REVIEW: Mechanisms of Amyloid Fibril Formation
N. V. Dovidchenko, E. I. Leonova, and O. V. Galzitskaya*
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: ogalzit@vega.protres.ru* To whom correspondence should be addressed.
Received June 10, 2014; Revision received July 28, 2014
Amyloid and amyloid-like aggregates are elongated unbranched fibrils consisting of β-structures of separate monomers positioned perpendicular to the fibril axis and stacked strictly above each other. In their physicochemical properties, amyloid fibrils are reminiscent of synthetic polymers rather than usual proteins because they are stable to the action of denaturing agents and proteases. Their mechanical stability can be compared to a spider’s web, that in spite of its ability to stretch, is stronger than steel. It is not surprising that a large number of diseases are accompanied with amyloid fibril depositing in different organs. Pathologies provoked by depositing of incorrectly folded proteins include Alzheimer’s, Parkinson’s, and Huntington’s diseases. In addition, this group of diseases involves mucoviscidosis, some types of diabetes, and hereditary cataracts. Each type of amyloidosis is characterized by aggregation of a certain type of protein that is soluble in general, and thus leads to specific distortions of functions of the corresponding organs. Therefore, it is important to understand the process of transformation of “native” proteins to amyloid fibrils to clarify how these molecules acquire such strength and what key elements of this process determine the pathway of erroneous protein folding. This review presents our analysis of complied information on the mechanisms of formation and biochemical properties of amyloid fibrils.
KEY WORDS: prion, stress granules, Alzheimer’s disease, oligomer particles, thioflavin T, aggregation kineticsDOI: 10.1134/S0006297914130057
Abbreviations: ALS, amyotrophic lateral sclerosis; ApoE, apolipoprotein E; APP, amyloid precursor protein; CJD, Creutzfeldt–Jakob disease; EWSR, Ewing sarcoma breakpoint region 1; TAF15, TATA box binding protein (TBP)-associated factor; FTD, frontotemporal degeneration; FUS, fused in sarcoma; PSF, PTB-associated splicing factor; SAP, serum amyloid P component; TDP-43, TAR DNA-binding protein-43; TIA1, cytotoxic granule-associated RNA binding protein.
The paradigm that one polypeptide chain can accept only one unique
three-dimension conformation became history long ago [1]. Convincing evidence of this are prions, which
illustrate that because of conformational changes, one protein can form
various structures including amyloids and amyloid-like fibrils. This
became especially relevant when it was clarified that incorrect packing
of some proteins could be a reason for pathological aggregations and
cause development of many neurodegenerative diseases such as
Alzheimer’s and Parkinson’s, type II diabetes, amyotrophic
lateral sclerosis (ALS), frontotemporal degeneration (FTD),
Huntington’s disease, etc. [2-5]. It should be noted that not all amyloids and
different amyloid-like fibrils are associated with neurodegenerative
diseases, and this property is inherent in a great number of proteins.
However, in spite of such diversity of proteins, at first glance the
formed amyloid fibrils are similar and look like antiparallel
β-structures stacked in a pile perpendicular to the fibril axis
[6]. Electron microscopy and X-ray analysis have
revealed a more accurate description of fibrils: amyloids are
antiparallel β-sheets folded in a helix with a cylinder-like
cavity formed within the latter [7]. Amyloid-like
structures are formed by yeast prions Sup35 that are rich in
asparagines and glutamines. Nevertheless, more profound analyses have
revealed a number of differences between amyloid fibrils depending on
the protein composition and conditions under which they are formed. For
example, α-synuclein, aggregates of which are specific for
Parkinson’s disease, forms single cylinders consisting of
β-sheets, while fibrils of Aβ-fragments specific for
Alzheimer’s disease consist of two- or three-cylinder
β-sheets [7].
The first amyloid depositions were detected in tissues upon iodine staining more than 150 years ago [8]. The depositions were stained blue analogous to starch grains in plants. Though the result lead to the erroneous conclusion of the existence in humans of starch grains like in plants (hence the name “amyloid” appeared), during the following 100 years data were accumulating on the occurrence of amyloids in different tissues. At the time of the discovery of amyloid depositions, researchers could use only a light microscope and various stains, and during the following “descriptive” period the revealed depositions were classified depending on the site of location, the extent of staining with a certain reagent, and the clinical case in which the amyloid depositions were found. Therefore, in the 1920s it was known that amyloids were stained with Congo red and therefore they could be attributed to active substances, and the extent of staining indicated their amount in the sample [9, 10]. For example, Congo red was once used as a test for amyloidosis, when the stain was applied intravenously, and after a specified time the portion of bound stain in serum was estimated [11, 12]. It was accepted that the estimate was proportional to the total amount of amyloids in the blood. Though the use of the test was discontinued because of the risk of developing anaphylactic shock, it was believed that the binding of amyloid aggregates is a specific peculiarity of the stain, and it was used for isolation of fibrils from depositions upon autopsy. However, at present the accurate mechanism of binding with Congo red remains unclear [13, 14]. Another frequently used stain for revealing amyloidosis is thioflavin T (ThT), which when incorporated in fibrils significantly increases the fluorescence quantum yield with a shift to longer wavelength of the spectrum. However, not all amyloid formations are stained with these preparations, including protein MAVS that is active in antiviral immunity [15], α-synuclein in Parkinson’s disease [16], τ-protein in Alzheimer’s disease [17], FUS in amyotrophic lateral sclerosis [18], and others. This can be explained by the fact that in water the short-wavelength excitation spectrum overlaps insignificantly the absorption spectrum of the stain, i.e. only weak absorption occurs on maximal excitation. Meanwhile, for a long-wavelength excitation spectrum, thioflavin T fluoresces in the same spectral range as the stain incorporated in fibrils, the solution of the stain and amyloid fibrils being usually a mixture of unbound stain and stain intercalated in the fibrils [19]. It was demonstrated that thioflavin T can bind to aromatic amino acid residues located in a particular order in β-sheets [20]. Although thioflavin T is widely used as a fluorescence probe, no agreement has been achieved on the mechanism of thioflavin T incorporation into amyloid fibrils [19].
