was used for transformation of E. coli BL21(DE3) cells, which were cultured under the following conditions: the bacterial culture was grown in LB media in the presence of a selective antibiotic to OD600 = 1.0, followed by addition of isopropyl β-D-1-thiogalactopyranoside to final concentration of 0.4 mM to induce protein expression during 4 h at 37°C. The bacterial culture was then centrifuged, and the bacterial cells in the pellet were lysed. Soluble protein was purified from the cytoplasmic fraction of the bacterial lysate using Ni2+-affinity chromatography on agarose conjugated with Ni-nitrilotriacetic acid (Invitrogen, USA) according to manufacturer’s protocol. The purified fraction of the hybrid protein was cleared of imidazole by dialysis and filtered under sterile conditions. Final protein concentration was measured using the BCA Protein Assay Kit (Pierce, USA) according to the manufacturer’s protocol.
In vivo imaging and immunohistochemistry. In vivo imaging of TNF expression in the affected joint of C57Bl/6 mice after induction of experimental arthritis was carried out as described earlier [13a] on the Kodak Carestream in vivo FX PRO device (Carestream Health, USA) with excitation at 570 nm and emission at 635 nm with 30 sec exposure. Snapshots were taken every 5 min following the intravenous administration 50 pM of the fluorescent TNF detector (Vhh41-Katushka). The immunohistochemistry of paraffin-embedded sections of affected and normal joints was carried out using secondary anti-Katushka-FITC antibodies, and images were generated using a Leica TCS SP5 confocal microscope. Tissue samples were collected following arthritis induction with collagen in C57Bl/6 mice. Fluorescent TNF detector (Vhh41-Katushka) was administered i.p. at 200 µg per mouse. After 40 min, the mice were euthanized and their joints collected for histology.
Collagen-induced arthritis. C57Bl/6 and TNF–/– females at the age 8-10 weeks were immunized with emulsion containing 100 µg of chicken collagen type II (Sigma, USA) in complete Freund’s adjuvant (Sigma). Dry collagen was dissolved in 10 mM acetic acid at 2 mg/ml for 10 h at 4°C with continuous shaking. Then the collagen solution was mixed at ratio 1 : 1 with complete Freund’s adjuvant that was additionally enriched with 8 mg/ml of killed M. tuberculosis H37RA (Thermo Fisher Scientific Inc., USA). The resulting suspension was thoroughly mixed, transferred to Eppendorf tubes at 0.8 ml per tube, and then chilled on ice. Contents of each test tube were subjected to disintegration using an ultrasonic homogenizer (BANDELIN SONOPULS, Germany) repeatedly three times for 5 sec with the test tubes embedded in ice to prevent the contents from overheating. The resulting mix was administered s.c. into the tail base at 100 µl per mouse. On day 21, mice were subjected to the second immunization. Pathology score was evaluated macroscopically every 3-4 days based on the following scale: 0 – normal joint; 1 – minor reddening and edema; 2 – mild edema and reddening of a few joints; 3 – strong edema of all joints of a paw. The total score for each mouse was calculated based on the sum of the scores for each of the four paws.
Collagen antibody-induced arthritis. For induction of arthritis with arthritogenic antibodies, a cocktail containing five monoclonal antibodies against collagen type II and LPS from E. coli 0111:B4 as adjuvant was injected in accordance with the manufacturer’s protocol (Chondrex, USA). On the first day of the experiment, BALB/c mice were injected i.p. or i.v. with 1.5 mg of 5-clone cocktail in 200 µl of PBS, and hTNFKI mice (on C57Bl/6 genetic background) were injected with 5 mg of 5-clone cocktail. The control group was injected with 200 µl of sterile phosphate buffer (PBS; Thermo Fisher Scientific, USA). After three days, each mouse was injected i.p. with 50 µg of LPS dissolved in 200 µl of PBS. Pathogenesis of arthritis was evaluated during 10 days following arthritogenic antibody injection. Pathology score for each paw was determined based on the following scale: 0 – no reddening and swelling (normal joints); 1 – minor reddening and/or swelling; 2 – mild reddening and swelling; 3 – strong reddening and swelling; 4 – severe reddening and swelling. The total score for each mouse was calculated based on the sum of the scores for each of the four paws.
