REVIEW: Induced Pluripotent Stem Cells: From Derivation to Application in Biochemical and Biomedical Research
E. V. Novosadova and I. A. Grivennikov*
Institute of Molecular Genetics, Russian Academy of Sciences, 2 Kurchatov Sq., 123182 Moscow, Russia; E-mail: igorag@img.ras.ru; grivigan@mail.ru* To whom correspondence should be addressed.
Received September 12, 2014
This review considers different methods for obtaining induced pluripotent stem (iPS) cells and their use in biochemical and biomedical research. Some viral and nonviral methods for obtaining iPS cells are described. Basic factors involved in reprogramming are considered. It is also demonstrated that the most suitable source of iPS cells are skin fibroblasts. Properties of iPS cells and embryonic stem cells are compared, and some advantages of iPS cells for biological and biomedical investigations are emphasized. The possibilities for application of iPS cells in the development of cell models of some neurodegenerative diseases, drug screening, and cell therapy are also considered.
KEY WORDS: induced pluripotent stem cells, embryonic stem cells, reprogramming, differentiation, transcriptional factors, cell therapy, human diseases, neurodegenerationDOI: 10.1134/S000629791413001X
Abbreviations: ES cells, embryonic stem cells; iPS cells, induced pluripotent stem cells; LIF, leukemia inhibitory factor; MEF, mouse embryonic fibroblasts; PD, Parkinson disease; SNCA, alpha-synuclein; TF, transcription factor.
The latest discoveries in the fields of molecular genetics and cell
biology offer new opportunities for the study of the molecular
framework underlying the progression of several serious diseases of
humans, including the neurodegenerative diseases, as well as for
development of approaches for cell therapy of these medical conditions.
Until recently, it was believed that adult somatic cells that have
undergone their developmental program during embryogenesis could not be
reverted to their initial undifferentiated state (Fig. 1).
Fig. 1. Schematic representation of differentiation of somatic cells during the development of an organism.
Different manipulations with somatic cells cause their partial dedifferentiation accompanied by partial or complete malignization, which is schematically represented in Fig. 1. However, data obtained in the last decade indicate the possibility of reversal of the process of differentiation of mammalian cells. The prime objective is to reprogram human somatic cells into pluripotent cells capable of yielding adult cell types upon differentiation for possible future application in cell therapy approaches for treatment of neurodegenerative diseases.
In 2006, Japanese researchers Takahashi and Yamanaka [1] reprogrammed embryonic and adult mouse fibroblasts into pluripotent stem cells by viral transduction of four genes encoding transcription factors (TF) Oct3/4, Sox2, c-Myc, and Klf4. They named these cells induced pluripotent stem (iPS) cells. The cells obtained by the described method shared growth properties and morphological characteristics with embryonic stem (ES) cells and expressed ES cell-specific markers. In as little as one year, Takahashi et al. [2] followed by Nakagawa et al. [3] from the same laboratory of Kyoto University reported successful reprogramming of adult human fibroblasts and derivation of human iPS cells using the same factors (Oct4, Sox2, c-Myc, and Klf4). In this article, we will review the methods for derivation of iPS cells and compare their properties with ES cells. We will discuss possible future applications of iPS cells for studying biochemical cues underlying several serious diseases of the human nervous system and for the development of cell therapy approaches for treatment of these diseases.
METHODS FOR DERIVATION AND PROPERTIES OF INDUCED PLURIPOTENT STEM
CELLS
Embryonic Stem Cells
First, we turn our attention to several properties of ES cells. In 1998, James Thompson and his colleagues from the University of Wisconsin-Madison reported the generation of the first human ES cell line [4]. These cells were derived from inner cell mass of the blastocyst stage embryos, and they were propagated on feeder layers of mouse embryonic fibroblasts (MEFs). It is known that MEFs secrete all growth factors that are essential for maintenance and self-renewal of ES cells (LIF, FGF, TGFβ, Activin, Wnt, etc.) [5, 6]. Subsequently, a feeder-free method was developed for propagation of human ES cells using Matrigel, CELLstart, and several other artificial substrates [7-9]. To date, the most popular and efficient protocols for propagation of human ES cells utilize either fibroblast feeder layers or Matrigel and mTeSR medium, which contain all components that are essential for maintenance of the pluripotent state of human ES cells. Under these conditions, ES cells grow as dense, usually round-shaped colonies. The size of the cells is approximately 20 µm. Human ES cells demonstrate high nucleus-to-cytoplasm ratio, distinct nucleoli, perinuclear localization of mitochondria, low ATP level, high levels of oxygen consumption, and low levels of mitochondrial DNA [10-13].
ES cells have a unique pattern of histone modifications: the extensive regions upstream of genes involved in early development are occupied by histone H3 trimethylated at lysine 27 (H3K27me3, a repressed chromatin mark) surrounded by less extended regions of histone H3 dimethylated at lysine 4 (H3K4me2, an active chromatin mark). Such special chromatin domains were named “bivalent”. This results in an intermediate state of early development genes that are neither activated nor completely repressed [14, 15]. Perhaps the hyperdynamic structure of chromatin in ES cells is explained by very short times of interaction of various histone modifications with chromatin (from several seconds or minutes) [16]. It is noteworthy that DNA domains with “bivalent” chromatin structure were also observed in somatic cells, but in this case the repressive H3K27me3 mark dominates over the H3K4me2 activator mark. ES cells have the capacity for indefinite in vitro proliferation, retaining their normal karyotype. In these cells the internal signal cascades are blocking differentiation and supporting the presence of active self-renewal promoting TFs.
Several primary TFs essential for maintenance of pluripotent state were identified in the end of the last century – namely Oct3/4 (POU5F1), Sox2, and Nanog [17-19]. These work closely with each other and with a variety of other genes. Some of these genes are responsible for maintenance of pluripotency of ES cells; others are involved in differentiation in the ectodermal, endodermal, and mesodermal directions as well as to extraembryonic lineages. The list of genes actively transcribed in ES cells includes properly the ones encoding Oct3/4, Sox2, and Nanog, the genes encoding TFs STAT3, Zic3, Hesx1, and Esrrb, as well as the genes encoding chromatin-remodeling proteins (SET, MYST3), neurogenesis inhibitors (Rest), and proteins related to telomeres (Rif2 complex with other proteins protects chromosome ends from degradation). The genes inactive in ES cells are particularly those involved in the differentiation process [20]. Despite the open chromatin organization, the DNA methylation level in ES cells is higher than in differentiated somatic cells [21]. ES cells are also distinct in their pattern of DNA methylation; in 99.98% of cases, the CpG islands are methylated in fibroblasts, while in ES cells only 75% of these are methylated.
Induced Pluripotent Stem Cells
The reprogramming of somatic cells using a set of TFs was first achieved by Japanese researchers Takahashi and Yamanaka in 2006 [1]. That discovery founded a new line of research in developmental biology. Yamanaka’s laboratory work was focused on a search for the factors involved in the maintenance of pluripotency in ES cells. Several dozen genes were identified whose activity was significantly elevated compared to the level in adult differentiated cells. It was already known that fusion of ES cell and a specialized adult cell could generate a pluripotent cell [22].
The Japanese scientists tested combinations of 24 TFs involved in maintenance of pluripotency in ES cells to select a set of factors essential for reprogramming of somatic cells. By infecting somatic cells with retroviruses bearing various combinations of TFs, it was found that four of these factors (Oct4, Sox2, Klf4, and c-Myc) are necessary and sufficient to induce the pluripotent state in MEF cells. As early as in 2007, Takahashi and Yamanaka published data on derivation of iPS cells from adult human skin fibroblasts [2]. The cells obtained by this method shared morphology, growth properties, and specific marker expression with traditional ES cells.
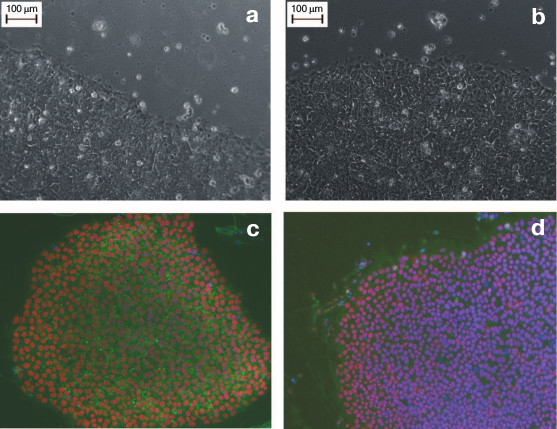
Figure 2 (a and b) shows the similar morphology of human ES and human iPS cells obtained from a healthy donor. The size of iPS cells is approximately 20 µm, and like ES cells, the iPS cells are characterized by high nucleus/cytoplasm ratio. The iPS cells grow as flat monolayer colonies with tight contacts between adjacent cells.
Fig. 2. Human ES and iPS cells in vitro. a) Human ES cell colony of HUES9 line. b) A colony of human iPS cells derived from fibroblasts of a healthy donor. Magnification ×200. c, d) Immunocytochemical staining of pluripotency markers in iPS cells; c) Oct4 (red), SSEA4 (green), DAPI (blue); d) Sox2 (red), TRA-1-61 (green), DAPI (green). Magnification ×100.
ES cells represent the “gold standard” for iPS cells; in fact, iPS cells are in vitro derived ES cells. Thus, the two types of cells demonstrate similarity, or even identity in their morphological, molecular, immunocytochemical, and functional characteristics. Undifferentiated iPS cells are characterized by expression of conventional surface markers such as proteoglycans TRA-1-60, TRA-1-81, glycolipid SSEA4, and, to a lesser extent, the glycolipid SSEA3 [2]. SSEA-4 and SSEA-3 are definitive only for human and primate pluripotent cells, while murine ES and iPS cells bear the specific surface antigen SSEA-1 [1].
The intracellular markers for the undifferentiated state are represented by TFs that are essential for maintenance of pluripotency, such as Oct4, Nanog, and Sox2 [1, 2]. ES and iPS cells are also similar in their epigenetic profiles. For instance, the promoters of the Oct4 and Nanog genes are demethylated in both types of pluripotent cells. Figures 2c and 2d show data on expression of several pluripotency-associated markers in human iPS cells.
Transcription factors involved in reprogramming. Here we briefly describe the properties of each of the four transcription factors that were identified by Yamanaka’s group [1, 2].
Transcription factor Oct. This TF, also known as Oct3/4(POU5F1), appears to be a crucial element in the regulation of pluripotency and in controlling cell differentiation [23]. The homeodomain-containing protein encoded by Oct4(pou5f1) gene binds the DNA sequence 5′-ATGCAAT-3′, causing either activation or suppression of transcription depending on sequences flanking the binding site. In human and mouse embryos, Oct4 expression is localized in the inner mass of the blastocyst [24], Oct4-knockout embryos die after the blastocyst stage. Pluripotent stem cells are sensitive to variations in protein Oct4 level. Down-regulation of Oct4 expression in ES cells causes spontaneous differentiation to trophoblast phenotype, while its increased expression induces formation of primitive endoderm and mesoderm [25-27]. Such influence is possibly explained by connection of Oct4 with its target genes. For example, Hand is responsible for development of early trophectoderm, Spp1 encodes osteopontin protein that is expressed in developing primitive ectoderm, Fbx15 and FGF4 are expressed in inner cell mass (ICM), etc. [28]. Oct4 is a classical protooncogene, its aberrant expression leading to dysplastic growth and formation of various types of tumors [29, 30]. Oct4 also influences the tumorigenicity of ES cells in vitro, since the elevation of Oct4 expression induces malignization in normal cells [25].