FORMATION OF AMYLOIDS BY THE PRION MECHANISM
Prions are proteins able to accept different conformations including amyloid fibrils that can serve as a matrix and “infect” other proteins, both within and between cells and between organisms [21-23]. Originally, prions were studied in connection with infectious diseases such as Kuru, Creutzfeldt–Jakob disease (CJD), scrapie, bovine spongiform encephalopathy, fatal familial insomnia, in which a prion protein was the pathological agent [24]. However, later it was found that yeast prions assist them in adaptation to diverse conditions of the environment [25]. In mammals they can both cause different diseases, sometimes even leading to death, as well as performing useful functions, for example, activate cell-based innate immunity (protein MAVS), provide for long-term memory (protein CPEB) [26, 27], and form stress granule assemblies under conditions unfavorable for the cell [28]. Many proteins contain prion-like domains that endow the protein with properties close to those of prions, including the ability for self-assembly. For example, the amino acid composition of prion-like domains of proteins fused in sarcoma (FUS), TAR DNA-binding protein-43 (TDP-43), and cytotoxic granule-associated RBA-binding protein (TIA1) is similar to prion domains of yeast proteins such as Sup35 and Ure2 [29]. The latter two proteins are characterized by a high content of polar amino acid residues, for example, asparagine, glutamine, tyrosine, and glycine. These domains enable the proteins to pass from the unfolded three-dimensional structure to intermediate states, which in their turn are predisposed to different conformational transitions including the formation of amyloid fibrils [18, 30, 31]. These proteins are generally in a dynamic equilibrium between two forms: unfolded soluble monomers and oligomers close to molten ones. Such oligomers can be involved in diverse conformational states (Fig. 1). According to one scenario, they can organize in structured amyloidogenic oligomers, then transform into pathological aggregates of a non-amyloid type or amyloid fibrils. The latter can serve as a “matrix” for incorrect folding by the prion type. According to another scenario, protein molecules can form amorphous aggregates consisting of both soluble monomers and molten oligomers organized as dynamic cross-β-structures with properties of fluids, and can also form gel-like structures [32-34]. The hydrogel state is crucial for the formation of various non-membrane structures such as stress granules, ribonucleic complexes, etc. [31, 32, 35, 36]. In other words, prion-like domains are required for aggregation at the stage of protein conformational transition from the fluid state to the gel-like state in order to perform some vital functions, but under certain conditions they can irreversibly transform into amyloid fibrils.
Fig. 1. Schematic representation of prions and prion-like domains that can lead to different conformational states and scenarios (modified and adapted from [37]).
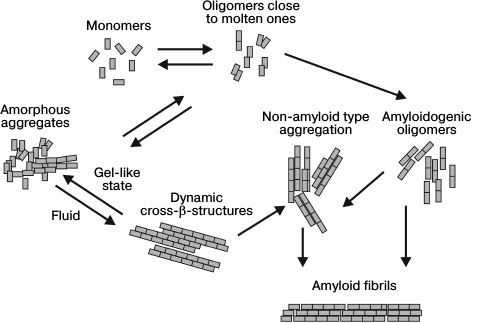
FORMATION OF AMYLOIDS BY THE PRION MECHANISM RESULTING IN
PATHOGENESIS
Recently, a new concept on the development of some neurodegenerative diseases has appeared. It includes the idea of the transition and multiplication of protein aggregates from cell-to-cell, from one cortex lobe to another during the disease progression. It has been shown both in vitro and in vivo that in different neurological diseases, pathological proteins can invade into a cell, find proteins similar to themselves, and then serve as a “matrix” for pathological aggregation [23, 38-41]. Thus, pathologic affection of cells spreads from the lesion focus. It should be noted that the cases when the disease broke out in different cortex lobes do not necessarily exclude the prion-like mechanism of its excrescence. In such a case, every lesion focus would spread the disease independently from the others. But what is the molecular basis for neurodegeneration spreading by the prion mechanism? How can these proteins acquire the conformation when the modified protein can serve as a “matrix” for “healthy” proteins, transforming the latter into an aggregated state?
The formation of insoluble β-amyloid fibrils in brain tissues accompanies many neurodegenerative diseases such as Alzheimer’s disease, Down’s syndrome, and others. The Aβ-peptide is formed as a result of proteolytic cleavage of the amyloid precursor protein (APP). The mRNA of APP frequently undergoes alternative splicing, and so it has several isoforms. In general, proteins perform important physiological functions, and the APP gene is expressed in almost all cells. A change in APP proteolysis results in accumulation of Aβ-fragments that associate in amyloid fibrils [42].
When prion-like domains were discovered in TDP-43 and FUS [18, 30, 31, 43], it became clear that mutations in these proteins can lead to pathological states. Thus in protein TDP-43, mutations Q331K and M337V located in the prion-like domain directly enhance incorrect folding. In protein FUS, mutations associated with amyotrophic lateral sclerosis (ALS) are positioned in the nuclear localization signal (NLS), which causes the protein to accumulate in the cytoplasm, thus facilitating pathological aggregation. Normally, the FUS protein is localized in the nucleus, though it sometimes moves to the cytoplasm and back [44-49]. Along with TDP-43 and FUS, prion-like domains have been revealed in 40 other RNA-binding proteins. It is not surprising that they will be also related to the development of ALS and similar neurodegenerative diseases. As has been determined, other members of the family such as TAF15 (TATA box-binding protein (TBP)-associated factor) and EWS (Ewing sarcoma breakpoint region 1) are also connected with ALS and FTD. Moreover, in the case of FTD another RNA-binding protein, PSF (PTB-associated splicing factor), is anomalously accumulated in the cytoplasm of oligodendrocytes and forms an insoluble structure [50]. Mutations in TIA1, which is an important protein for the formation of stress granules, have been detected in Welander distal myopathy (a slowly progressing muscle dystrophy) [51]. In RNA-binding proteins with prion-like domains, other mutations associated with different neurodegenerative diseases were also revealed [52]. This suggests the important role of this domain in pathogenesis.
FORMATION OF AMYLOIDS BY THE PRION MECHANISM IS REQUIRED FOR
NORMAL CELL FUNCTIONING
Living organisms widely use the property of protein molecules to form amyloid structures for various specific purposes. Normally, some organisms form amyloid fibrils for performing various functions. One of the best-studied examples of such functional amyloids is protein curlin, which is used by E. coli to colonize inert surfaces and is a mediator upon binding with proteins of other organisms. Like other amyloid structures, such fibrils are 6-12 nm thick in diameter, have a large portion of β-structure (as shown with CD), and bind thioflavin T and Congo red [53]. Another example is the bacterium Streptomyces coelicolor, which due to the formation of amyloid fibrils with chaplin proteins form hyphae used for spreading spores [54].
In these examples, the process of amyloid nucleation that will initiate aggregate growth depends on the ambient conditions and is controlled by a definite cascade of reactions. Controlled formation of functional amyloid aggregates occurs in mammals as well. For example, melanosomes are organelles differentiating into melanocytes responsible for melanin biogenesis in skin cells, containing fibril formations on which melanin granules are formed. Such fibril formations have much in common with amyloids; they are formed from a proteolytically cleaved domain of the membrane protein Pmel17 [55].
Long-term memory is also provided by the principle of fibril formation in which protein CPEB (an RNA-binding protein capable of controlling local translation of mRNAs in dendrites) plays an essential role. This protein can stimulate mRNA polyadenylation, and its aggregation activates translation of the “silent” mRNA accumulated in synaptic end-feet [56]. The N-domain of CPEB is rich in asparagines and glutamines, which is specific for prion-like domains.
Thus, protein MAVS located on the surface of mitochondrial membranes activates innate antiviral cell immunity. When aggregated, it can interact with cytoplasmic receptors recognizing patterns specific for most pathogens, which activates a cascade of reactions leading to the synthesis of β-interferon [57, 58]. This protein can also aggregate by the prion mechanism.