Statistical analysis. For statistical analysis of the data, we used the GraphPad Prism 6 program (GraphPad Software). Statistically significant difference was established using Student’s test. Data are presented in the form of average values and standard error for each group of mice (n = 5). Distinctions between groups were considered statistically significant with P value less or equal to 0.05.
RESULTS AND DISCUSSION
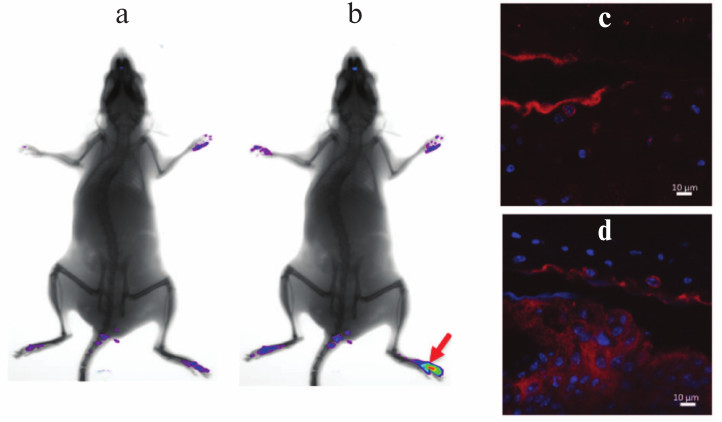
Induction of arthritis in C57Bl/6 mice with collagen immunization. Pathogenesis of collagen-induced arthritis carries many common features with the course of RA in patients and, in particular, is characterized by synovial hyperplasia, infiltration of the affected tissue with mononuclear cells, and cartilage degradation [14]. Predisposition to the development of collagen-induced arthritis, just as in the case of RA, depends on the presence of certain alleles in the MHC class II gene locus [15], which results in varying sensitivity of different inbred strains of mice to experimental arthritis. The preferred standard model for study of collagen-induced arthritis is DBA/1 mice, which are highly sensitive to the established disease induction protocol [16, 17]. However, optimization of experimental conditions allows arthritis induction in other inbred strains, in particular, in mice on the C57Bl/6 genetic background, which are known to be highly resistant to collagen-induced arthritis. It should be pointed out that the majority of mice with genetic modifications (such as knockout or transgenic mice) were generated on C57Bl/6 genetic background. Just as in the case of RA, this experimental model is characterized by TNF expression in inflamed joints (Fig. 1), and the level of its expression is so high that in vivo imaging during disease onset using fluorescently labeled detectors is possible (Figs. 1a and 1b).
Fig. 1. Increased TNF expression in inflamed tissue of a mouse during collagen-induced arthritis. In vivo imaging of TNF expression in the affected joint after induction of autoimmune arthritis was carried out on a Kodak Carestream in vivo FX PRO device (a) prior to injection and (b) after intravenous injection of 50 pM of the fluorescent TNF detector (Vhh41-Katushka). The image was taken 50 min after the injection of Vhh41-Katushka. Collagen-induced arthritis protocol was applied to C57Bl/6 mice according to the standard protocol followed with 200-µg i.p. injection of the fluorescent TNF detector (Vhh41-Katushka). After 40 min, the mice were euthanized and joints of back paws were collected for histology. The immunohistochemistry of paraffin-embedded sections was carried out with using secondary anti-Katushka-FITC antibodies: (c) normal joint and (d) joint affected by arthritis, which is characterized by the increased TNF expression level in synovial tissue.
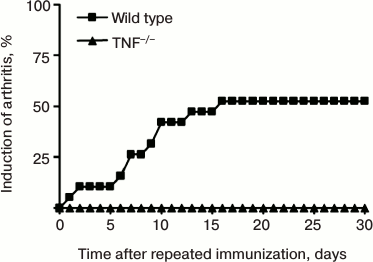
TNF-deficient mice are resistant to collagen-induced arthritis. For induction of the disease, the first and the second (after three weeks) immunizations were carried out with chicken collagen type II in complete Freund’s adjuvant via subcutaneous injection [16, 17]. However, in comparison with collagen-induced arthritis in DBA/1 mice, the disease onset in C57Bl/6 mice or mice on C57Bl/6 genetic background is characterized by lower pathology score, in particular, reduced swelling of the paws, and smaller frequency of disease induction. This type of experimental arthritis can be induced in about 50-60% of C57Bl/6 mice, in comparison with 90-100% DBA/1 mice [17]. Mice with complete TNF ablation [11] are resistant to collagen-induced arthritis (Fig. 2). Immunopathogenesis of collagen-induced arthritis relies on both T-cell-specific and B-cell responses [18-22].