Transcription factor Sox2. Sox2 (SRY, sex-determining region Y-box2) belongs to Sox family of TFs involved in regulation of various stages of cell development and differentiation. Proteins of this family bear a highly conserved DNA-binding domain known as HMG (high-mobility group), which on average consists of 80 amino acid residues. This group of proteins is involved in regulation of transcription and chromatin architecture. Sox2 forms a physical complex with Oct4 that is involved in regulation of expression of UTF1, Fgf4, and Fbx15 genes that are essential for maintenance of pluripotency of ES cells [19, 31, 32]. Homozygous Sox2–/– embryos do not survive to the epiblast stage and die at the implantation stage. Sox2 knockout in ES cells induces their differentiation into various cell types, including trophectoderm, which indicates an important role of Sox2 in maintenance of pluripotency [33, 34]. Sox2 is also a protooncogene. Aberrant expression of this gene can lead to development of breast cancer, small-cell lung cancer, and prostate cancer [35, 36].
Transcription factor Klf4. Klf4 belongs to of the Kruppel-like family of transcription factors. Proteins of this family are involved in various processes including embryonic development, cell proliferation, differentiation, and apoptosis [37]. In experiments on mice, a sequential expression of this gene was demonstrated in embryonic development. Thus, initially the expression of Klf4 is detectable in extraembryonic tissues, then in digestive tract, and finally in developing layers of embryonic skin [38-40].
In adult animals, Klf4 expression is observed in digestive tract and skin as well as in terminally differentiated epithelial tissues [39, 40]. Of interest, Klf4 demonstrates high expression level in non-dividing cells and nearly zero expression in actively proliferating cells [41]. Murine embryos with Klf4 gene knockout develop normally but die soon after birth because of a defect in skin protective function [39].
Decreased Klf4 protein level does not manifest itself phenotypically; however, simultaneous inhibition of several members of the Klf family induces differentiation of ES cells. Based on these data, it was proposed that other proteins of the Klf family could act redundantly to mitigate the absence of Klf4 expression [28, 42]. In contrast to Oct4 and Sox2, Klf4 was identified both as a potential oncogene and as tumor suppressor. Klf4 is associated with development of esophageal, intestinal, and breast cancer [43-47].
Transcription factor c-Myc. c-Myc is a multidomain TF involved in processes of proliferation, differentiation, and cell growth [48, 49]. It was demonstrated that c-Myc is involved in regulation of more than 10% of all known genes [48, 50]. Consequently, approximate calculations reveal more than 2500 possible genomic binding sites for c-Myc [51]. Besides its involvement in transcription of genes encoding specific proteins, c-Myc also participates in regulation of noncoding genes of microRNA [52, 53].
c-Myc knockout murine embryos normally proceed through early stages, but at later stages abnormalities in development of the neural tube, heart, and blood vessels emerge, which leads to embryonic lethality on day 10 [54, 55]. Curiously, ES cells bearing c-myc knockout proliferate normally and are capable of self-renewal in vitro [54]. c-Myc, like all other TFs listed above, is a protooncogene. Its elevated expression is observed in more than 70% of various human tumors, making c-myc one of the most commonly detected tumor markers [48].
It should be noted that iPS cells could be generated not only by Oct4, Sox2, Klf4, and c-Myc, but also by other TF combinations. Thus, soon after the first communication on generation of iPS cells by “Yamanaka cocktail”, data were published on possible substitution of Klf4 and c-Myc with Nanog and Lin28 [56], while Sox2 and Klf4 can be replaced with Sox1 and Klf2 [3].
The following developments in the field of iPS cell generation were pointed, in particular, towards improvement of effectiveness of reprogramming and the search for new reprogramming approaches that preclude the use of a genome-integrating vector based on lentiviral and retroviral sequences.
iPS Cell Derivation Methods
Most of the established methods for iPS cell derivation are presented in Fig. 3.
Fig. 3. Several methods for reprogramming of mammalian cells.
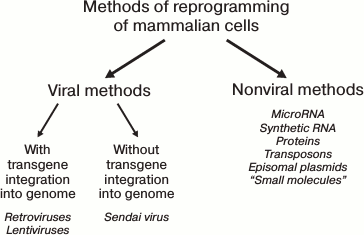
All known reprogramming methods can be conventionally divided into viral-based and nonviral-based. In turn, the viral-based methods can utilize genome-integrating and non-integrating vectors. Historically, the viral-based methods were the first to be developed, utilizing retroviral [1, 2] or lentiviral [56] vectors. With these methods, transgenes are integrated into random regions of the target cell genomic DNA with random copy number. Because of their expression, reprogramming is induced. Subsequently, as the cells progress through the stages of the reprogramming process, the transgenic expression should become silenced due to induction of corresponding genes of the target cell and activation of histone methyltransferases [57]. The transgene silencing is expected to occur in a strictly defined period. In the case of early down-regulation, the reprogramming would not take place at all, or it will not proceed to completion, generating partially reprogrammed cells that depend on exogenic expression [58]. At the same time, the constitutively active transgenes can affect the differentiation potential of the derived iPS cells and cause the emergence of tumors in chimeric animals [59]. One approach to solve the problem of transgenic silencing at the appropriate time is the use of inducible vectors. For instance, the widely used Dox-inducible system based on lentiviral vectors of TFs allows controlling the level of transgenic TF expression by doxycycline added to the culture medium. This method has its specific limitations. First, “leaking” is common to inducible the expression system, with some transgenic expression observed even in the absence of activator. Second, virtually all cell lines require empirical identification of doxycycline concentration and optimal time for transgene silencing. For fibroblasts and keratinocytes, the optimal durations are established, being 16 and 10 days, respectively [60]. By using the Dox-system in mice, chimeras were obtained in which the transgenes were silenced in all tissues. The cells of the mice that were deficient in one of the essential transgenes were used for screening of small molecules that can substitute for the corresponding TF in the reprogramming process [61].
The solution of the integration problem can be solved by using DNA recombinase enzymes. Viral vectors were constructed in which the transgenes were flanked by LoxP sites [62, 63]. The DNA sequence between two direct LoxP repeats can be excised by Cre-mediated recombination. The Cre recombinase can be delivered into the cell nucleus by using Pseudomonas aeruginosa bacteria, which cause injection of proteins with specific N-terminal sequence into the cells followed by the transport of the protein into the nucleus [64].
Vector systems based on episomal plasmids do not require being integrated into the genome, and, thus, they are considered potentially safer for generation of iPS cells to be used for cell therapy of human diseases. The use of the combination of plasmids bearing Oct3/4, Sox2, Klf4, c-Myc, Lin-28, and small interfering RNA in combination with EBNA1 (Epstein–Barr nuclear antigen 1, essential for amplification of episomal vectors) improves several-fold the reprogramming efficiency [65]. Recently, the use of single-stranded RNA Sendai virus, of the Paramyxoviridae family, has become an increasingly popular method of reprogramming [66-68]. The Sendai virus does not integrate itself into the genomic DNA of the cell. Moreover, it can be removed from iPS cells by incubating them at 39°C. Completely reprogrammed cells are usually obtained 25 days after infection. As with the other methods, the effectiveness of reprogramming greatly varies depending on the cell type used in the experiment. For example, for fibroblasts the effectiveness of reprogramming on average is 1%, which presents a rather good index, and for blood cells is 10 times lower, about 0.1% [68].
Another nonviral method of delivery of genes, which is suitable for various types of somatic cells, is the use of DNA-transposons. DNA-transposons are mobile genetic elements capable of moving inside the genome, and they also possess the capacity to “cut and paste” using specific transposase enzyme [69]. Transposons consist of insertion DNA segments able to move inside the genome as a whole element, carrying together the interjacent genes. To date, several transposon systems for delivery of genes into mammalian cells have been described: Sleeping Beauty (SB), SB100X, PiggiBag (PB), and Tol2 [70-72]. The use of the PiggiBag-based vector bearing a DNA cassette encoding four TFs (Oct4, c-Myc, Klf4, Sox2), plus Rarg (retinoic acid receptor-gamma) and LRH-1 (liver receptor homolog 1), allowed successful generation of both mouse and human iPS cells. The last two receptor genes were included not by accident; LRH-1 is an important element in regulation of transcription of genes, and also an essential factor in maintenance of pluripotency. Rarg and LRH-1 cooperatively act to induce the expression of Oct4. All these effects contributed to improvement of the reprogramming efficiency.
Another approach to overcome the limitations associated with introduction of viral sequences into reprogrammed cells, as well as with excessive activity of pluripotency-inducing genes, is the use of microRNA [73, 74]. It is known that several microRNAs are highly expressed in ES cells and play an important role in control of the activity of the genes associated with maintenance of pluripotency. For example, microRNA from the miR-302 family interacts with p52 and promotes progression through G1/S phase of the cell cycle. miR-195 is known to associate with WEE1 kinase (a negative regulator of cyclin B/CDK complex) and facilitates progression through G2/M phase [75, 76]. Utilization of microRNA, in particular miR-93 that is able to suppress the TGF-β II receptor, considerably improves reprogramming efficiency [77]. Combined use of TFs and microRNAs of the miR-302 family dramatically improves the efficiency of reprogramming of human fibroblasts [74]. Mioshi et al. achieved reprogramming of human and mouse cells by mature microRNA and named the resulting cells “miRIPSC” [78]. The use of a cocktail of miRNAs (miR-302-367) without introduction of additional TFs also yielded iPS cells [79].
Another method for producing iPS cells without interference with cell genomic DNA is the use of RNA of corresponding TFs. Warren et al. reprogrammed human neonatal fibroblasts with 1.4% efficiency and were able to further elevate it to 4.4% by adding another TF, Lin28, to a standard cocktail of four TFs and combining it with a methyltransferase inhibitor (valproic acid). For this purpose, the reprogramming of fibroblasts was performed at lowered O2 level (5%), which is a hypoxic condition [80]. However, this method has not become common in practice due its technical difficulty (the use of modified RNA) and its cost.
Several laboratories have successfully reprogrammed human [81] and mouse [82] fibroblasts utilizing recombinant proteins of corresponding TFs, carrying “tat” peptide of HIV-1 and polyarginine, which are necessary for delivery of these proteins into the cell [82]. The reprogramming efficiency under those conditions was rather low (0.006% for mouse and 0.001% for human fibroblasts). Technical difficulties and low efficiency leave in doubt the development of this method in the near future.