These examples demonstrate that even in highly organized organisms, the formation of amyloids located in a strictly defined place and rigidly controlled can be beneficial physiologically for performing specific and specialized biological functions.
BIOCHEMICAL CHARACTERISTICS OF AMYLOID FIBRILS
In the first stages of studies, chemical analysis of amyloids was complicated because both in human and animal samples the formed fibrils are frequently coated with other tissue components resulting in their nonsusceptibility to the extraction procedure. More vigorous methods of extraction, such as the use of strong bases and acids, destroy amyloid structures. The crucial point in chemical identification of amyloid proteins was the fact that intact amyloid fibrils can be quantitatively extracted from human and animal tissues using a distillate and buffers with low ionic strength [10]. The procedure is as follows: the tissue samples are repeatedly extracted with a saline solution. When the supernatant stops showing significant optical density at 280 nm, the granules are kept in the distillate until the formation of an opalescent solution that upon precipitation with salt leaves a precipitate of pure fibrils associated with Congo red when viewed in an electron microscope. Then the fibrils can be denatured in 8 M urea or 6 M guanidine chloride and layered onto a chromatography column for further purification to analyze the amino acid sequence [59, 60]. It is evident that any product soluble in the saline solution will be lost upon washing, and so will impede the determination of critically important molecules that lead to amyloidogenesis or toxicity. The chemical identification of specific precursors allowed the development of immunohistochemical reagents that can identify the chemical nature of amyloids in tissue samples without chemical extraction.
The first identified and chemically characterized amyloid was an aggregate of light chains of human immunoglobulin isolated from tissues of a patient with so-called primary amyloidosis and was not associated with any other disease [61]. Subsequent studies showed that the same class of proteins is a precursor of amyloid depositions in multiple myeloma [62]. L-chains and their fragments included in amyloid have a similar amino acid sequence with circulating in serum and/or monoclonal light chains isolated from the same patient. The depositions in tissues consist of intact L-chains and/or C-terminal fragments of such chains, which shows that the fragments in vivo subjected to proteolysis are more amyloidogenic than intact chains or the intact chain depositions are the object of proteolysis in situ [63]. Biosynthetic experiments with bone marrow cells revealed that in some cases truncated L-chains can be products of the synthesis, but these experiments were not strict enough to exclude the hypothesis of a fast postsynthetic digestion [64]. Subsequent experiments demonstrated that fragments of some isolated L-chains obtained by treatment with proteases in vitro formed amyloid-like fibrils in a test tube [59, 65]. The basic result of these experiments is the finding that the amyloidogenic potential is associated with the amino acid sequence of the protein.
The following observations revealed some structural peculiarities rendering some members of a given class of proteins more amyloidogenic as compared to other members [66]. Experiments with recombinant versions of the variable region of the amyloidogenic IgL-chain and non-amyloidogenic IgL-chain (obtained from an amyloidogenic molecule by mutating some amino acids) also showed differences that can lead to the propensity to form fibrils [67]. From the biophysical point of view, the multiplicity of sequences of amino acid residues in L-chains brings about four potentially soluble forms when the concentration of any single L-chain molecule increases by cloning upon the immune response during the disease.
Some human L-chains of immunoglobulins (about 15% of all possible versions of L-chains, predominantly from the λ-class) are exceptionally amyloidogenic and form deposits leading to dysfunction of many organs, especially kidney, heart, stomach, and peripheral nerves [63].
Other L-chains can form non-fibrillar aggregates, and this is a menace for organs just like the fibril-forming L-chains [63]. Cases of depositions of the two types of the same protein in one person are rare, though having been registered, and this suggests that the conformation acquired in vivo can be associated with the environment of the affected tissue [68].
Still other L-chains have a specific Bence-Jones feature (aggregation at specific temperatures, usually 56°C, with subsequent dissociation at higher temperatures), and this is associated with aggregation in renal tubules where the protein concentration is higher while pH is lower, and this can lead to kidney dysfunction [69].
Another fraction of L-chains of human immunoglobulin probably does not precipitate in physiological conditions and does not aggregate in vivo [70]. The exact structural features responsible for the wide multiplicity of biophysical peculiarities of proteins so similar under physiological conditions are not known.
It is no surprise that taking into account the homology of Ig domains, rare cases have been reported when solitary heavy chains served as precursors for amyloid aggregates upon monoclonal B-cell production [71]. The initial non-immunoglobulin amyloid was isolated from depositions in a monkey with chronic inflammation. The protein has an amino acid sequence dissimilar either to the sequence of an amyloid formed by immunoglobulin L-chains or to the sequence of any other protein sequenced to date [72]. At the same time, a similar protein was isolated and sequenced from the kidney of an Israeli citizen with a periodic disease and secondary (or associated with inflammation) amyloidosis [60]. The two proteins had similar amino acid sequence, it being identical from the N-terminus and varying amino acids not deviating from the frequency characteristic of residues of normal polymorphisms. Later it was found that the given protein also forms amyloids under long-term rheumatoid arthritis (another chronic inflammatory disease) and is a basic component of fibrils in murine and rat models [73].
More than 30 amyloid proteins are now recognized by the International Nomenclature Committee on Amyloidosis [33, 50, 74, 75]. The precursors of most of these proteins are apolipoproteins A (1, 2), serum amyloid A, as well as different proteohormones and immunoglobulins [76]. It has been found that depositions contain also other molecules such as apolipoprotein E (ApoE), SAP (serum amyloid P component), and the proteoglycan perlecan [77-80]. Under AA-amyloidosis (see below), fibrils are generated from the cleaved product SAA whose concentration increases upon development of chronic inflammations and other systemic diseases [81, 82]. This protein is involved in lipometabolism in macrophages participating in inflammations and can also realize other functions, for example, play a part in atherogenesis associated with inflammation [74]. SAA was the first of the five proteins (the other proteins being IAPP, Aβ-peptide, prion, and protein Bri2 whose depositions were found at fatal familial insomnia [83-86]) in which amyloid depositions provide evidence of a normally functioning soluble precursor, identified either after preparation of appropriate antibodies to fibrils and the binding of these antibodies to normal protein, or after identification using cDNA of the corresponding gene in normal tissues.
However, of more importance is that these investigations allowed developing isolation methods that can be used for studying fibrils from any tissue or organism; for example, the methods were used to isolate proteins from congophilic vascular and cerebral deposits of Aβ [87, 88]. Extracellular molecules of matrix and membrane components like laminin and tenascin were also present in some depositions. Such molecules may contribute to accumulation and formation of depositions in tissues. It has been proposed that additional molecules can play a part in stabilization of fibril structure or provide conditions for enhancement of fibrogenesis.