Fig. 2. Development of experimental arthritis induced by immunization with collagen in C57Bl/6 mice and TNF-deficient (TNF–/–) mice. The disease was induced by double immunization with type II collagen in complete Freund’s adjuvant additionally enriched with M. tuberculosis (5 mg/ml) with a three-week interval between the immunizations. Arthritis in C57Bl/6 mice developed in 50% of cases, while TNF-deficient mice were resistant to arthritis.
Experimental arthritis induced by transfer of autoimmune collagen-specific antibodies. Induction of the acute form of collagen-induced arthritis depends on the presence of arthritogenic antibodies in the serum. Following this notion, a fast and reproducible model for autoimmune arthritis induction is based on the transfer of antibodies specific to collagen. The important role of B-lymphocytes in the development of arthritis has been demonstrated in a number of experiments as well as in clinical data. In particular, treatment of RA with anti-CD20 can successfully be applied [23-25]. In view of an established role of B-cells and autoimmune antibodies in the development of RA, some experimental models of arthritis are based on transfer of autoimmune antibodies including those specific to collagen or serum containing antibodies to glucose-6-phosphate isomerase [26-30]. Autoantibodies can also serve as markers for diagnosis of the disease.
In the model of arthritis caused by transfer of collagen-specific antibodies, the disease is induced by systemic injection of a cocktail of monoclonal antibodies specific to certain epitopes of the collagen type II molecule with subsequent injection of LPS as an adjuvant. Injection of LPS reduces the quantity of antibodies necessary for induction of arthritis [31]. Signs of the developing disease are very similar to those of RA. Mice develop synovitis, the joints are infiltrated by leukocytes, pannus is formed, and the cartilage degradation and bone erosion are observed [32, 33]. Though it was shown that collagen-specific antibodies bind to the cartilage surface, the exact mechanism of development of the disease in this model has not been established [34, 35]. It was assumed that antibodies as a part of immune complexes are capable of inducing inflammatory reactions either by activating the complement system [36, 37] or by interacting and activating Fc-receptor-expressing cells [36, 38]. Components of the complement system, in turn, are involved in opsonization, which increases the activity of phagocytes and autoantigen presentation. Moreover, some of the components (C5a, C4a, C3a) can serve as activators of inflammation, inducing increased permeability of vessels and attracting and activating phagocytes. It was shown that the complement system components C3, C5, and B-factor as well as C5a-receptor are necessary for the induction of disease in recipient mice [39-42]. It has also been shown that C3a and C5a increase the translation of IL-1 and TNF in monocytes [43, 44], which may play a pathogenic role.
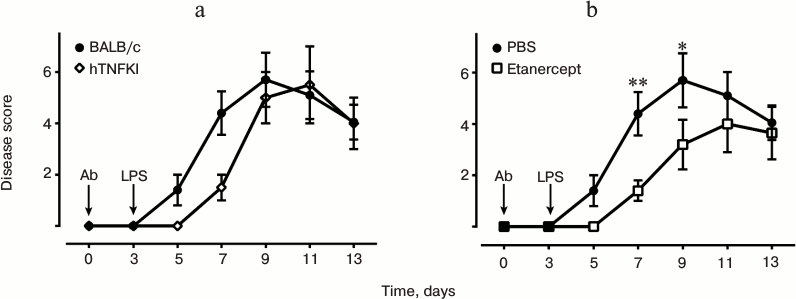
In this study, we established an arthritis model using BALB/c mice (Fig. 3) and in “humanized” hTNFKI mice generated on C57Bl/6 genetic background (Fig. 4a). We were also able to demonstrate the therapeutic effect of TNF inhibitors in this model (Fig. 4b). For induction of disease, we used a cocktail consisting of five monoclonal antibodies to collagen type II (Chondrex). In BALB/c mice, arthritis was induced intraperitoneal or by intravenous administration of 1.5-3.0 mg of five-component cocktail of antibodies, while for the induction of the protocol in mice resistant to the development of arthritis, such as hTNFKI mice on C57Bl/6 genetic background, the amount of antibodies required for arthritis induction was 5 mg/mouse (Fig. 4a). Disease pathogenesis was assessed during the two weeks after the initiation of the experiment. However, the first manifestations of arthritis were already noticeable on the next day following LPS injection (Fig. 3). The peak of disease pathogenesis fell between 7-10 days of the experiment, after which the symptoms decreased. Induction of arthritis in BALB/c mice was blocked by injection of anti-TNF preparation used in the clinic, i.e. etanercept (Fig. 4b), which unlike all other clinically available inhibitors is active against murine TNF.