Attempts have been made to find so-called “small molecules” that can improve the efficiency and time of reprogramming, many of which influence epigenetic status of the cells and promote decondensation of chromatin [83]. For instance, it was shown that the simultaneous application of Bix-01294 (an inhibitor of histone methyltransferase) and BayK8644 (a calcium channel activator) makes it possible to exclude two TFs and to use vectors bearing only the Oct4 and Klf4 genes in the reprogramming procedure [84]. It was found that it is sufficient to use only valproic acid (histone deacetylase inhibitor) in combination with Oct4 and Sox2 to generate iPS cells from human fibroblasts [85]. Valproic acid also improved the efficiency of reprogramming during iPS cell preparation by recombinant proteins of proper TFs, as indicated above [82]. Molecules SB432542 and PD0325901, which are inhibitors of the TGFβ and MEK signaling pathways, respectively, can promote the efficiency of reprogramming up to 100-fold when used in combination with the four main TFs (Oct4, Sox2, Klf4, c-Myc) [86].
In 2013, the reprogramming of mouse fibroblasts was accomplished utilizing exclusively “small molecules”. The effectiveness of this method was about 0.2%, which is comparable to the effectiveness obtained with viral vectors. The generated cells were named “chemically-induced pluripotent stem” (CiPS) cells [87].
For a rather long period of time, it was unclear why the effectiveness of reprogramming is so low (from 0.002 to 2-4%) even when an excess of reprogramming factors (viruses, plasmids, RNAs, small molecules, etc.) are applied to cells. Only recently, researchers in Israel were able to reach nearly 100% effectiveness of reprogramming of human fibroblasts [88]. It was found that an essential requirement of successful reprogramming depends on suppression of the nucleosome remodeling and deacetylation (NuRD) protein complex, which is expressed in all somatic cells [89]. Mbd3 protein is one of the subunits of this complex. It is possible that Mbd3/NuRD is a specific epigenetic regulator that limits the expression of key pluripotency genes. Thus, it was shown that increased expression of Mbd3 prevents the derivation of iPS cells. This is caused by deacetylation of Lysine 27 in the histone H3 molecule by the above complex. As a result, another complex, called PRC2 (polycomb repressive complex 2), causes trimethylation of this lysine residue of H3 histone, leading to inhibition of several pluripotency-related genes including Oct4 and Nanog [90]. On the other hand, the suppression of Mbd3 promotes the reprogramming efficiency and favors the derivation of pluripotent stem cells able to contribute to viable chimeric mice, even in the absence of c-Myc and Sox2 [91].
Hanna and colleagues showed that using the four classic TFs (Oct3, Sox2, Klf4, c-Myc) in combination with inhibition of Mbd3 expression permits determined and synchronized reprogramming of skin cells of mouse and human into iPS cells in seven days with almost 100% efficiency [89]. Therefore, we conclude that the problem of effective reprogramming of mammalian cells is solved. It should be noted, however, that even with reprogramming at low efficiency of 0.01 to 0.1% (the first works on derivation of iPS cells) by using, for example, 1,000,000 fibroblasts in viral transfection, from 100 to 1000 independent iPS cell clones can be generated, which is more than sufficient for further experiments.
Sources for Derivation of iPS Cells
Although fibroblasts are the most popular source for preparation of iPS cells, it must be noted that other somatic cells can be successfully reprogrammed as well. Thus, iPS cells have been derived from neural stem cells [92], endothelial cells [93], keratinocytes [60], and terminally differentiated lymphocytes [94], hepatocytes [60], epithelial cells of stomach [95], hair follicles [96], cells of conjunctiva [97], and mononuclear cells of peripheral blood [98]. The present variety of sources for iPS cell preparation shows that iPS cells can be derived from any type of somatic cells of an adult organism, but the preference, especially in the case of humans, would rely on accessibility of the corresponding donor material. In this case, skin fibroblasts and keratinocytes are beyond competition.
One important question related to reprogramming is whether the reversion of pluripotency of a cell is a stochastic process, or is it related to a sequence of events? To answer that question, corresponding investigations on reversion of the pluripotent state of the cells were carried out. The changes occurring in fibroblast cells during reprogramming were analyzed at different stages. It was found that after three days of viral infection, the silencing of fibroblast-specific genes ensued with simultaneous activation of ES cell-specific genes (alkaline phosphatase, SSEA1, Fbx5). From 10 to 15 days after transfection, endogenous expression of Oct4, Sox2, and Nanog can be observed. Simultaneously with increased expression of these genes, reactivations of telomerase and of the second X chromosome in female cells occurs [99]. At this stage, the reprogramming process becomes less dependent on viral expression of transgenes; however, these cells are still not completely reprogrammed, and a decreased level of transgenic expression leads to inevitable reversal to a differentiated fibroblast phenotype [99]. When the level of endogenous Oct4, Sox2, and Nanog will increase, the stable functioning of the autoregulatory loop of endogenous pluripotency-related genes is established, and reactivation of the ES-specific transcriptional network occurs along with transgene silencing.
Based on the above-mentioned and other publications, Scheper and Copray proposed their own model of reprogramming in their review, which was named “two-stage switch” [100]. In the first stage, the suppression of expression of lineage-specific genes and removal of repressive epigenetic marks from pluripotency-associated genes occurs. In the second stage of reprogramming, reactivation of an endogenous autoregulatory loop occurs, as well as the induction of the main transcriptional network underlying the pluripotent state. Exogenous Oct4 and Sox2 became able to activate endogenous regulatory sites of Oct4, Sox2, and Nanog. Full epigenetic remodeling of other pluripotency genes leads to their activation and restoration of the transcriptional network, thus characterizing the pluripotent state of cells. Finally, complete silencing of transgenes takes place, and the pluripotency becomes dependent on the autoregulatory loop of the endogenes. Thus, it was demonstrated that overexpression of a limited number of key factors can drive cells into a new stable state associated with changes in activity of hundreds of genes in the process of the reprogramming. During this, the telomeres of chromosomes elongate, and their normal shortening during differentiation of iPS cells back into fibroblasts occurs [101].
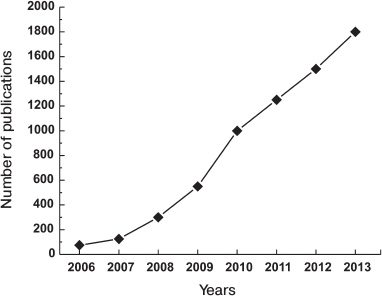
The discovery of Yamanaka and his colleagues was the most important fundamental discovery in biology in the beginning of XXI century, and thus it was recognized by the Nobel Prize in 2012. The importance and the potential of the current findings are shown by the growth of the number of publications on this subject, which is represented in Fig. 4.
Fig. 4. Growth of number of iPS cell related publications.
Similarities and Differences between ES and iPS Cells
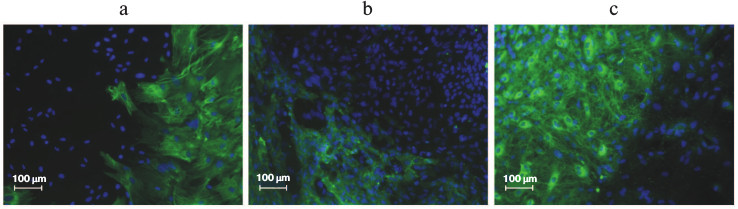
As mentioned above, completely reprogrammed cells and ES cells express the same pluripotency markers. In addition to protein markers, there are functional assays for pluripotency. The most common functional assay is the spontaneous in vitro differentiation of ES cells. After cultivation of human ES cells in the absence of feeder of mouse embryonic fibroblasts, the formation of embryoid bodies occurs, and during their further cultivation the expression of markers of endoderm, mesoderm, and ectoderm can be detected in the differentiating cells [102]. The same picture is observed during in vitro differentiation of iPS cells. Figure 5 depicts immunofluorescent detection of ectodermal (a), mesodermal (b), and endodermal (c) markers after spontaneous differentiation of iPS cells in vitro.
Fig. 5. Spontaneous differentiation of iPS cells in vitro with formation of representatives of three germ layers: ectoderm (a), mesoderm (b), endoderm (c).
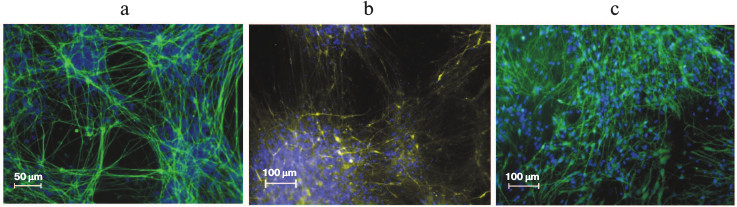
The only possible in vivo assay for pluripotency for human ES and iPS cells is teratoma formation after injection of cells into immunodeficient SCID mice. In the derived teratomas, differentiated tissues such as gut epithelium (endoderm), cartilage, bone and smooth muscle (mesoderm), as well as neural epithelium (ectoderm) can be observed [2, 103]. Both ES and iPS cells obtained from differentiated mouse cells, aside from their ability to contribute to somatic lineages in chimeric animals, can also participate in formation of germ line [82, 104]. The iPS cells completely replicate the capacity of ES cells for in vitro differentiation into specialized cell types. Thus, Fig. 6 shows neurons that were derived from human iPS cells by differentiation under specific conditions.
Fig. 6. Neural differentiation of iPS cells in vitro. Immunofluorescence detection: a) DAPI (blue), βIII-tubulin (green); b) DAPI (blue), dopaminergic neurons (yellow); c) DAPI (blue), GABAergic neurons (green).
As noted above, ES and iPS cells are almost identical in morphological and functional characteristics; however, the social aspects of these two pluripotent types of cells, as well as their potential applications, are significantly different. ES cells have several drawbacks that limit their potential use in medical and research applications. Some ES and iPS cell-specific attributes are presented in the table.
Comparison of human ES and iPS cells
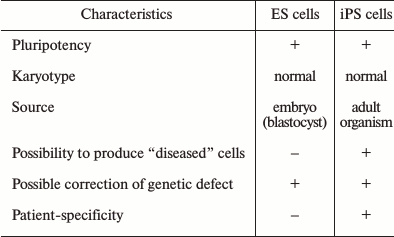
Indeed, both ES and iPS cells possess pluripotency – the capability to differentiate into all known cell types of an adult organism. Both cell types can grow indefinitely under specific in vitro conditions, and what is important, retain their normal karyotype. The principal differences between the two cell types lie in their sources: ES cells are derived from blastocysts, while iPS cells are derived from cells of an adult organism. In addition, of course, an important advantage of iPS cells is the possibility to derive “diseased” cells, which are the cells from patients with various health problems, including hereditary diseases. In addition, iPS cells provide individualization and patient-specificity, which is very important for the development of approaches for individual cell therapy.