EXPERIMENTAL MODELING OF AMYLOIDOSIS IN ANIMALS
Since the 1960s, animal models have become one of the means to study amyloidosis. By introducing a particular dose of casein or Freund’s adjuvant into mice or rabbits, an acute inflammatory process was provoked which, in turn, was concomitant with accumulation of deposits stained with Congo red in the liver, kidneys, and spleen [89]. It was found that as a result of inflammation, the generation of cytokines promotes formation of the amyloid precursor serum amyloid A (SAA) [90]. This model completely mirrors human AA amyloidosis generated during inflammations and is frequently used for studying pathogenesis. A high percentage of amyloidosis in humans is associated with AA amyloidosis, so-called secondary or reactive amyloidosis, when as a response to any chronic inflammation, acute phase proteins (precursors of serum amyloid A – SAA) are synthesized in the liver [76]. Another interesting feature of the model is that spleen extracts of an already infected animal, when transferred to another animal, can competitively accelerate the process of fibril deposition. Such acceleration was explained by the presence of fibril fragments that can serve as beginnings for genesis and deposition of newly formed amyloid precursors [91]. By associating monomers to the termini, these seeds can form elongated protofibrils, which later can form amyloid fibrils. Of interest is that the murine model of amyloidogenesis can be activated by incorporating seeds of xenogenous origin, which shows that the seeds have comparable structural elements [92, 93]. A similar phenomenon was found for the model of spontaneous amyloidogenesis in SAMP1 mice. This line is characterized by the existence of an isoform of protein ApoA2(C) with the following substitutions: Gln for Pro in position 5, Ala for Val in position 26, and Met for Val in position 38 [94]. The rate of amyloid accumulation in the animals is directly proportional to their age and, as has been revealed, amyloid accumulation in young animals can be accelerated by introduction of preparations obtained from aged animals [95, 96]. These models are characterized by the requirement of a sufficient amount of the amyloid precursor, caused genetically in one case (ApoA2-AA amyloidosis) and by artificially stimulated inflammation in the other case of AA amyloidosis. Mutual effect of ApoA2-AA is also likely upon either inhibition or acceleration of fibril formation [97].
Protein ApoE is no less important in the formation of amyloid fibrils. The progeny produced by cross breeding of apoe-(ApoE–/–) knockout animals with transgenic mice with hyperexpression of mutant human protein APP-(PDAPP+/+) contained much less cerebral Aβ deposits compared to the transgenic parents (PDAPP+/+ ApoE+/+) [98, 99]. As a result of these experiments, it was suggested that protein ApoE might be used for the treatment of Alzheimer’s disease [100]. However, further experiments with apoe knockout mice demonstrated that such animals are sensitive to amyloidosis caused by IAPP to the same extent as the wild-type mice. There are conflicting data on the increase or decrease in depositions in inflammation-induced AA amyloidoses [101-103]. It was shown that protein SAP in vitro inhibits cleavage of amyloid fibrils not because of direct inhibition of the process, but rather because of direct binding to fibrils, thus shielding potential cleavage sites from proteases [80]. The sap knockout mice had a lower rate of amyloidogenesis in response to inflammatory stimulation, which agrees with the above observations, but these experiments do not indicate that protein SAP is required for fibrogenesis [104, 105]. The absence of sustainable models with knockout of the gene encoding perlecan prevents experiments like those with SAP protein. At the same time, the observations of expression of the gene encoding perlecan before the production of amyloid depositions in a murine model of AA-amyloidosis in tissues, as well as the experiments in vitro, demonstrated the acceleration of fibrillogenesis by the action of both the core protein perlecan (a heparan-sulfate proteoglycan) and associated with it chains of heparan sulfate and chondroitin sulfate. In particular, the formation of amyloids in transgenic mice with hyperexpression of heparanase was inhibited. Moreover, other sulfated glucosoaminoglycans, such as heparin, dermatan sulfate, dextran sulfate, and pentosan, augmented the formation of amyloid fibrils [106-109].
The use of these models shows that homomolecular aggregation associated with nucleation and the subsequent growth of fibrils occurs in connection with heteromolecular interactions, some of which are amyloidogenic and others are anti-amyloidogenic.
Monitoring of these models permitted posing the important question whether all amyloids are prion-like (or possess infectious activity) and whether this phenomenon can hold true for some forms of amyloids causing neurodegenerative diseases capable of transmission over the whole organism [110]. However, one should take into account the difference between cell-to-cell transmission of amyloids in a restricted space where autologous proteins and genetic compatibility for direct cell-to-cell transmission exist and the spread of infectious diseases from the environment, which imposes strict requirements on the amount and quantity of interactions between the seed and further monomers of the aggregate. The difference between prions existing predominantly as fibrils and infectious prions as well as the visible interleaving of the aggregation and neurotoxicity phases during crisis may reflect in some way acquisition or selection of conformational compatibility required for infectivity or toxicity [111, 112].
CHARACTERIZATION OF KINETICS OF AMYLOIDOGENESIS
To make the description of amyloidogenesis complete, it is necessary to find all possible conformational states of the protein during its aggregation as well as possible oligomeric structures formed during amyloidogenesis. Moreover, the description would not be complete without a corresponding determination of kinetic and thermodynamic parameters for all potential forms and states of the protein in the course of aggregation. It should be noted that these requirements include also the analysis of aggregation on the molecular level for subsequent revealing of the regions vital for the whole process of amyloidogenesis. It is accepted that this process has a nucleation stage (see Fig. 2). The aggregation of protein monomers into a fibril can be divided in two stages: the lag-time when seeds are generated, and the time of transition of all monomers into an aggregate. Notice that the latter phase varies depending on the protein and can be either linear or exponential [113]. Reactions having a nucleation stage have been studied quite well from the theoretical and experimental points of view for a wide range of substances, mostly in connection with crystallization of large and small molecules [114].
Fig. 2. Schematic representation of amyloidogenesis. km+ is the rate constant of monomer association with the oligomer; km– is the rate constant of monomer dissociation from the oligomer; kn+ is the rate constant of transition of the seed (an oligomer consisting of s monomers) from state α to state β; kp is the polymerization rate constant; kexp is the rate constant of exponential increase of the ends of a growing fibril.
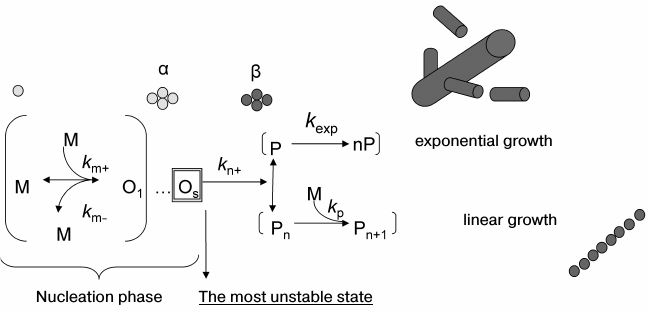
Like for other processes with a nucleation phase (including crystallization), the addition of primes from the seeds already formed during amyloidogenesis practically levels off the lag-period because the aggregation rate is no longer limited by the nucleation stage [115, 116]. It was demonstrated that introduction of particular mutations to aggregating proteins or certain changes in experimental conditions could also level off the lag-period, assuming that nucleation is not a limiting stage of the process any longer [117-119]. The absence of a lag-period is not necessarily related to the fact that the description of the aggregation mechanism with a nucleation stage is no longer applicable. Rather, it shows that the time required for fibril formation is quite large relative to the rate of seed formation, and so nucleation is not already a rate-limiting stage during the transition of the protein from the soluble form to an amyloid. Though no fibrils appear in the lag-period, it is clear that this is an essential stage for the formation of different oligomers including those that would serve as a seed for the formation of mature fibrils.