Fig. 3. Assessment of arthritis score caused by transfer of arthritogenic antibodies. For induction of arthritis with arthritogenic antibodies, BALB/c mice were injected i.p. with 1.5 mg of five-clone antibody cocktail in 200 µl of PBS. After three days, each mouse was injected i.p. with 50 µg of LPS dissolved in 200 µl of PBS. Pathogenesis of arthritis was evaluated during 10 days following arthritogenic antibody transfer. Pathology score for each paw was determined based on the following scale: 0 – no reddening or swelling (normal paw); 1 – minor reddening and/or swelling; 2 – mild reddening and swelling; 3 – strong reddening and/or swelling; 4 – severe reddening and/or swelling.
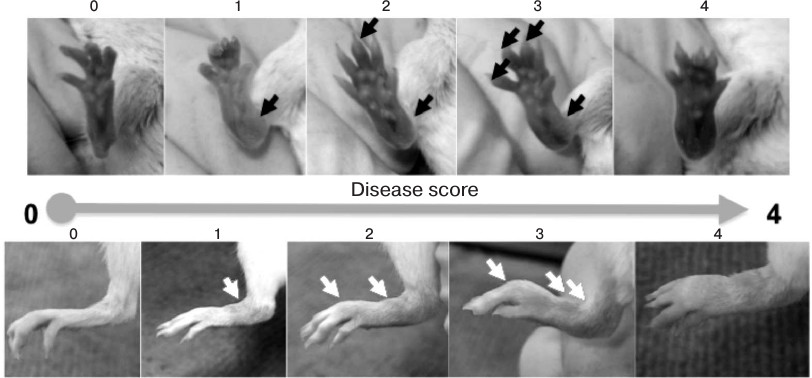
Fig. 4. Development of experimental arthritis induced by antibodies to collagen in BALB/c mice and tnf-“humanized” mice (hTNFKI) on C57Bl/6 genetic background. For induction of arthritis with arthritogenic antibodies (Ab), BALB/c mice were injected i.p. 1.5 mg of five-clone antibody cocktail in 200 µl of PBS, and hTNFKI mice were injected with 5 mg of five-clone antibody cocktail. The control group was injected with 200 µl of sterile phosphate buffer. After three days, each mouse was injected i.p. with 50 µg of LPS dissolved in 200 µl of PBS (a). Development of arthritis in BALB/c mice was considerably reduced upon treatment with etanercept (200 µg per mouse), which is a clinically approved TNF inhibitor (b). Data are presented in the form of average values and standard error for each group (n = 5). Statistically significant differences were determined using Student’s test with * P ≤ 0.05 and ** P ≤ 0.01.
Based on the literature as well as our own data, the overall picture of RA progression can be presented as follows: during the initial stage of RA, inflammation of the synovial surface of a joint occurs. This develops as a result of migration to the affected area and/or local activation of a large number of T- and B-cells, antibody-producing plasma cells, dendritic cells, macrophages, and mast cells, and the induction of neoangiogenesis. As a result, the synovial lining becomes multilayered, and the synovial membrane extends and forms extrusions. The osteoclast-rich site of synovial surface, or pannus, is the main source for bone destruction, and the enzymes released by neutrophils, synoviocytes, and chondrocytes lead to cartilage degradation [45, 46] (Fig. 5). In addition to local symptoms in the joints, approximately 50% of RA patients also exhibit systemic manifestations of the disease, which may include development of rheumatic nodules in the skin, vasculitis, pericarditis, uveitis, rheumatic lung, anemia, cardiovascular diseases, osteoporosis, chronic fatigue, and depression [47-49].