Concerning cell therapy, one of the main drawbacks of ES cells should be noted – the possible development of immune response to a transplant. Since a limited number of ES cell lines exist, it might be not possible to find a transplant that would match a host in all leukocyte antigens (HLAs). A possible way to overcome that limitation is the use of human ES cells, genetically modified with recipient HLA genes. However, this approach is complex and labor intensive, and it is not known how many clones would be necessary. Another possible alternative is the generation of a patient-specific ES cell line by nuclear transfer from a donor’s somatic cell to an enucleated oocyte. This method bypasses the problem of immune rejection of a transplant; however, it is inevitably associated with a generation of a human embryo, and its further disruption for generation of ES cells. Thus, the current method combines both human cloning and human embryo destruction, two highly questionable procedures for ethical and political concerns [105]. The legal status of the human embryo and possibility or impossibility of destroying it are discussed by politicians, experts in bioethics, human right activists, and publicists over long periods of time. In several countries, manipulations with human embryo are legally forbidden.
Finally, the iPS cells open the opportunity (as well as ES cells) to correct genetic defects associated with several hereditary diseases by the use of homologous recombination.
SEVERAL ASPECTS OF APPLICATION OF INDUCED PLURIPOTENT STEM CELLS
IN BIOCHEMICAL AND BIOMEDICAL RESEARCH
Let us now review the problems related to the use of iPS cells in biological and medical research. Figure 7 schematically illustrates the stages of iPS cell creation, along with some of their possible applications [106].
Fig. 7. Generation and various applications of iPS cells (original figure from [106] is presented with some modifications).
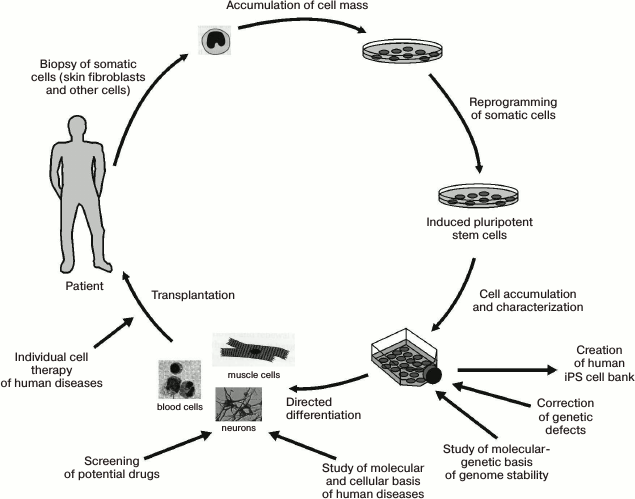
First, iPS cells represent a unique object for creation of a cell bank that can be used for basic research, and for future considerations, for individualized cell therapy of a specific patient. In addition, after the development of standardized protocols these cells will also find their use in test systems for screening of novel therapeutic drugs. Currently, works on creation of iPS cell cryobanks are underway in Japan, USA, Russia, and several other countries [107].
Testing of Novel Pharmaceutical Drugs
One pressing problem of modern pharmacology and medicine is the creation of safe and effective drugs for treatment of specific medical conditions. For their creation, adequate, relatively inexpensive, and reproducible high-throughput technologies for screening of potential drugs are necessary. The fact that reprogramming technology permits the generation of iPS cells from individual differentiated somatic cells of healthy donors and patients with hereditary and acquired diseases highlights its advantages over ES cell technology. The development of such technology will allow significant reduction in the number of animal experiments in drug screening. The capacity to differentiate iPS cells into cardiomyocytes, hepatocytes, fibroblasts, and neurons will allow investigators to perform targeted preclinical toxicological in in vitro trials [108-110].
Cell Models of Human Diseases
In the recent years, a large number of publications have described the production of iPS cells from patients with different diseases, in particular, with hereditary diseases. After directed differentiation, these cells demonstrate specific changes characteristic for a given disease [111-115]. The modeling of various neurodegenerative diseases of the central nervous system is of special interest. This is for several reasons. First, the obtaining of biopsy material from human brain is impossible in most conditions except for specific surgical operations typically related to oncology. Second, it is virtually impossible to propagate adult neurons in vitro. The human brain contains a huge diversity of neural cells that differ in their ergicity and specific functions. However, one can overcome the difficulties indicated above using iPS cells. Since iPS cells can be virtually indefinitely propagated in vitro, it is possible to obtain from them the necessary number of cells for all kinds of biological experiments. To date, sufficiently effective protocols have been developed for differentiation of these cell into neurons of specific ergicity, as well as glial cells [116-118].
As an example, let us refer to certain results obtained during studies of pathophysiological mechanisms underlying Parkinson disease (PD) by using iPS cells. PD is a chronic progressive degenerative disease of the central nervous system that is accounted for by the loss of dopaminergic neurons and decreased levels of the neurotransmitter dopamine in the striatum [119]. This disease can be either hereditary or sporadic. For hereditary forms of this disease, several genes involved in the progression of the disease have been identified (Park8, Park2, Pink1, Snca). Sanchez-Danes et al. [120] undertook a comprehensive study on the pathogenesis of PD using iPS cells derived from seven patients with the sporadic from of the disease, four patients bearing the G2019S mutation in the Lrrk2 gene, and four patients with no previous history of neurodegenerative diseases. The efficiency of formation of iPS cell clones varied between the donors, but it was not associated with diagnosis of PD or age of donor. Upon differentiation, adult dopaminergic neurons of predominantly A9 subtype were obtained. That particular group of neurons forms the substantia nigra pars compacta, which undergoes massive degeneration in PD.
Alpha-synuclein (SNCA) is another protein related to the development of PD. The exact function of this protein is still not clear. There is evidence for its role as a molecular chaperone that regulates protein–protein and protein–lipid interactions. SNCA might also play an important role in synaptic vesicles, in storage and compartmentalization of neurotransmitters, and, most significantly, of dopamine [116]. It is well known that protofibrils of SNCA are the main component of Levi bodies in PD. This indicates the important role of aggregation of SNCA in pathogenesis of PD. A study was undertaken to reveal the role of SNCA in differentiated dopaminergic neurons generated from iPS cells that were derived from patients with various forms of PD as well as from healthy donors. It was found that neurons from patients bearing the G2019S mutation in Lrrk2 gene have anomalous accumulation of SNCA compared to dopaminergic neurons from healthy donors and of patients with the sporadic form of PD [120]. Another team demonstrated a high level of SNCA in dopaminergic neurons derived from patients bearing mutations in the Lrrk2 gene [121]. Thus, these results not only support the hypothesis of a mutual influence of Lrrk2 and Snca mutations, but also give an opportunity to use the obtained iPS cells as a model of the monogenic form of PD. It is known that PD develops over several years, sometimes even decades. Long-term (65-75 days) culture of iPS cell-derived dopaminergic neurons was principal (patients with sporadic form of disease, patients bearing Lrrk2 mutations, or healthy donors) [120]. Notably, such long-term culture was accomplished on a mouse postnatal astrocyte monolayer. It was shown that dopaminergic neurons from healthy donors are morphologically homogenous with adult neuron phenotype with well-developed neurites. At the same time, the neurons of PD patients had different and significant changes in morphology (decreased length and number of neurites, full absence of neurites, fragmented nuclei, and vacuolization). It is important that such differences were not observed under standard 30-day cell culture conditions. After 75 days in vitro, the dopaminergic neurons derived from patients bearing the mutant Lrrk2 gene and a sporadic form of PD had a higher percentage of apoptotic cells compared to normal cells. Another team of scientists derived iPS cells from patients with a hereditary from of PD bearing mutations in the Pink1 gene. It was demonstrated that the current mutation does not influence the reprogramming process and differentiation of iPS cells into dopaminergic neurons. However, in adult neurons that bear the above mutation, in stress conditions the mobilization of Parkin to damaged mitochondria is disturbed, while neurons from healthy donors do not demonstrate such disturbances [122]. Nguyen et al. [121] showed that cells bearing a mutant Lrrk2 gene express elevated levels of oxidative stress-associated genes in response to various damaging agents such as hydrogen peroxide, 6-hydroxydopamine, and the proteasome inhibitor MG-132. These data suggest that cells bearing mutant genes and cells from healthy donors might have different responses to pharmacological drugs, and in particular, to the concentration of a drug that is administered. A promising approach in studying the molecular mechanisms underlying the origin and development of Parkinsonism is the construction of maps of metabolic pathways that involve the products of mutant genes [123].
Development of cell-therapy approaches to human diseases
Several teams are now conducting transplantation experiments with dopaminergic neurons derived from iPS cells in animal models. Thus, differentiated cells were successfully transplanted into a rat PD model induced by 6-hydroxydopamine. After implantation of donor cells, a significant improvement in motor functions was observed [124-126]. At the same time, the application of iPS cells in cell therapy is limited by several problems. So far, no studies have been performed to reveal the correlation between cellular characteristics of cultured neurons and clinical parameters of disease in donor patients. In this regard, the potential of these cultures as adequate cellular models of neurodegenerative diseases is not completely clear. The role of derivatives of iPS cells in the development of informative vital diacritical and prognostic biomarkers of PD and other neurodegenerative diseases has not yet been investigated [127]. Individual publications have illustrated that neural cultures derived from iPS cells can be used as an adequate biological matrix for studying biomolecular aspects and pathochemical sequences of development of the neurodegenerative process: its staging and possibilities for prevention and/or therapy [126, 128, 129]. Nevertheless, investigations in the field of potential applications of iPS cells in transplantology are developing quite rapidly. Thus, it is planned to begin clinical trials of iPS cell derivatives for correction of several diseases such as PD, platelet deficiency, cardiovascular diseases, multiple sclerosis, retinal lesions, and spinal cord traumas [130-134].
CONCLUSION
The development of technology for reprogramming of somatic cells and production of mammalian iPS cells, including human cells, has revealed new prospects in transplantology and in vitro study of the molecular and cellular basis of serious human diseases [1, 2]. The development of this technology has also opened possibilities for creation of models for several serious human pathologies, including neurodegenerative diseases, as well as for creation of test systems that permit large-scale screening of drugs for targeted treatment of specific human diseases in vitro, considering the phenotypic traits of the patient. The fact, reprogramming technology provides an opportunity to generate iPS cells from individual differentiated somatic cells of healthy donors and patients with various medical conditions, giving it a clear advantage over ES cell technology, which in turn opens broad perspectives for the development of personalized medicine. The development of such technology will allow significant reduction in the number of animal experiments in drug screening by directly conducing assays on human cells. The capacity to differentiate iPS cells into cardiomyocytes, hepatocytes, fibroblasts, neurons, etc. would allow investigators to perform preclinical toxicological in vitro trials.
The iPS cells generated by various methods, especially by those that do not interfere with the structure of genes in somatic cells (nonviral reprogramming methods), can become a key for their wide use in cell therapy of different serious diseases of humans that require the restoration of cell or organs of the patient that were damaged in the pathological process. In recent years, papers devoted to this important question have emerged, and it can be expected that in the near future their number will increase [135-139].
However, the wide application of iPS cells in cell therapy is still limited by several problems, of which their possible malignant transformation in vivo appears the most serious. For another thing, in case of diseases of the brain, considering the exclusive complexity of the human nervous system, several additional problems arise in using iPS cells for transplantation. First, it is the generation of a sufficient number of neurons of correct ergicity and the maintenance of their survival after transplantation. Second, the correct integration of transplanted cells in the appropriate brain regions, including their differentiation, and the establishment of correct contacts between neurons. Moreover, in the third place, it is a functional activity of transplanted iPS cells.