The efficiency of the fibrils formed in stimulation of aggregation as a seed can strongly decrease on an increase in the differences of the primary structure [120-122]. It was demonstrated for the example of immunoglobulin domains with different primary structure that co-aggregation of various types of domains does not occur when the identity of the protein primary structure is below 30-40% [121]. Bioinformatics analysis of homologous sequential domains in large multidomain proteins revealed identity of less than 40%, which may be evidence of the primary structure of such proteins being composed to avoid aggregation.
During recent decades, significant attempts have been made to identify, isolate, and describe oligomeric particles formed in solution before fibrils appear. This interest in oligomers can be explained by two reasons: apparently, the formation of such particles is a vital stage in amyloidogenesis, and one has every reason to believe that oligomers are the greatest menace for pathogenesis, i.e. they are toxic. As an example, we can take the formation of amyloids by the Aβ-peptide. The aggregation of this peptide is preceded by the development of a number of metastable non-fibril formations observed using the AFM and TEM techniques [123-126]. Some of the formations look like small spherical beads 2-5-nm in diameter, and others look like small beads on a string with individual beads having also the diameter of 2-5 nm, and the third form ring structures that have evidently appeared as a result of closure of the structures similar to the beads on a string. All these assemblies, called protofibrils by the authors who were the first to detect them [123-126] should not be confused however with protofilaments that are singular threads of mature fibrils. Protofibrils, which consist of the Aβ peptide, can bind Congo red and thioflavin T [126], contain a large portion of β-structure and are composed of about 20 molecules forming small spherical particles.
Similar spherical and bead-like protofibril formations were also detected in other systems including α-synuclein [127], amylin [128], immunoglobulin light chains [129], transthyretin [130], poly-Q [128], β2-microglobulin [131], lysozyme [132], acylphosphatase from Sulfolobus solfataricus [133], and the SH3 domain [134]. These formations can be characterized as particles rich in β-structure with sufficiently high orderliness allowing for binding Congo red and thioflavin T. A peculiarity of the formations is the affinity of specific antibodies to protofibrils obtained from different sources, the absence of affinity to monomer units and mature particles, which suggests the presence of definite similar structural elements in soluble oligomer formations.
In some cases protofibrils may be off pathway aggregates [131, 135], but there are data showing that protofibrils represent a state that the protein overcomes during amyloidogenesis [116, 123]. It was found that the protofibril → mature fibril transition of peptide (109-122) from Syrian hamster prion protein proceeds through the adjustment of originally non-aligned β-regions forming a potential fibril [136]. This alignment includes isolation of β-regions with their subsequent inclusion into the potential fibril; however, internal rearrangement of β-regions is also possible, which is realized subject to conditions [137]. Summarizing the above, it can be stated that independent of the particular role of protofibrils in the formation of amyloid fibrils, the determination of the mechanism of formation of such oligomer particles as well as their structure is extremely important especially because it is believed that amyloid fibrils are the basic toxic formations involved in neurodegenerative diseases.
Further investigations of oligomer particles preceding the formation of fibrils have become feasible through the method of photoinduced coupling of unmodified proteins (PICUP). Thus, it has been found that soluble oligomers formed by the Aβ-peptide (both versions 1-40 and 1-42) exist in dynamic equilibrium with the monomer form. These oligomers consist of 2-4 monomers for Aβ 1-40 and 5-6 monomers for Aβ 1-42 and, as shown by CD methods, are relatively unstructured [138]. The interest in oligomer formations of Aβ was driven, in particular, by detection of such particles in the brains of patients with Alzheimer’s disease [139] and also in lysates and the solution concentrate with cells expressing the precursor protein of Aβ [140, 141].
Region NM of yeast prion Sup35p is liable also to fast formation of unstructured oligomers. Transition with a growing β-structure takes place only after the formation of such oligomers, and then after transformation they can serve as seeding for fibrils [116]. The transition can occur due to covalent dimerization of NM molecules if the residues in the head of particle N (25-38) are cross-linked. Moreover, provided the oligomers are kept in an oxygen-enriched medium, intramolecular disulfide bridges form more promptly for the molecules in which cysteines are in the N region. These results apparently show that the interaction of the two head regions of molecule N generates a seed for an amyloid-like structure within the aggregate.
Similar behavior was detected for aggregation of denatured yeast phosphoglycerate kinase at low pH using dynamic light scattering and CD spectroscopy [142].
In this case, stabilization of β-structure occurred with growing dimensions of the aggregate. When a critical mass is accumulated, oligomers stick together and form short irregular protofibrils similar to those formed by Aβ and α-synuclein [142]. Moreover, the unfolding of the SH3 domain of bovine phosphatidylinositol kinase leads to fast formation of unstructured particles of different dimensions, which then transform sequentially into thin irregular protofibrils that bind thioflavin T [134]. So, the experimental data show that structured protofibrils can form either by sticking to each other or through rearrangement of small relatively unstructured oligomers formed at the very beginning of aggregation.
CONCLUSION
Many natural proteins can form amyloid fibrils. Nevertheless, the molecular mechanism underlying the formation of amyloids is still unclear. This is because a vast number of factors can affect the conformational transition from a “native” protein into pathological aggregates, including high protein concentration, specific proteolytic cleavage, mutations, interaction with ligands, and many other factors not yet determined. At present, many questions concerning the process of protein aggregation remain unanswered. A striking circumstance is that amyloidogenesis is specific for all living beings from microorganisms to humans. Based on this, it can be concluded that formation of amyloid fibrils has been tested by evolution and has preserved a conservative structure. The unique physicochemical properties of amyloids and the fact that many proteins form them with an ability to regulate the process as well as its being widespread in nature highlight the biological requirement for such formations. In spite of a great variety of neurodegenerative and other diseases associated with the formation of pathological protein aggregations, it can be proposed that the ability of proteins to form amyloid aggregates is connected first of all with multiple functions of proteins, and therefore the library of proteins generating “functional” amyloids will widen. Further studies are required to clarify the detailed mechanism of fibril amyloidogenesis and to reveal certain determinants affecting the kinetics of the process. But first and foremost is determination of possible key factors for controlling the process of aggregation to prevent development of pathologies and diseases caused by them, or on the contrary to use them for amelioration of health, for example, for improving antiviral immunity or memory strength.
This study was supported by the Russian Science Foundation No. 14-14-00536.
REFERENCES
1.Anfinsen, C. B. (1973) Principles that govern the
folding of protein chains, Science, 181, 223-230.