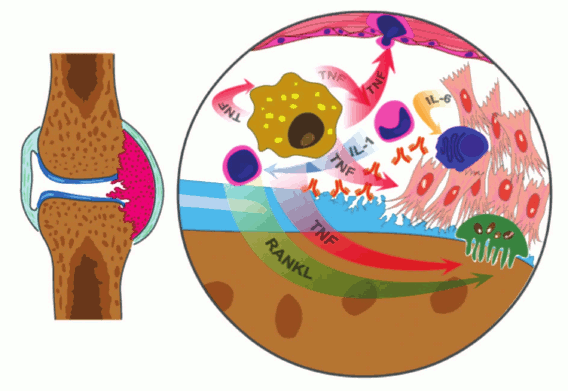
Fig. 5. Schematic representation the joint affected with RA, which is characterized by inflammation of the synovial lining. Macrophages, the main source of TNF, activate endothelial cells, which leads to increased local infiltration and activation of monocytes and T-cells, increased angiogenesis, and proliferation. TNF has a direct impact on osteoclasts, which leads to their activation and an increase in bone resorption with assistance from activated T-cells producing RANK ligand. The activated synovial fibroblasts are the main source of IL-6, which promotes differentiation of B-cells towards plasma cells producing large quantities of autoreactive antibodies.
When studying the mechanisms leading to the development of RA in humans and when testing new therapeutics, the contribution of experimental animal models is invaluable. There are a number of RA animal models available; however, due to a large variety of factors influencing the development of this disease, it is not possible to choose a uniform and universal model. In this article, we considered the existing experimental approaches to study anti-cytokine therapy in the treatment of autoimmune diseases. We established two experimental models of arthritis whose pathogenesis is dependent on TNF, including mice with the “humanized” tnf gene generated on C57Bl/6 genetic background. Mice with complete TNF ablation are resistant to collagen-induced arthritis. Studying the “humanized” hTNFKI mice in the context of experimental arthritis will in future allow more adequate assessment of the efficacy and side effects of different TNF inhibitors (existing, as well as newly developed). Treatment with TNF inhibitors significantly reduces the symptoms of the disease both in the model of arthritis using collagen-specific antibodies, as well as in collagen-induced arthritis. Finally, we developed and tested a new fluorescent TNF detector suitable for in vivo imaging of TNF expression in mice.
This work was supported by a Russian Federation government grant and the Russian Ministry of Education and Science grant (Contract No. 14.Z50.31.0008 of 19.03.2014 and Agreement No. 8493 of 07.09.2012), and also the grants of the Russian Foundation for Basic Research No. 13-04-91458 (S. A. Nedospasov) and No. 14-04-01656 (G. A. Efimov) and the President’s grant No. MD-3044.2014.4 (D. M. Chudakov).
REFERENCES
1.Kollias, G., Douni, E., Kassiotis, G., and
Kontoyiannis, D. (1999) The function of tumor necrosis factor and
receptors in models of multi-organ inflammation, rheumatoid arthritis,
multiple sclerosis and inflammatory bowel disease, Ann. Rheum.
Dis., 58, Suppl. 1, 132-139.
2.Probert, L., Keffer, J., Corbella, P., Cazlaris,
H., Patsavoudi, E., Stephens, S., Kaslaris, E., Kioussis, D., and
Kollias, G. (1993) Wasting, ischemia, and lymphoid abnormalities in
mice expressing T cell-targeted human tumor necrosis factor transgenes,
J. Immunol., 151, 1894-1906.
3.Probert, L., Plows, D., Kontogeorgos, G., and
Kollias, G. (1995) The type I interleukin-1 receptor acts in series
with tumor necrosis factor (TNF) to induce arthritis in TNF-transgenic
mice, Eur. J. Immunol., 25, 1794-1797.
4.Kontoyiannis, D., Pasparakis, M., Pizarro, T. T.,
Cominelli, F., and Kollias, G. (1999) Impaired on/off regulation of TNF
biosynthesis in mice lacking TNF AU-rich elements: implications for
joint and gut-associated immunopathologies, Immunity, 10,
387-398.
5.Kruglov, A. A., Kuchmiy, A., Grivennikov, S. I.,
Tumanov, A. V., Kuprash, D. V., and Nedospasov, S. A. (2008)
Physiological functions of tumor necrosis factor and the consequences
of its pathologic overexpression or blockade: mouse models, Cytokine
Growth Factor Rev., 19, 231-244.
6.Silva, L. C., Ortigosa, L. C., and Benard, G.
(2010) Anti-TNF-alpha agents in the treatment of immune-mediated
inflammatory diseases: mechanisms of action and pitfalls,
Immunotherapy, 2, 817-833.