Nonetheless, the iPS cell technology appears currently as the most promising for study of molecular mechanisms of cell pathologies under personalized conditions, the creation of efficient test systems for search and screening of pharmacological drugs, as well as for development of methods for cell therapy of various human diseases [140].
We thank Dr. O. S. Lebedeva for the preparation of figures and Dr. S. A. Antonov for linguistic help in preparation of the manuscript.
This work was supported in part by the grants from the programs “Molecular and Cell Biology” and “Fundamental Investigations for the Development of Biomedical Technologies” of the Russian Academy of Sciences, a grant of the Russian Foundation for Basic Research (No. 14-08-01089-a), and grant from the Russian Ministry for Education and Science (No. 14.604.21.0115).
REFERENCES
1.Takahashi, K., and Yamanaka, S. (2006) Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors, Cell, 126, 663-676.
2.Takahashi, K., Tanabe, K., Ohnuki, M.,
Narita, M., Ichisaka, T., Tomoda, K., and Yamanaka, S. (2007)
Induction of pluripotent stem cells from adult human fibroblasts by
defined factors, Cell, 131, 861-872.
3.Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi,
K., Ichisaka, T., Aoi, T., Okita, K., Mochiduki, Y., Takizawa, N., and
Yamanaka, S. (2008) Generation of induced pluripotent stem cells
without Myc from mouse and human fibroblasts, Nature
Biotechnol., 26, 101-106.
4.Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S.
S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., and Jones, J. M.
(1998) Embryonic stem cell lines derived from human blastocysts,
Science, 282, 1145-1147.
5.Prowse, A. B., McQuade, L. R., Bryant, K. J.,
Marcal, H., and Gray, P. P. (2007) Identification of potential
pluripotency determinants for human embryonic stem cells following
proteomic analysis of human and mouse fibroblast conditioned media,
J. Proteome Res., 6, 3796-3807.
6.Williams, R. L., Hilton, D. J., Pease, S., Willson,
T. A., Stewart, C. L., Gearing, D. P., Wagner, E. F., Metcalf, D.,
Nicola, N. A., and Gough, N. M. (1988) Myeloid leukemia inhibitory
factor maintains the developmental potential of embryonic stem cells,
Nature, 336, 684-687.
7.Nakagawa, M., Taniguchi, Y., Senda, S., Takizawa,
N., Ichisaka, T., Asano, K., Morizane, A., Doi, D., Takahashi, J.,
Nishizawa, M., Yoshida, Y., Toyoda, T., Osafune, K., Sekiguchi, K., and
Yamanaka, S. (2014) A novel efficient feeder-free culture system for
the derivation of human induced pluripotent stem cells, Sci.
Rep., 4, 1-7.
8.Chen, K. G., Mallon, B. S., McKay, R. D., and
Robey, P. G. (2014) Human pluripotent stem cell culture: considerations
for maintenance, expansion, and therapeutics, Cell Stem Cell,
14, 13-26.
9.Dolley-Sonneville, P. J., Romeo, L. E., and
Melkoumian, Z. K. (2013) Synthetic surface for expansion of human
mesenchymal stem cells in xeno-free, chemically defined culture
conditions, PLoS ONE, 8, e70263.
10.Parker, G. C., Acsadi, G., and Brenner, C. A.
(2009) Mitochondria: determinants of stem cell fate? Stem Cells
Devel., 18, 803-806.
11.Facucho-Oliveira, J. M., and St John, J. C.
(2009) The relationship between pluripotency and mitochondrial DNA
proliferation during early embryo development and embryonic stem cell
differentiation, Stem Cell Rev. Rep., 5, 140-158.
12.Mattout, A., and Meshorer, E. (2010) Chromatin
plasticity and genome organization in pluripotent embryonic stem cells,
Curr. Opin. Cell Biol., 22, 334-341.
13.Park, S. H., Kook, M. C., Kim, E. Y., Park, S.,
and Lim, J. (2004) Ultrastructure of human embryonic stem cells and
spontaneous and retinoic acid-induced differentiating cells,
Ultrastruct. Pathol., 28, 229-238.
14.Bernstein, B. E., Mikkelsen, T. S., Xie, X.,
Kamal, M., Huebert, D. J., Cuff, J., Fry, B., Meissner, A., Wernig, M.,
Plath, K., Jaenisch, R., Wagschal, A., Feil, R., Schreiber, S. L., and
Lander, E. S. (2006) A bivalent chromatin structure marks key
developmental genes in embryonic stem cells, Cell, 125,
315-326.
15.Zhao, X. D., Han, X., Chew, J. L., Liu, J., Chiu,
K. P., Choo, A., Orlov, Y. L., Sung, W. K., Shahab, A., Kuznetsov, V.
A., Bourque, G., Oh, S., Ruan, Y., Ng, H. H., and Wei, C. L. (2007)
Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals
distinct genomic compartments in human embryonic stem cells, Cell
Stem Cell, 1, 286-298.
16.Meshorer, E., Yellajoshula, D., George, E.,
Scambler, P. J., Brown, D. T., and Misteli, T. (2006) Hyperdynamic
plasticity of chromatin proteins in pluripotent embryonic stem cells,
Devel. Cell, 10, 105-116.
17.Chambers, I., Colby, D., Robertson, M., Nichols,
J., Lee, S., Tweedie, S., and Smith, A. (2003) Functional expression
cloning of Nanog, a pluripotency sustaining factor in embryonic stem
cells, Cell, 113, 643-655.
18.Scholer, H. R., Hatzopoulos, A. K., Balling, R.,
Suzuki, N., and Gruss, P. (1989) A family of octamer-specific proteins
present during mouse embryogenesis: evidence for germline-specific
expression of an Oct factor, EMBO J., 8, 2543-2550.
19.Yuan, H., Corbi, N., Basilico, C., and Dailey, L.
(1995) Developmental-specific activity of the FGF-4 enhancer requires
the synergistic action of Sox2 and Oct-3, Genes Devel.,
9, 2635-2645.
20.Scheper, W., and Copray, S. (2009) The molecular
mechanism of induced pluripotency: a two-stage switch, Stem Cell
Rev. Rep., 5, 204-223.
21.Lister, R., Pelizzola, M., Dowen, R. H., Hawkins,
R. D., Hon, G., Tonti-Filippini, J., Nery, J. R., Lee, L., Ye, Z., Ngo,
Q. M., Edsall, L., Antosiewicz-Bourget, J., Stewart, R., Ruotti, V.,
Millar, A. H., Thomson, J. A., Ren, B., and Ecker, J. R. (2009) Human
DNA methylomes at base resolution show widespread epigenomic
differences, Nature, 462, 315-322.
22.Tada, M., Takahama, Y., Abe, K., Nakatsuji, N.,
and Tada, T. (2001) Nuclear reprogramming of somatic cells by in
vitro hybridization with ES cells, Curr. Biol., 11,
1553-1558.
23.Boiani, M., and Scholer, H. R. (2005) Regulatory
networks in embryo-derived pluripotent stem cells, Nature Rev. Mol.
Cell Biol., 6, 872-884.
24.Nichols, J., Zevnik, B., Anastassiadis, K., Niwa,
H., Klewe-Nebenius, D., Chambers, I., Scholer, H., and Smith, A. (1998)
Formation of pluripotent stem cells in the mammalian embryo depends on
the POU transcription factor Oct4, Cell, 95, 379-391.
25.Gidekel, S., Pizov, G., Bergman, Y., and
Pikarsky, E. (2003) Oct-3/4 is a dose dependent oncogenic fate
determinant, Cancer Cell, 4, 361-370.
26.Niwa, H., Miyazaki, J., and Smith, A. G. (2000)
Quantitative expression of Oct-3/4 defines differentiation,
dedifferentiation or self-renewal of ES cells, Nature Genet.,
24, 372-376.
27.Zaehres, H., Lensch, M. W., Daheron, L., Stewart,
S. A., Itskovitz-Eldor, J., and Daley, G. Q. (2005) High-efficiency RNA
interference in human embryonic stem cells, Stem Cells,
23, 299-305.
28.Nakatake, Y., Fukui, N., Iwamatsu, Y., Masui, S.,
Takahashi, K., Yagi, R., Yagi, K., Miyazaki, J., Matoba, R., Ko, M. S.,
and Niwa, H. (2006) Klf4 cooperates with Oct3/4 and Sox2 to activate
the Lefty1 core promoter in embryonic stem cells, Mol. Cell.
Biol., 26, 7772-7782.
29.Trosko, J. E. (2006) From adult stem cells to
cancer stem cells: Oct-4 gene, cell–cell communication,
and hormones during tumor promotion, Ann. N.Y. Acad. Sci.,
1089, 36-58.
30.Cheng, L., Sung, M. T., Cossu-Rocca, P., Jones,
T. D., MacLennan, G. T., De Jong, J., Lopez-Beltran, A., Montironi, R.,
and Looijenga, L. H. (2007) OCT4: biological functions and clinical
applications as a marker of germ cell neoplasia, J. Pathol.,
211, 1-9.
31.Miyagi, S., Saito, T., Mizutani, K., Masuyama,
N., Gotoh, Y., Iwama, A., Nakauchi, H., Masui, S., Niwa, H., Nishimoto,
M., Muramatsu, M., and Okuda, A. (2004) The Sox-2 regulatory regions
display their activities in two distinct types of multipotent stem
cells, Mol. Cell. Biol., 24, 4207-4220.
32.Tokuzawa, Y., Kaiho, E., Maruyama, M., Takahashi,
K., Mitsui, K., Maeda, M., Niwa, H., and Yamanaka, S. (2003) Fbx15 is a
novel target of Oct3/4 but is dispensable for embryonic stem cell
self-renewal and mouse development, Mol. Cell. Biol., 23,
2699-2708.
33.Masui, S., Nakatake, Y., Toyooka, Y., Shimosato,
D., Yagi, R., Takahashi, K., Okochi, H., Okuda, A., Matoba, R., Sharov,
A. A., Ko, M. S., and Niwa, H. (2007) Pluripotency governed by Sox2 via
regulation of Oct3/4 expression in mouse embryonic stem cells,
Nature Cell Biol., 9, 625-635.
34.Chew, J. L., Loh, Y. H., Zhang, W., Chen, X.,
Tam, W. L., Yeap, L. S., Li, P., Ang, Y. S., Lim, B., Robson, P., and
Ng, H. H. (2005) Reciprocal transcriptional regulation of Pou5f1 and
Sox2 via the Oct4/Sox2 complex in embryonic stem cells, Mol. Cell.
Biol., 25, 6031-6046.
35.Kelberman, D., Rizzoti, K., Avilion, A.,
Bitner-Glindzicz, M., Cianfarani, S., Collins, J., Chong, W. K., Kirk,
J. M., Achermann, J. C., Ross, R., Carmignac, D., Lovell-Badge, R.,
Robinson, I. C., and Dattani, M. T. (2006) Mutations within Sox2/SOX2
are associated with abnormalities in the hypothalamo-pituitary-gonadal
axis in mice and humans, J. Clin. Investig., 116,
2442-2455.