2.Neumann, M., Rademakers, R., Roeber, S., Baker, M.,
Kretzschmar, H. A., and Mackenzie, I. R. A. (2009) A new subtype of
frontotemporal lobar degeneration with FUS pathology, Brain J.
Neurol., 132, 2922-2931.
3.Doi, H., Koyano, S., Suzuki, Y., Nukina, N., and
Kuroiwa, Y. (2010) The RNA-binding protein FUS/TLS is a common
aggregate-interacting protein in polyglutamine diseases, Neurosci.
Res., 66, 131-133.
4.Kwiatkowski, T. J., Jr., Bosco, D. A., Leclerc, A.
L., Tamrazian, E., Vanderburg, C. R., Russ, C., Davis, A., Gilchrist,
J., Kasarskis, E. J., Munsat, T., Valdmanis, P., Rouleau, G. A.,
Hosler, B. A., Cortelli, P., de Jong, P. J., Yoshinaga, Y., Haines, J.
L., Pericak-Vance, M. A., Yan, J., Ticozzi, N., Siddique, T.,
McKenna-Yasek, D., Sapp, P. C., Horvitz, H. R., Landers, J. E., and
Brown, R. H., Jr. (2009) Mutations in the FUS/TLS gene on chromosome 16
cause familial amyotrophic lateral sclerosis, Science,
323, 1205-1208.
5.Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K.
J., Nishimura, A. L., Sreedharan, J., Hu, X., Smith, B., Ruddy, D.,
Wright, P., Ganesalingam, J., Williams, K. L., Tripathi, V., Al-Saraj,
S., Al-Chalabi, A., Leigh, P. N., Blair, I. P., Nicholson, G., de
Belleroche, J., Gallo, J.-M., Miller, C. C., and Shaw, C. E. (2009)
Mutations in FUS, an RNA processing protein, cause familial amyotrophic
lateral sclerosis type 6, Science, 323, 1208-1211.
6.Sunde, M., and Blake, C. (1997) The structure of
amyloid fibrils by electron microscopy and X-ray diffraction, Adv.
Protein Chem., 50, 123-159.
7.Perutz, M. F., Finch, J. T., Berriman, J., and
Lesk, A. (2002) Amyloid fibers are water-filled nanotubes, Proc.
Natl. Acad. Sci. USA, 99, 5591-5595.
8.Virchow, R. (1857) Neue beobacktungen uber amyloid
degeneration, Virchows Arch. Pathol. Anat. Physiol., 11,
188-189.
9.Divry, P., and Florkin, M. (1927) Sur les
proprietes optiques de l’amyloide, Comptes Rendus. Soc.
Biol., 97, 1808-1810.
10.Pras, M., Schubert, M., Zucker-Franklin, D.,
Rimon, A., and Franklin, E. C. (1968) The characterization of soluble
amyloid prepared in water, J. Clin. Invest., 47,
924-933.
11.Bennhold, H. (1923) Uber die Ausscheidung
intravenous einverbleibten Kongorotes bei den verschiedensten
Erkrankungen insbesondere bei amyloidosis, Dtsch. Arch. Klin.
Med., 142, 32-46.
12.Ouchi, E., Nomura, N., Watabe, S., Seiji, K., and
Sato, J. (1976) Clinical significance of Congo red test, Tohoku J.
Exp. Med., 118, 191-198.
13.Linke, R. (2006) Congo red staining of amyloid:
improvements and practical guide for a more precise diagnosis of
amyloid and the different amyloidoses, in Protein Reviews, Vol.
4, Protein Misfolding, Aggregation and Conformational Diseases
(Uversky, V., and Fink, A., eds.) Springer, pp. 239-276.
14.Groenning, M. (2010) Binding mode of thioflavin T
and other molecular probes in thecontext of amyloid fibrils-current
status, J. Chem. Biol., 3, 1-18.
15.Xu, H., He, X., Zheng, H., Huang, L. J., Hou, F.,
Yu, Z., de la Cruz, M. J., Borkowski, B., Zhang, X., Chen, Z. J., and
Jiang, Q.-X. (2014) Structural basis for the prion-like MAVS filaments
in antiviral innate immunity, eLife, 3, e01489.
16.Narkiewicz, J., Giachin, G., and Legname, G.
(2014) In vitro aggregation assays for the characterization of
α-synuclein prion-like properties, Prion, 8,
19-32.
17.Sydow, A., and Mandelkow, E.-M. (2010)
“Prion-like” propagation of mouse and human τ
aggregates in an inducible mouse model of tauopathy, Neurodegener.
Dis., 7, 28-31.
18.Kato, M., Han, T. W., Xie, S., Shi, K., Du, X.,
Wu, L. C., Mirzaei, H., Goldsmith, E. J., Longgood, J., Pei, J.,
Grishin, N. V., Frantz, D. E., Schneider, J. W., Chen, S., Li, L.,
Sawaya, M. R., Eisenberg, D., Tycko, R., and McKnight, S. L. (2012)
Cell-free formation of RNA granules: low complexity sequence domains
form dynamic fibers within hydrogels, Cell, 149,
753-767.
19.Sulatskaya, A., and Kuznetsova, I. (2010)
Interaction of thioflavin T with amyloid fibrils as an instrument for
study their structure, Tsitologiya, 52, 955-959.
20.Biancalana, M., and Koide, S. (2010) Molecular
mechanism of thioflavin T binding to amyloid fibrils, Biochim.
Biophys. Acta, 1804, 1405-1412.
21.Gendoo, D. M. A., and Harrison, P. M. (2011)
Origins and evolution of the HET-s prion-forming protein: searching for
other amyloid-forming solenoids, PloS One, 6, e27342.
22.King, O. D., Gitler, A. D., and Shorter, J.
(2012) The tip of the iceberg: RNA-binding proteins with prion-like
domains in neurodegenerative disease, Brain Res., 1462,
61-80.
23.Aguzzi, A., and Rajendran, L. (2009) The
transcellular spread of cytosolic amyloids, prions, and prionoids,
Neuron, 64, 783-790.
24.Prusiner, S. B., Scott, M. R., DeArmond, S. J.,
and Cohen, F. E. (1998) Prion protein biology, Cell, 93,
337-348.
25.Halfmann, R., Alberti, S., and Lindquist, S.
(2010) Prions, protein homeostasis, and phenotypic diversity, Trends
Cell Biol., 20, 125-133.
26.Hou, F., Sun, L., Zheng, H., Skaug, B., Jiang,
Q.-X., and Chen, Z. J. (2011) MAVS forms functional prion-like
aggregates to activate and propagate antiviral innate immune response,
Cell, 146, 448-461.
27.Bailey, C. H., Kandel, E. R., and Si, K. (2004)
The persistence of long-term memory: a molecular approach to
self-sustaining changes in learning-induced synaptic growth,
Neuron, 44, 49-57.
28.Gilks, N., Kedersha, N., Ayodele, M., Shen, L.,
Stoecklin, G., Dember, L. M., and Anderson, P. (2004) Stress granule
assembly is mediated by prion-like aggregation of TIA-1, Mol. Biol.
Cell, 15, 5383-5398.