7.Ehrenstein, M. R., Evans, J. G., Singh, A., Moore,
S., Warnes, G., Isenberg, D. A., and Mauri, C. (2004) Compromised
function of regulatory T cells in rheumatoid arthritis and reversal by
anti-TNFalpha therapy, J. Exp. Med., 200, 277-285.
8.Hess, A., Axmann, R., Rech, J., Finzel, S., Heindl,
C., Kreitz, S., Sergeeva, M., Saake, M., Garcia, M., Kollias, G.,
Straub, R. H., Sporns, O., Doerfler, A., Brune, K., and Schett, G.
(2011) Blockade of TNF-alpha rapidly inhibits pain responses in the
central nervous system, Proc. Natl. Acad. Sci. USA, 108,
3731-3736.
9.Bean, A. G., Roach, D. R., Briscoe, H., France, M.
P., Korner, H., Sedgwick, J. D., and Britton, W. J. (1999) Structural
deficiencies in granuloma formation in TNF gene-targeted mice underlie
the heightened susceptibility to aerosol Mycobacterium tuberculosis
infection, which is not compensated for by lymphotoxin, J.
Immunol., 162, 3504-3511.
10.Jacobs, M., Togbe, D., Fremond, C., Samarina, A.,
Allie, N., Botha, T., Carlos, D., Parida, S. K., Grivennikov, S.,
Nedospasov, S., Monteiro, A., Le Bert, M., Quesniaux, V., and Ryffel,
B. (2007) Tumor necrosis factor is critical to control tuberculosis
infection, Microbes Infect., 9, 623-628.
11.Kuprash, D. V., Tumanov, A. V., Liepinsh, D. J.,
Koroleva, E. P., Drutskaya, M. S., Kruglov, A. A., Shakhov, A. N.,
Southon, E., Murphy, W. J., Tessarollo, L., Grivennikov, S. I., and
Nedospasov, S. A. (2005) Novel tumor necrosis factor-knockout mice that
lack Peyer’s patches, Eur. J. Immunol., 35,
1592-1600.
12.Efimov, G. A., Khlopchatnikova, Z. V., Sazikin,
A. Y., Drutskaya, M. S., Kruglov, A. A., Shilov, E. S., Kuchmiy, A. A.,
Nedospasov, S. A., and Tillib, S. B. (2012) Isolation and
characteristics of a new recombinant single domain antibody that
specifically binds to human TNF, Russ. J. Immunol., 6,
337-345.
13.Shcherbo, D., Merzlyak, E. M., Chepurnykh, T. V.,
Fradkov, A. F., Ermakova, G. V., Solovieva, E. A., Lukyanov, K. A.,
Bogdanova, E. A., Zaraisky, A. G., Lukyanov, S., and Chudakov, D. M.
(2007) Bright far-red fluorescent protein for whole-body imaging,
Nat. Methods, 4, 741-746.
13a.Kuchmiy, A. A., Efimov, G. A., and Nedospasov, S. A. (2012) Methods
for in vivo molecular imaging, Biochemistry (Moscow),
77, 1339-1353.
14.Bevaart, L., Vervoordeldonk, M. J., and Tak, P.
P. (2010) Collagen-induced arthritis in mice, Methods Mol.
Biol., 602, 181-192.
15.Holmdahl, R., Jansson, L., Andersson, M., and
Jonsson, R. (1992) Genetic, hormonal and behavioral influence on
spontaneously developing arthritis in normal mice, Clin. Exp.
Immunol., 88, 467-472.
16.Brand, D. D., Latham, K. A., and Rosloniec, E. F.
(2007) Collagen-induced arthritis, Nat. Protoc., 2,
1269-1275.
17.Campbell, I. K., Hamilton, J. A., and Wicks, I.
P. (2000) Collagen-induced arthritis in C57BL/6 (H-2b) mice: new
insights into an important disease model of rheumatoid arthritis,
Eur. J. Immunol., 30, 1568-1575.
18.Londei, M., Savill, C. M., Verhoef, A., Brennan,
F., Leech, Z. A., Duance, V., Maini, R. N., and Feldmann, M. (1989)
Persistence of collagen type II-specific T-cell clones in the synovial
membrane of a patient with rheumatoid arthritis, Proc. Natl. Acad.
Sci. USA, 86, 636-640.