36.Dong, C., Wilhelm, D., and Koopman, P. (2004)
Sox genes and cancer, Cytogenet. Genome Res., 105,
442-447.
37.Dang, D. T., Pevsner, J., and Yang, V. W. (2000)
The biology of the mammalian Kruppel-like family of transcription
factors, Int. J. Biochem. Cell Biol., 32, 1103-1121.
38.Wei, D., Kanai, M., Huang, S., and Xie, K. (2006)
Emerging role of KLF4 in human gastrointestinal cancer,
Carcinogenesis, 27, 23-31.
39.Segre, J. A., Bauer, C., and Fuchs, E. (1999)
Klf4 is a transcription factor required for establishing the barrier
function of the skin, Nature Genet., 22, 356-360.
40.Conkright, M. D., Wani, M. A., Anderson, K. P.,
and Lingrel, J. B. (1999) A gene encoding an intestinal-enriched member
of the Kruppel-like factor family expressed in intestinal epithelial
cells, Nucleic Acids Res., 27, 1263-1270.
41.Shields, J. M., Christy, R. J., and Yang, V. W.
(1996) Identification and characterization of a gene encoding a
gut-enriched Kruppel-like factor expressed during growth arrest, J.
Biol. Chem., 271, 20009-20017.
42.Chen, X., Johns, D. C., Geiman, D. E., Marban,
E., Dang, D. T., Hamlin, G., Sun, R., and Yang, V. W. (2001)
Kruppel-like factor 4 (gut-enriched Kruppel-like factor) inhibits cell
proliferation by blocking G1/S progression of the cell cycle, J.
Biol. Chem., 276, 30423-30428.
43.Ohnishi, S., Ohnami, S., Laub, F., Aoki, K.,
Suzuki, K., Kanai, Y., Haga, K., Asaka, M., Ramirez, F., and Yoshida,
T. (2003) Downregulation and growth inhibitory effect of
epithelial-type Kruppel-like transcription factor KLF4, but not KLF5,
in bladder cancer, Biochem. Biophys. Res. Commun., 308,
251-256.
44.Katz, J. P., Perreault, N., Goldstein, B. G.,
Actman, L., McNally, S. R., Silberg, D. G., Furth, E. E., and Kaestner,
K. H. (2005) Loss of Klf4 in mice causes altered proliferation and
differentiation and precancerous changes in the adult stomach,
Gastroenterology, 128, 935-945.
45.Foster, K. W., Frost, A. R., McKie-Bell, P., Lin,
C. Y., Engler, J. A., Grizzle, W. E., and Ruppert, J. M. (2000)
Increase of GKLF messenger RNA and protein expression during
progression of breast cancer, Cancer Res., 60,
6488-6495.
46.Wang, N., Liu, Z. H., Ding, F., Wang, X. Q.,
Zhou, C. N., and Wu, M. (2002) Downregulation of gut-enriched
Kruppel-like factor expression in esophageal cancer, World J.
Gastroenterol., 8, 966-970.
47.Zhao, W., Hisamuddin, I. M., Nandan, M. O.,
Babbin, B. A., Lamb, N. E., and Yang, V. W. (2004) Identification of
Kruppel-like factor 4 as a potential tumor suppressor gene in
colorectal cancer, Oncogene, 23, 395-402.
48.Dang, C. V., O’Donnell, K. A., Zeller, K.
I., Nguyen, T., Osthus, R. C., and Li, F. (2006) The c-Myc
target gene network, Semin. Cancer Biol., 16,
253-264.
49.Lebofsky, R., and Walter, J. C. (2007) New
Myc-anisms for DNA replication and tumorigenesis? Cancer Cell,
12, 102-103.
50.Patel, J. H., Loboda, A. P., Showe, M. K., Showe,
L. C., and McMahon, S. B. (2004) Analysis of genomic targets reveals
complex functions of MYC, Nature Rev. Cancer, 4,
562-568.
51.Cawley, S., Bekiranov, S., Ng, H. H., Kapranov,
P., Sekinger, E. A., Kampa, D., Piccolboni, A., Sementchenko, V.,
Cheng, J., Williams, A. J., Wheeler, R., Wong, B., Drenkow, J.,
Yamanaka, M., Patel, S., Brubaker, S., Tammana, H., Helt, G., Struhl,
K., and Gingeras, T. R. (2004) Unbiased mapping of transcription factor
binding sites along human chromosomes 21 and 22 points to widespread
regulation of noncoding RNAs, Cell, 116, 499-509.
52.Cowling, V. H., and Cole, M. D. (2006) Mechanism
of transcriptional activation by the Myc oncoproteins, Semin. Cancer
Biol., 16, 242-252.
53.Chang, T. C., Yu, D., Lee, Y. S., Wentzel, E. A.,
Arking, D. E., West, K. M., Dang, C. V., Thomas-Tikhonenko, A., and
Mendell, J. T. (2008) Widespread microRNA repression by Myc contributes
to tumorigenesis, Nature Genet., 40, 43-50.
54.Davis, A. C., Wims, M., Spotts, G. D., Hann, S.
R., and Bradley, A. (1993) A null c-myc mutation causes
lethality before 10.5 days of gestation in homozygotes and reduced
fertility in heterozygous female mice, Genes Devel., 7,
671-682.
55.Baudino, T. A., McKay, C., Pendeville-Samain, H.,
Nilsson, J. A., Maclean, K H., White, E. L., Davis, A. C., Ihle, J. N.,
and Cleveland, J. L. (2002) c-Myc is essential for vasculogenesis and
angiogenesis during development and tumor progression, Genes
Devel., 16, 2530-2543.
56.Yu, J., Vodyanik, M. A., Smuga-Otto, K.,
Antosiewicz-Bourget, J., Frane, J. L., Tian, S., Nie, J., Jonsdottir,
G. A., Ruotti, V., Stewart, R., Slukvin, I. I., and Thomson, J. A.
(2007) Induced pluripotent stem cell lines derived from human somatic
cells, Science, 318, 1917-1920.
57.Matsui, T., Leung, D., Miyashita, H., Maksakova,
I. A., Miyachi, H., Kimura, H., Tachibana, M., Lorincz, M. C., and
Shinkai, Y. (2010) Proviral silencing in embryonic stem cells requires
the histone methyltransferase ESET, Nature, 464,
927-931.
58.Sridharan, R., Tchieu, J., Mason, M. J.,
Yachechko, R., Kuoy, E., Horvath, S., Zhou, Q., and Plath, K. (2009)
Role of the murine reprogramming factors in the induction of
pluripotency, Cell, 136, 364-377.
59.Okita, K., Ichisaka, T., and Yamanaka, S. (2007)
Generation of germline-competent induced pluripotent stem cells,
Nature, 448, 313-317.
60.Maherali, N., Ahfeldt, T., Rigamonti, A., Utikal,
J., Cowan, C., and Hochedlinger, K. (2008) A high-efficiency system for
the generation and study of human induced pluripotent stem cells,
Cell Stem Cell, 3, 340-345.
61.Markoulaki, S., Hanna, J., Beard, C., Carey, B.
W., Cheng, A. W., Lengner, C. J., Dausman, J. A., Fu, D., Gao, Q., Wu,
S., Cassady, J. P., and Jaenisch, R. (2009) Transgenic mice with
defined combinations of drug-inducible reprogramming factors, Nature
Biotechnol., 27, 169-171.
62.Kaji, K., Norrby, K., Paca, A., Mileikovsky, M.,
Mohseni, P., and Woltjen, K. (2009) Virus-free induction of
pluripotency and subsequent excision of reprogramming factors,
Nature, 458, 771-775.
63.Soldner, F., Hockemeyer, D., Beard, C., Gao, Q.,
Bell, G. W., Cook, E. G., Hargus, G., Blak, A., Cooper, O., Mitalipova,
M., Isacson, O., and Jaenisch, R. (2009) Parkinson’s disease
patient-derived induced pluripotent stem cells free of viral
reprogramming factors, Cell, 136, 964-977.
64.Bichsel, C., Neeld, D. K., Hamazaki, T., Wu, D.,
Chang, L. J., Yang, L., Terada, N., and Jin S. (2011) Bacterial
delivery of nuclear proteins into pluripotent and differentiated cells,
PLoS One, 6, e16465.
65.Okita, K., Yamakawa, T., Matsumura. Y., Sato, Y.,
Amano, N., Watanabe, A., Goshima, N., and Yamanaka, S. (2013) An
efficient non-viral method to generate integration-free human iPS cells
from cord blood and peripheral blood cells, Stem Cells,
31, 458-466.
66.Nakanishi, M., and Otsu, M. (2012) Development of
Sendai virus vectors and their potential applications in gene therapy
and regenerative medicine, Curr. Gene Therapy, 12,
410-416.
67.Macarthur, C. C., Fontes, A., Ravinder, N.,
Kuninger, D., Kaur, J., Bailey, M., Taliana, A., Vemuri, M. C., and
Lieu, P. T. (2012) Generation of human-induced pluripotent stem cells
by a nonintegrating RNA Sendai virus vector in feeder-free or xeno-free
conditions, Stem Cells Int., 2012, Article ID 564612;
doi: 10.1155/2012/564612.
68.Fusaki, N., Ban, H., Nishiyama, A., Saeki, K.,
and Hasegawa, M. (2009) Efficient induction of transgene-free human
pluripotent stem cells using a vector based on Sendai virus, an RNA
virus that does not integrate into the host genome, Proc. Jap. Acad.
Ser. B: Phys. Biol. Sci., 85, 348-362.
69.Golubovsky, M. D. (2011) Gene instability and
mobile elements: a history of its research and discovery, Stud.
History Biol., 4, 60-78.
70.Woltjen, K., Michael, I. P., Mohseni, P., Desai,
R., Mileikovsky, M., Hamalainen, R., Cowling, R., Wang, W., Liu, P.,
Gertsenstein, M., Kaji, K., Sung, H. K., and Nagy, A. (2009) piggyBac
transposition reprograms fibroblasts to induced pluripotent stem cells,
Nature, 458, 766-770.
71.Hackett, P. B., Jr., Aronovich, E. L., Hunter,
D., Urness, M., Bell, J. B., Kass, S. J., Cooper, L. J., and
McIvor, S. (2011) Efficacy and safety of sleeping beauty
transposon-mediated gene transfer in preclinical animal studies,
Curr. Gene Therapy, 11, 341-349.
72.Belay, E., Dastidar, S., VandenDriessche, T., and
Chuah, M. K. (2011) Transposon-mediated gene transfer into adult and
induced pluripotent stem cells, Curr. Gene Therapy, 11,
406-413.
73.Mallanna, S. K., and Rizzino, A. (2010) Emerging
roles of microRNAs in the control of embryonic stem cells and the
generation of induced pluripotent stem cells, Devel. Biol.,
344, 16-25.