29.Shorter, J., and Lindquist, S. (2004) Hsp104
catalyzes formation and elimination of self-replicating Sup35 prion
conformers, Science, 304, 1793-1797.
30.Halfmann, R., Alberti, S., Krishnan, R., Lyle,
N., O’Donnell, C. W., King, O. D., Berger, B., Pappu, R. V., and
Lindquist, S. (2011) Opposing effects of glutamine and asparagine
govern prion formation by intrinsically disordered proteins, Mol.
Cell, 43, 72-84.
31.Han, T. W., Kato, M., Xie, S., Wu, L. C.,
Mirzaei, H., Pei, J., Chen, M., Xie, Y., Allen, J., Xiao, G., and
McKnight, S. L. (2012) Cell-free formation of RNA granules: bound RNAs
identify features and components of cellular assemblies, Cell,
149, 768-779.
32.Sun, Z., Diaz, Z., Fang, X., Hart, M. P., Chesi,
A., Shorter, J., and Gitler, A. D. (2011) Molecular determinants and
genetic modifiers of aggregation and toxicity for the ALS disease
protein FUS/TLS, PLoS Biol., 9, e1000614.
33.Wickner, R. B., Taylor, K. L., Edskes, H. K., and
Maddelein, M. L. (2000) Prions: portable prion domains, Curr. Biol.
CB, 10, 335-337.
34.Li, L., and Lindquist, S. (2000) Creating a
protein-based element of inheritance, Science, 287,
661-664.
35.Kaiser, T. E., Intine, R. V., and Dundr, M.
(2008) De novo formation of a subnuclear body, Science,
322, 1713-1717.
36.Shevtsov, S. P., and Dundr, M. (2011) Nucleation
of nuclear bodies by RNA, Nat. Cell Biol., 13,
167-173.
37.Li, Y. R., King, O. D., Shorter, J., and Gitler,
A. D. (2013) Stress granules as crucibles of ALS pathogenesis, J.
Cell Biol., 201, 361-372.
38.Cushman, M., Johnson, B. S., King, O. D., Gitler,
A. D., and Shorter, J. (2010) Prion-like disorders: blurring the divide
between transmissibility and infectivity, J. Cell Sci.,
123, 1191-1201.
39.Frost, B., and Diamond, M. I. (2010) Prion-like
mechanisms in neurodegenerative diseases, Nat. Rev. Neurosci.,
11, 155-159.
40.Braak, H., Rub, U., Gai, W. P., and Del Tredici,
K. (2003) Idiopathic Parkinson’s disease: possible routes by
which vulnerable neuronal types may be subject to neuroinvasion by an
unknown pathogen, J. Neural Transm., 110, 517-536.
41.Ravits, J. M., and La Spada, A. R. (2009) ALS
motor phenotype heterogeneity, focality, and spread: deconstructing
motor neuron degeneration, Neurology, 73, 805-811.
42.Selkoe, D. J. (1999) Translating cell biology
into therapeutic advances in Alzheimer’s disease, Nature,
399, 23-31.
43.Alberti, S., Halfmann, R., King, O., Kapila, A.,
and Lindquist, S. (2009) A systematic survey identifies prions and
illuminates sequence features of prionogenic proteins, Cell,
137, 146-158.
44.Dormann, D., Rodde, R., Edbauer, D., Bentmann,
E., Fischer, I., Hruscha, A., Than, M. E., Mackenzie, I. R. A., Capell,
A., Schmid, B., Neumann, M., and Haass, C. (2010) ALS-associated fused
in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import,
EMBO J., 29, 2841-2857.
45.Gal, J., Zhang, J., Kwinter, D. M., Zhai, J.,
Jia, H., Jia, J., and Zhu, H. (2011) Nuclear localization sequence of
FUS and induction of stress granules by ALS mutants, Neurobiol.
Aging, 32, 27-40.
46.Ito, D., Seki, M., Tsunoda, Y., Uchiyama, H., and
Suzuki, N. (2011) Nuclear transport impairment of amyotrophic lateral
sclerosis-linked mutations in FUS/TLS, Ann. Neurol., 69,
152-162.
47.Kino, Y., Washizu, C., Aquilanti, E., Okuno, M.,
Kurosawa, M., Yamada, M., Doi, H., and Nukina, N. (2011) Intracellular
localization and splicing regulation of FUS/TLS arevariably affected by
amyotrophic lateral sclerosis-linked mutations, Nucleic Acids
Res., 39, 2781-2798.
48.Niu, C., Zhang, J., Gao, F., Yang, L., Jia, M.,
Zhu, H., and Gong, W. (2012) FUS-NLS/transportin 1 complex structure
provides insights into the nuclear targeting mechanism of FUS and the
implications in ALS, PloS One, 7, e47056.
49.Zhang, Z. C., and Chook, Y. M. (2012) Structural
and energetic basis of ALS-causing mutations in the atypical
proline-tyrosine nuclear localization signal of the fused in sarcoma
protein (FUS), Proc. Natl. Acad. Sci. USA, 109,
12017-12021.
50.Seyfried, N. T., Gozal, Y. M., Donovan, L. E.,
Herskowitz, J. H., Dammer, E. B., Xia, Q., Ku, L., Chang, J., Duong, D.
M., Rees, H. D., Cooper, D. S., Glass, J. D., Gearing, M., Tansey, M.
G., Lah, J. J., Feng, Y., Levey, A. I., and Peng, J. (2012)
Quantitative analysis of the detergent-insoluble brain proteome in
frontotemporal lobar degeneration using SILAC internal standards, J.
Proteome Res., 11, 2721-2738.
51.Klar, J., Sobol, M., Melberg, A., Mabert, K.,
Ameur, A., Johansson, A. C. V., Feuk, L., Entesarian, M., Orlen, H.,
Casar-Borota, O., and Dahl, N. (2013) Welander distal myopathy caused
by an ancient founder mutation in TIA1 associated with perturbed
splicing, Hum. Mutat., 34, 572-577.
52.Sawaya, M. R., Sambashivan, S., Nelson, R.,
Ivanova, M. I., Sievers, S. A., Apostol, M. I., Thompson, M. J.,
Balbirnie, M., Wiltzius, J. J. W., McFarlane, H. T., Madsen, A. O.,
Riekel, C., and Eisenberg, D. (2007) Atomic structures of amyloid
cross-β spines reveal varied steric zippers, Nature,
447, 453-457.
53.Chapman, M. R., Robinson, L. S., Pinkner, J. S.,
Roth, R., Heuser, J., Hammar, M., Normark, S., and Hultgren, S. J.
(2002) Role of Escherichia coli curli operons in directing
amyloid fiber formation, Science, 295, 851-855.
54.Claessen, D., Rink, R., de Jong, W., Siebring,
J., de Vreugd, P., Boersma, F. G. H., Dijkhuizen, L., and Wosten, H. A.
B. (2003) A novel class of secreted hydrophobic proteins is involved in
aerial hyphae formation in Streptomyces coelicolor by forming
amyloid-like fibrils, Genes Dev., 17, 1714-1726.