19.Kim, H. Y., Kim, W. U., Cho, M. L., Lee, S. K.,
Youn, J., Kim, S. I., Yoo, W. H., Park, J. H., Min, J. K., Lee, S. H.,
Park, S. H., and Cho, C. S. (1999) Enhanced T cell proliferative
response to type II collagen and synthetic peptide CII (255-274) in
patients with rheumatoid arthritis, Arthritis Rheum., 42,
2085-2093.
20.Terato, K., Shimozuru, Y., Katayama, K.,
Takemitsu, Y., Yamashita, I., Miyatsu, M., Fujii, K., Sagara, M.,
Kobayashi, S., Goto, M., Nishioka, K., Miyasaka, N., and Nagai, Y.
(1990) Specificity of antibodies to type II collagen in rheumatoid
arthritis, Arthritis Rheum., 33, 1493-1500.
21.Kim, W. U., Yoo, W. H., Park, W., Kang, Y. M.,
Kim, S. I., Park, J. H., Lee, S. S., Joo, Y. S., Min, J. K., Hong, Y.
S., Lee, S. H., Park, S. H., Cho, C. S., and Kim, H. Y. (2000) IgG
antibodies to type II collagen reflect inflammatory activity in
patients with rheumatoid arthritis, J. Rheumatol., 27,
575-581.
22.Watson, W. C., Tooms, R. E., Carnesale, P. G.,
and Dutkowsky, J. P. (1994) A case of germinal center formation by
CD45RO T and CD20 B lymphocytes in rheumatoid arthritic subchondral
bone: proposal for a two-compartment model of immune-mediated disease
with implications for immunotherapeutic strategies, Clin. Immunol.
Immunopathol., 73, 27-37.
23.Edwards, J. C., and Cambridge, G. (2001)
Sustained improvement in rheumatoid arthritis following a protocol
designed to deplete B lymphocytes, Rheumatology (Oxford),
40, 205-211.
24.Edwards, J. C., Szczepanski, L., Szechinski, J.,
Filipowicz-Sosnowska, A., Emery, P., Close, D. R., Stevens, R. M., and
Shaw, T. (2004) Efficacy of B-cell-targeted therapy with rituximab in
patients with rheumatoid arthritis, N. Engl. J. Med.,
350, 2572-2581.
25.Emery, P., Fleischmann, R., Filipowicz-Sosnowska,
A., Schechtman, J., Szczepanski, L., Kavanaugh, A., Racewicz, A. J.,
van Vollenhoven, R. F., Li, N. F., Agarwal, S., Hessey, E. W., Shaw, T.
M., and Group, D. S. (2006) The efficacy and safety of rituximab in
patients with active rheumatoid arthritis despite methotrexate
treatment: results of a phase IIB randomized, double-blind,
placebo-controlled, dose-ranging trial, Arthritis Rheum.,
54, 1390-1400.
26.Stuart, J. M., and Dixon, F. J. (1983) Serum
transfer of collagen-induced arthritis in mice, J. Exp. Med.,
158, 378-392.
27.Terato, K., Hasty, K. A., Reife, R. A., Cremer,
M. A., Kang, A. H., and Stuart, J. M. (1992) Induction of arthritis
with monoclonal antibodies to collagen, J. Immunol., 148,
2103-2108.
28.Williams, R. O., Inglis, J. J., Simelyte, E.,
Criado, G., and Sumariwalla, P. F. (2005) Analyzing the effect of novel
therapies on cytokine expression in experimental arthritis, Int. J.
Exp. Pathol., 86, 267-278.
29.Svensson, L., Jirholt, J., Holmdahl, R., and
Jansson, L. (1998) B cell-deficient mice do not develop type II
collagen-induced arthritis (CIA), Clin. Exp. Immunol.,
111, 521-526.
30.Nandakumar, K. S., Svensson, L., and Holmdahl, R.
(2003) Collagen type II-specific monoclonal antibody-induced arthritis
in mice: description of the disease and the influence of age, sex, and
genes, Am. J. Pathol., 163, 1827-1837.
31.Terato, K., Harper, D. S., Griffiths, M. M.,
Hasty, D. L., Ye, X. J., Cremer, M. A., and Seyer, J. M. (1995)
Collagen-induced arthritis in mice: synergistic effect of E.
coli lipopolysaccharide bypasses epitope specificity in the
induction of arthritis with monoclonal antibodies to type II collagen,
Autoimmunity, 22, 137-147.
32.Staines, N. A., and Wooley, P. H. (1994) Collagen
arthritis – what can it teach us? Br. J. Rheumatol.,
33, 798-807.