74.Subramanyam, D., Lamouille, S., Judson, R. L.,
Liu, J. Y., Bucay, N., Derynck, R., and Blelloch, R. (2011) Multiple
targets of miR-302 and miR-372 promote reprogramming of human
fibroblasts to induced pluripotent stem cells, Nature
Biotechnol., 29, 443-448.
75.Qi, J., Yu, J.-Y., Shcherbata, H. R., Mathieu,
J., Wang, A. J., Seal, S., Zhou, W., Stadler, B. M., Bourgin, D., Wang,
L., Nelson, A., Ware, C., Raymond, C., Lim, L. P., Magnus, J.,
Ivanovska, I., Diaz, R., Ball, A., Cleary, M. A., and Ruohola-Baker, H.
(2009) microRNAs regulate human embryonic stem cell division, Cell
Cycle, 8, 3729-3741.
76.Dolezalova, D., Mraz, M., Barta, T., Plevova, K.,
Vinarsky, V., Holubcova, Z., Jaros, J., Dvorak, P., Pospisilova, S.,
and Hampl, A. (2012) microRNAs regulate p21(Waf1/Cip1) protein
expression and the DNA damage response in human embryonic stem cells,
Stem Cells, 30, 1362-1372.
77.Li, Z., Yang, C. S., Nakashima, K., and Rana, T.
M. (2011) Small RNA-mediated regulation of generation iPS cell, EMBO
J., 30, 823-834.
78.Miyoshi, N., Ishii, H., Nagano, H., Haraguchi,
N., Dewi, D. L., Kano, Y., Nishikawa, S., Tanemura, M., Mimori, K.,
Tanaka, F., Saito, T., Nishimura, J., Takemasa, I., Mizushima, T.,
Ikeda, M., Yamamoto, H., Sekimoto, M., Doki, Y., and Mori, M. (2011)
Reprogramming of mouse and human cells to pluripotency using mature
microRNAs, Cell Stem Cell, 8, 633-638.
79.Liao, B., Bao, X., Liu, L., Feng, S., Zovoilis,
A., Liu, W., Xue, Y., Cai, J., Guo, X., Qin, B., Zhang, R., Wu, J.,
Lai, L., Teng, M., Niu, L., Zhang, B., Esteban, M. A., and Pei, D.
(2011) MicroRNA cluster 302-367 enhances somatic cell reprogramming by
accelerating a mesenchymal-to-epithelial transition, J. Biol.
Chem., 286, 17359-17364.
80.Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y.
H., Li, H., Lau, F., Ebina, W., Mandal, P. K., Smith, Z. D., Meissner,
A., Daley, G. Q., Brack, A. S., Collins, J. J., Cowan, C., Schlaeger,
T. M., and Rossi, D. J. (2010) Highly efficient reprogramming to
pluripotency and directed differentiation of human cells with synthetic
modified mRNA, Cell Stem Cell, 7, 618-630.
81.Kim, D., Kim, C. H., Moon, J. I., Chung, Y. G.,
Chang, M. Y., Han, B. S., Ko, S., Yang, E., Cha, K. Y., Lanza, R., and
Kim, K. S. (2009) Generation of human induced pluripotent stem cells by
direct delivery of reprogramming proteins, Cell Stem Cell,
4, 472-476.
82.Zhou, H., Wu, S., Joo, J. Y., Zhu, S., Han, D.
W., Lin, T., Trauger, S., Bien, G., Yao, S., Zhu, Y., Siuzdak, G.,
Scholer, H. R., Duan, L., and Ding, S. (2009) Generation of induced
pluripotent stem cells using recombinant proteins, Cell Stem
Cell, 4, 381-384.
83.Efe, J. A., and Ding, S. (2011) The evolving
biology of small molecules: controlling cell fate and identity,
Philosoph. Transact. Roy. Soc. B: Biol. Sci., 366,
2208-2221.
84.Shi, Y., Desponts, C., Do, J. T., Hahm, H. S.,
Scholer, H. R., and Ding, S. (2008) Induction of pluripotent stem cells
from mouse embryonic fibroblasts by Oct4 and Klf4 with small molecule
compounds, Cell Stem Cell, 3, 568-574.
85.Duinsbergen, D., Eriksson, M., ‘t Hoen, P.
A., Frisen, J., and Mikkers, H. (2008) Induced pluripotency with
endogenous and inducible genes, Exp. Cell Res., 314,
3255-3263.
86.Lin, T., Ambasudhan, R., Yuan, X., Li, W.,
Hilcove, S., Abujarour, R., Lin, X., Hahm, H. S., Hao, E., Hayek, A.,
and Ding, S. (2009) A chemical platform for improved induction of human
iPSCs, Nature Methods, 6, 805-808.
87.Hou, P., Li, Y., Zhang, X., Liu, C., Guan, J.,
Li, H., Zhao, T., Ye, J., Yang, W., Liu, K., Ge, J., Xu, J., Zhang, Q.,
Zhao, Y., and Deng, H. (2013) Pluripotent stem cells induced from mouse
somatic cells by small-molecule compounds, Science, 341,
651-654.
88.Rais, Y., Zviran, A., Geula, S., Gafni, O.,
Chomsky, E., Viukov, S., Mansour, A. A., Caspi, I., Krupalnik, V.,
Zerbib, M., Maza, I., Mor, N., Baran, D., Weinberger, L., Jaitin, D.
A., Lara-Astiaso, D., Blecher-Gonen, R., Shipony, Z., Mukamel, Z.,
Hagai, T., Gilad, S., Amann-Zalcenstein, D., Tanay, A., Amit, I.,
Novershtern, N., and Hanna, J. H. (2013) Deterministic direct
reprogramming of somatic cells to pluripotency, Nature,
502, 65-70.
89.Kaji, K., Nichols, J., and Hendrich, B. (2007)
Mbd3, a component of the NuRD co-repressor complex, is required for
development of pluripotent cells, Development, 134,
1123-1132.
90.Reynolds, N., Salmon-Divon, M., Dvinge, H.,
Hynes-Allen, A., Balasooriya, G., Leaford, D., Behrens, A., Bertone,
P., and Hendrich, B. (2012) NuRD-mediated deacetylation of H3K27
facilitates recruitment of polycomb repressive complex 2 to direct gene
repression, EMBO J., 31, 593-605.
91.Luo, M., Ling, T., Xie, W., Sun, H., Zhou, Y.,
Zhu, Q., Shen, M., Zong, L., Lyu, G., Zhao, Y., Ye, T., Gu, J., Tao,
W., Lu, Z., and Grummt, I. (2013) NuRD blocks reprogramming of mouse
somatic cells into pluripotent stem cell, Stem Cells, 31,
1278-1286.
92.Eminli, S., Utikal, J., Arnold, K., Jaenisch, R.,
and Hochedlinger, K. (2008) Reprogramming of neural progenitor cells
into induced pluripotent stem cells in the absence of exogenous Sox2
expression, Stem Cells, 26, 2467-2474.
93.Lagarkova, M. A., Shutova, M. V., Bogomazova, A.
N., Vassina, E. M., Glazov, E. A., Zhang, P., Rizvanov, A. A.,
Chestkov, I. V., and Kiselev, S. L. (2010) Induction of pluripotency in
human endothelial cells resets epigenetic profile on genome scale,
Cell Cycle, 9, 937-946.
94.Hanna, J., Markoulaki, S., Schorderet, P., Carey,
B. W., Beard, C., Wernig, M., Creyghton, M. P., Steine, E. J., Cassady,
J. P., Foreman, R., Lengner, C. J., Dausman, J. A., and Jaenisch, R.
(2008) Direct reprogramming of terminally differentiated mature B
lymphocytes to pluripotency, Cell, 133, 250-264.
95.Aoi, T., Yae, K., Nakagawa, M., Ichisaka, T.,
Okita, K., Takahashi, K., Chiba, T., and Yamanaka, S. (2008) Generation
of pluripotent stem cells from adult mouse liver and stomach cells,
Science, 321, 699-702.
96.Muchkaeva, I. A., Dashinimaev, E. B., Artyuhov,
A. S., Myagkova, E. P., Vorotelyak, E. A., Yegorov, Y. Y., Vishnyakova,
K. S., Kravchenko, I. E., Chumakov, P. M., Terskikh, V. V.,
and Vasiliev, A. V. (2014) Generation of iPS cells from human hair
follicle dermal papilla cells, Acta Naturae, 6,
45-53.
97.Yang, J., Li, Y., Erol, D., Wu, W. H., Tsai, Y.
T., Li, X. R., Davis, R. J., and Tsang, S. H. (2014) Generation of
induced pluripotent stem cells from conjunctiva, Graefe’s
Arch. Clin. Exp. Ophthalmol., 252, 423-431.
98.Churko, J. M., Burridge, P. W., and Wu, J. C.
(2013) Generation of human iPSCs from human peripheral blood
mononuclear cells using non-integrative Sendai virus in chemically
defined conditions, Methods Mol. Biol., 1036, 81-88.
99.Stadtfeld, M., Maherali, N., Breault, D., and
Hochedlinger, K. (2008) Defining molecular cornerstones during
fibroblast to iPS cell reprogramming in mouse, Cell Stem Cell,
2, 230-240.
100.Scheper, W., and Copray, S. (2009) The
molecular mechanism of induced pluripotency: a two-stage switch,
Stem Cell Rev. Rep., 5, 204-223.
101.Yehezkel, S., Rebibo-Sabbah, A., Segev, Y.,
Tzukerman, M., Shaked, R., Huber, I., Gepstein, L., Skorecki, K., and
Selig, S. (2011) Reprogramming of telomeric regions during the
generation of human induced pluripotent stem cells and subsequent
differentiation into fibroblast-like derivatives, Epigenetics,
6, 63-75.
102.Sun, N., Lee, A., and Wu, J. C. (2009) Long
term non-invasive imaging of embryonic stem cells using reporter genes,
Nature Protocols, 4, 1192-2001.
103.Brambrink, T., Foreman, R., Welstead, G. G.,
Lengner, C. J., Wernig, M., Suh, H., and Jaenisch, R. (2008) Sequential
expression of pluripotency markers during direct reprogramming of mouse
somatic cells, Cell Stem Cell, 7, 151-159.
104.Maherali, N., Sridharan, R., Xie, W., Utikal,
J., Eminli, S., Arnold, K., Stadtfeld, M., Yachechko, R., Tchieu, J.,
Jaenisch, R., Plath, K., and Hochedlinger, K. (2007) Directly
reprogrammed fibroblasts show global epigenetic remodeling and
widespread tissue contribution, Cell Stem Cell, 1,
55-70.
105.O’Mathuna, D. P. (2002) What to call
human cloning: the technical terminology increasingly used in the
cloning debate sidesteps the ethical questions raised, EMBO
Rep., 3, 502-505.
106.Grivennikov, I. A. (2008) Embryonic stem cells
and the problem of directed differentiation, Biochemistry
(Moscow), 73, 1438-1452.
107.Turner, M., Leslie, S., Martin, N. G.,
Peschanski, M., Rao, M., Taylor, C. J., Trounson, A., Turner, D.,
Yamanaka, S., and Wilmut, I. (2013) Toward the development of a global
induced pluripotent stem cell library, Cell Stem Cell,
13, 382-384.