33.Holmdahl, R., Andersson, M. E., Goldschmidt, T.
J., Jansson, L., Karlsson, M., Malmstrom, V., and Mo, J. (1989)
Collagen induced arthritis as an experimental model for rheumatoid
arthritis. Immunogenetics, pathogenesis and autoimmunity, APMIS,
97, 575-584.
34.Holmdahl, R., Mo, J. A., Jonsson, R., Karlstrom,
K., and Scheynius, A. (1991) Multiple epitopes on cartilage type II
collagen are accessible for antibody binding in vivo,
Autoimmunity, 10, 27-34.
35.Mo, J. A., Scheynius, A., Nilsson, S., and
Holmdahl, R. (1994) Germline-encoded IgG antibodies bind mouse
cartilage in vivo: epitope- and idiotype-specific binding and
inhibition, Scand. J. Immunol., 39, 122-130.
36.Abbas, A. K., Lichtman, A. H., and Pober, J. S.
(1997) Immune-mediated tissue injury and disease, in Cellular and
Molecular Immunology, Saunders, Philadelphia, pp. 423-438.
37.Colten, H. R. (1994) Immunology. Drawing a
double-edged sword, Nature, 371, 474-475.
38.Ravetch, J. V., and Clynes, R. A. (1998)
Divergent roles for Fc receptors and complement in vivo,
Annu. Rev. Immunol., 16, 421-432.
39.Hietala, M. A., Nandakumar, K. S., Persson, L.,
Fahlen, S., Holmdahl, R., and Pekna, M. (2004) Complement activation by
both classical and alternative pathways is critical for the effector
phase of arthritis, Eur. J. Immunol., 34, 1208-1216.
40.Wang, Y., Rollins, S. A., Madri, J. A., and
Matis, L. A. (1995) Anti-C5 monoclonal antibody therapy prevents
collagen-induced arthritis and ameliorates established disease,
Proc. Natl. Acad. Sci. USA, 92, 8955-8959.
41.Wang, Y., Kristan, J., Hao, L., Lenkoski, C. S.,
Shen, Y., and Matis, L. A. (2000) A role for complement in
antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant
to collagen-induced arthritis, J. Immunol., 164,
4340-4347.
42.Grant, E. P., Picarella, D., Burwell, T.,
Delaney, T., Croci, A., Avitahl, N., Humbles, A. A., Gutierrez-Ramos,
J. C., Briskin, M., Gerard, C., and Coyle, A. J. (2002) Essential role
for the C5a receptor in regulating the effector phase of synovial
infiltration and joint destruction in experimental arthritis, J.
Exp. Med., 196, 1461-1471.
43.Schindler, R., Gelfand, J. A., and Dinarello, C.
A. (1990) Recombinant C5a stimulates transcription rather than
translation of interleukin-1 (IL-1) and tumor necrosis factor:
translational signal provided by lipopolysaccharide or IL-1 itself,
Blood, 76, 1631-1638.
44.Takabayashi, T., Vannier, E., Clark, B. D.,
Margolis, N. H., Dinarello, C. A., Burke, J. F., and Gelfand, J. A.
(1996) A new biologic role for C3a and C3a desArg: regulation of
TNF-alpha and IL-1 beta synthesis, J. Immunol., 156,
3455-3460.
45.Choy, E. (2012) Understanding the dynamics:
pathways involved in the pathogenesis of rheumatoid arthritis,
Rheumatology (Oxford), 51, Suppl. 5, 3-11.
46.Smolen, J. S., and Steiner, G. (2003) Therapeutic
strategies for rheumatoid arthritis, Nat. Rev. Drug Discov.,
2, 473-488.
47.Hochberg, M. C., Johnston, S. S., and John, A. K.
(2008) The incidence and prevalence of extra-articular and systemic
manifestations in a cohort of newly-diagnosed patients with rheumatoid
arthritis between 1999 and 2006, Curr. Med. Res. Opin.,
24, 469-480.
48.Dayer, J. M., and Choy, E. (2010) Therapeutic
targets in rheumatoid arthritis: the interleukin-6 receptor,
Rheumatology (Oxford), 49, 15-24.
49.Pollard, L., Choy, E. H., and Scott, D. L. (2005)
The consequences of rheumatoid arthritis: quality of life measures in
the individual patient, Clin. Exp. Rheumatol., 23,
43-52.