108.Mackay-Sim, A. (2013) Patient-derived stem
cells: pathways to drug discovery for brain diseases, Front. Cell.
Neurosci., 7, 1-10.
109.Grskovic, M., Javaherian, A., Strulovici, B.,
and Daley, G. Q. (2011) Induced pluripotent stem cells –
opportunities for disease modeling and drug discovery, Nature Rev.
Drug Discov., 10, 915-929.
110.Maury, Y., Gauthier, M., Peschanski, M., and
Martinat, C. (2012) Human pluripotent stem cells for disease modeling
and drug screening, Bioessays, 34, 61-71.
111.Park, I. H., Arora, N., Huo, H., Maherali, N.,
Ahfeldt, T., Shimamura, A., Lensch, M. W., Cowan, C., Hochedlinger, K.,
and Daley, G. Q. (2008) Disease-specific induced pluripotent stem
cells, Cell, 134, 877-886.
112.Ebert, A. D., Yu, J., Rose, F. F., Jr., Mattis,
V. B., Lorson, C. L., Thomson, J. A., and Svendsen, C. N. (2009)
Induced pluripotent stem cells from a spinal muscular atrophy patient,
Nature, 457, 277-280.
113.Raya, A., Rodriguez-Piza, I., Guenechea, G.,
Vassena, R., Navarro, S., Barrero, M. J., Consiglio, A., Castella, M.,
Rio, P., Sleep, E., Gonzalez, F., Tiscornia, G., Garreta, E., Aasen,
T., Veiga, A., Verma, I. M., Surralles, J., Bueren, J., and Izpisua
Belmonte, J. C. (2009) Disease-corrected hematopoietic progenitors from
Fanconi anemia induced pluripotent stem cells, Nature,
460, 53-59.
114.Zhang, D., Jiang, W., Liu, M., Sui, X., Yin,
X., Chen, S., Shi, Y., and Deng, H. (2009) Highly efficient
differentiation of human ES and iPS cells into mature pancreatic
insulin-producing cells, Cell Res., 19, 429-438.
115.Nekrasov, E. D., Lebedeva, O. S., Vasina, E.
M., Bogomazova, A. N., Chestkov, I. V., Kiselev, S. L., Lagarkova, M.
A., Klyushnikov, S. A., Illarioshkin, S. N., and Grivennikov, I. A.
(2012) A platform for studies of Huntington’s disease on the
basis of induced pluripotent stem cells, Ann. Clin. Exp. Neurol.
6, 30-35.
116.Byers, B., Lee, H. L., and Reijo Pera, R.
(2012) Modeling Parkinson’s disease using induced pluripotent
stem cells, Curr. Neurol. Neurosci. Rep., 12,
237-242.
117.Yoshikawa, T., Samata, B., Ogura, A., Miyamoto,
S., and Takahashi, J. (2013) Systemic administration of valproic acid
and zonisamide promotes differentiation of induced pluripotent stem
cell-derived dopaminergic neurons, Front. Cell. Neurosci.,
7, 1-10.
118.Narytnyk, A., Verdon, B., Loughney, A.,
Sweeney, M., Clewes, O., Taggart, M. J., and Sieber-Blum, M. (2014)
Differentiation of human epidermal neural crest stem cells (hEPI-NCSC)
into virtually homogenous populations of dopaminergic neurons, Stem
Cell Rev. Rep., 10, 316-326.
119.Illarioshkin, S. N. (2003) Conformational
Diseases of Brain [in Russian], Janus-K, Moscow.
120.Sanchez-Danes, A., Richaud-Patin, Y.,
Carballo-Carbajal, I., Jimenez-Delgado, S., Caig, C., Mora, S., Di
Guglielmo, C., Ezquerra, M., Patel, B., Giralt, A., Canals, J. M.,
Memo, M., Alberch, J., Lopez-Barneo, J., Vila, M., Cuervo, A. M.,
Tolosa, E., Consiglio, A., and Raya, A. (2012) Disease-specific
phenotypes in dopamine neurons from human iPS-based models of genetic
and sporadic Parkinson’s disease, EMBO Mol. Med.,
4, 380-395.
121.Nguyen, H. N., Byers, B., and Cord, B. (2011)
LRRK2 mutant iPSC-derived DA neurons demonstrate increased
susceptibility to oxidative stress, Cell Stem Cell, 8,
267-280.
122.Seibler, P., Graziotto, J., Jeong, H.,
Simunovic, F., Klein, C., and Krainc, D. (2011) Mitochondrial Parkin
recruitment is impaired in neurons derived from mutant PINK1 induced
pluripotent stem cells, J. Neurosci., 31, 5970-5976.
123.Buchel, F., Saliger, S., Drager, A., Hoffmann,
S., Wrzodek, C., Zell, A., and Kahle, P. J. (2013) Parkinson’s
disease: dopaminergic nerve cell model is consistent with experimental
finding of increased extracellular transport of α-synuclein,
BMC Neurosci., 14, 136-147.
124.Hargus, G., Cooper, O., and Deleidi, M. (2010)
Differentiated Parkinson patient-derived induced pluripotent stem cells
grow in the adult rodent brain and reduce motor asymmetry in
Parkinsonian rats, Proc. Natl. Acad. Sci. USA, 107,
15921-15926.
125.Cai, J., Yang, M., and Poremsky, E. (2010)
Dopaminergic neurons derived from human induced pluripotent stem cells
survive and integrate into 6-OHDA-lesioned rats, Stem Cells
Devel., 19, 1017-1023.
126.Martinez-Morales, P. L., and Liste, I. (2012)
Stem cells as in vitro model of Parkinson’s disease,
Stem Cells Int., 2012, Article ID 980941; doi:
10.1155/2012/980941.
127.Abdulkadir, A., Ronneberger, O., Wolf, R. C.,
Pfleiderer, B., Saft, C., and Kloppel, S. (2013) Functional and
structural MRI biomarkers to detect pre-clinical neurodegeneration,
Curr. Alzheimer Res., 10, 125-134.
128.Lebedeva, O. S., Lagar’kova, M. A.,
Kiselev, S. L., Mukhina, I. V., Vedunova, M. V., Usova, O. V.,
Stavrovskaya, A. V., Yamshchikova, N. G., Fedotova, E. Yu.,
Grivennikov, I. A., Khaspekov, L. G., and Illarioshkin, S. N. (2013)
The morphofunctional properties of induced pluripotent stem cells
derived from human skin fibroblasts and differentiated to dopaminergic
neurons, Neurochem. J., 30, 233-241.
129.Imamura, K., and Inoue, H. (2012) Research on
neurodegenerative diseases using induced pluripotent stem cells,
Psychogeriatrics, 12, 115-119.
130.Okano, H., Nakamura, M., Yoshida, K., Okada,
Y., Tsuji, O., Nori, S., Ikeda, E., Yamanaka, S., and Miura, K. (2013)
Steps toward safe cell therapy using induced pluripotent stem cells,
Circ. Res., 112, 523-533.
131.Kamao, H., Mandai, M., Okamoto, S., Sakai, N.,
Suga, A., Sugita, S. J., Kiryu, J. M., and Takahashi, M. (2014)
Characterization of human induced pluripotent stem cell-derived retinal
pigment epithelium cell sheets aiming for clinical application, Stem
Cell Rev. Rep., 2, 1-14.
132.Doi, D., Samata, B., Katsukawa, M., Kikuchi,
T., Morizane, A., Ono, Y., Sekiguchi, K., Nakagawa, M., Parmar, M., and
Takahashi, J. (2014) Isolation of human induced pluripotent stem
cell-derived dopaminergic progenitors by cell sorting for successful
transplantation, Stem Cell Rev. Rep., 2, 337-350.
133.Kawamura, M., Miyagawa, S., Miki, K., Saito,
A., Fukushima, S., Higuchi, T., Kawamura, T., Kuratani, T., Daimon, T.,
Shimizu, T., Okano, T., and Sawa, Y. (2012) Feasibility, safety, and
therapeutic efficacy of human induced pluripotent stem cell-derived
cardiomyocyte sheets in a porcine ischemic cardiomyopathy model,
Circulation, 126, S29-S37.
134.Nakamura, S., Takayama, N., Hirata, S., Seo,
H., Endo, H., Ochi, K., Fujita, K. I., Koike, T., Harimoto, K. I.,
Dohda, T., Watanabe, A., Okita, K., Takahashi, N., Sawaguchi, A.,
Yamanaka, S., Nakauchi, H., Nishimura, S., and Eto, K. (2014)
Expandable megakaryocyte cell lines enable clinically applicable
generation of platelets from human induced pluripotent stem cells,
Cell Stem Cell, 14, 535-548.
135.Reinhardt, P., Schmid, B., Burbulla, L. F.,
Schondorf, D. C., Wagner, L., Glatza, M., Hoing, S., Hargus, G., Heck,
S. A., Dhingra, A., Wu, G., Muller, S., Brockmann, K., Kluba, T.,
Maisel, M., Kruger, R., Berg, D., Tsytsyura, Y., Thiel, C. S.,
Psathaki, O. E., Klingauf, J., Kuhlmann, T., Klewin, M., Muller, H.,
Gasser, T., Scholer, H. R., and Sterneckert, J. (2013) Genetic
correction of a LRRK2 mutation in human iPSCs links parkinsonian
neurodegeneration to ERK-dependent changes in gene expression, Cell
Stem Cell, 12, 354-367.
136.Xie, F., Ye, L., Chang, J. C., Beyer, A. I.,
Wang, J., Muench, M. O., and Kan, Y. W. (2014) Seamless gene correction
of β-thalassemia mutations in patient-specific iPSCs using
CRISPR/Cas9 and piggyBac, Genome Res., 24, 1526-1533.
137.Suzuki, K., Yu, C., Qu, J., Li, M., Yao, X.,
Yuan, T., Goebl, A., Tang, S., Ren, R., Aizawa, E., Zhang, F., Xu, X.,
Soligalla, R. D., Chen, F., Kim, J., Kim, N. Y., Liao, H. K., Benner,
C., Esteban, C. R., Jin, Y., Liu, G. H., Li, Y., and Izpisua Belmonte,
J. C. (2014) Targeted gene correction minimally impacts whole-genome
mutational load in human-disease-specific induced pluripotent stem cell
clones, Cell Stem Cell, 15, 31-36.
138.Mukherjee, S., and Thrasher, A. J. (2014) Gene
correction of induced pluripotent stem cells derived from a murine
model of X-linked chronic granulomatous disorder, Methods Mol.
Biol., 1114, 427-440.
139.Howden, S. E., and Thomson, J. A. (2014) Gene
targeting of human pluripotent stem cells by homologous recombination,
Methods Mol. Biol., 1114, 37-55.
140.Rao, M., and Gottesfeld, J. M. (2014)
Introduction to thematic mini-review series: development of human
therapeutics based on induced pluripotent stem cell (iPSC) technology,
J. Biol. Chem., 289, 4553-4554.