55.Berson, J. F., Theos, A. C., Harper, D. C.,
Tenza, D., Raposo, G., and Marks, M. S. (2003) Proprotein convertase
cleavage liberates a fibrillogenic fragment of a resident glycoprotein
to initiate melanosome biogenesis, J. Cell Biol., 161,
521-533.
56.Tompa, P., and Friedrich, P. (1998) Prion
proteins as memory molecules: an hypothesis, Neuroscience,
86, 1037-1043.
57.Onoguchi, K., Onomoto, K., Takamatsu, S., Jogi,
M., Takemura, A., Morimoto, S., Julkunen, I., Namiki, H., Yoneyama, M.,
and Fujita, T. (2010) Virus-infection or 5′ppp-RNA activates
antiviral signal through redistribution of IPS-1 mediated by MFN1,
PloS Pathog., 6, e1001012.
58.Jacobs, J. L., and Coyne, C. B. (2013) Mechanisms
of MAVS regulation at the mitochondrial membrane, J. Mol. Biol.,
425, 5009-5019.
59.Glenner, G. G., Ein, D., Eanes, E. D., Bladen, H.
A., Terry, W., and Page, D. L. (1971) Creation of “amyloid”
fibrils from Bence Jones proteins in vitro, Science,
174, 712-714.
60.Levin, M., Franklin, E. C., Frangione, B., and
Pras, M. (1972) The amino acid sequence of a major non-immunoglobulin
component of some amyloid fibrils, J. Clin. Invest., 51,
2773-2776.
61.Glenner, G. G., Terry, W., Harada, M., Isersky,
C., and Page, D. (1971) Amyloid fibril proteins: proof of homology with
immunoglobulin light chains by sequence analyses, Science,
172, 1150-1151.
62.Terry, W. D., Page, D. L., Kimura, S., Isobe, T.,
Osserman, E. F., and Glenner, G. G. (1973) Structural identity of Bence
Jones and amyloid fibril proteins in a patient with plasma cell
dyscrasia and amyloidosis, J. Clin. Invest., 52,
1276-1281.
63.Buxbaum, J. (1992) Mechanisms of disease:
monoclonal immunoglobulin deposition. Amyloidosis, light chain
deposition disease, and light and heavy chain deposition disease,
Hematol. Oncol. Clin. North Am., 6, 323-346.
64.Buxbaum, J. (1986) Aberrant immunoglobulin
synthesis in light chain amyloidosis. Free light chain and light chain
fragment production by human bone marrow cells in short-term tissue
culture, J. Clin. Invest., 78, 798-806.
65.Linke, R. P., Zucker-Franklin, D., and Franklin,
E. D. (1973) Morphologic, chemical, and immunologic studies of
amyloid-like fibrils formed from Bence Jones proteins by proteolysis,
J. Immunol. Baltim. Md 1950, 111, 10-23.
66.Stevens, F. J. (2000) Four structural risk
factors identify most fibril-forming κ light chains, Amyloid
Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis,
7, 200-211.
67.Hurle, M. R., Helms, L. R., Li, L., Chan, W., and
Wetzel, R. (1994) A role for destabilizing amino acid replacements in
light-chain amyloidosis, Proc. Natl. Acad. Sci. USA, 91,
5446-5450.
68.Kaplan, B., Vidal, R., Kumar, A., Ghiso, J.,
Frangione, B., and Gallo, G. (1997) Amino-terminal identity of
co-existent amyloid and non-amyloid immunoglobulin κ light chain
deposits. A human disease to study alterations of protein conformation,
Clin. Exp. Immunol., 110, 472-478.
69.Putnam, F. W., Easley, C. W., Lynn, L. T.,
Ritchie, A. E., and Phelps, R. A. (1959) The heat precipitation of
Bence–Jones proteins. I. Optimum conditions, Arch. Biochem.
Biophys., 83, 115-130.
70.Solomon, A., Weiss, D. T., and Kattine, A. A.
(1991) Nephrotoxic potential of Bence–Jones proteins, N. Engl.
J. Med., 324, 1845-1851.
71.Eulitz, M., Weiss, D. T., and Solomon, A. (1990)
Immunoglobulin heavy-chain-associated amyloidosis, Proc. Natl. Acad.
Sci. USA, 87, 6542-6546.
72.Benditt, E. P., Eriksen, N., Hermodson, M. A.,
and Ericsson, L. H. (1971) The majorproteins of human and monkey
amyloid substance: common properties including unusual N-terminal amino
acid sequences, FEBS Lett., 19, 169-173.
73.Sletten, K., and Husby, G. (1974) The complete
amino-acid sequence of non-immunoglobulin amyloid fibril protein AS in
rheumatoid arthritis, Eur. J. Biochem. FEBS, 41,
117-125.
74.Manley, P. N., Ancsin, J. B., and Kisilevsky, R.
(2006) Rapid recycling of cholesterol: the joint biologic role of
C-reactive protein and serum amyloid A, Med. Hypotheses,
66, 784-792.
75.Chapman, M. R., Robinson, L. S., Pinkner, J. S.,
Roth, R., Heuser, J., Hammar, M., Normark, S., and Hultgren, S. J.
(2002) Role of Escherichia coli curli operons in directing
amyloid fiber formation, Science, 295, 851-855.
76.Rocken, C., and Shakespeare, A. (2002) Pathology,
diagnosis and pathogenesis of AA amyloidosis, Virchows Arch. Int. J.
Pathol., 440, 111-122.
77.Skinner, M., and Cohen, A. S. (1988) Amyloid P
component, Methods Enzymol., 163, 523-536.
78.Ancsin, J. B. (2003) Amyloidogenesis: historical
and modern observations point to heparan sulfate proteoglycans as a
major culprit, Amyloid Int. J. Exp. Clin. Investig. Off. J. Int.
Soc. Amyloidosis, 10, 67-79.
79.Gallo, G., Wisniewski, T., Choi-Miura, N. H.,
Ghiso, J., and Frangione, B. (1994) Potential role of apolipoprotein-E
in fibrillogenesis, Am. J. Pathol., 145, 526-530.
80.Tennent, G. A., Lovat, L. B., and Pepys, M. B.
(1995) Serum amyloid P component prevents proteolysis of the amyloid
fibrils of Alzheimer disease and systemic amyloidosis, Proc. Natl.
Acad. Sci. USA, 92, 4299-4303.
81.Husby, G., and Natvig, J. B. (1974) A serum
component related to nonimmunoglobulin amyloid protein AS, a possible
precursor of the fibrils, J. Clin. Invest., 53,
1054-1061.
82.Husby, G., Husebekk, A., Skogen, B., Sletten, K.,
Marhaug, G., Magnus, J., and Syversen, V. (1988) Serum amyloid A (SAA)
– the precursor of protein AA in secondary amyloidosis, Adv.
Exp. Med. Biol., 243, 185-192.
83.Westermark, P., Wernstedt, C., Wilander, E.,
Hayden, D. W., O’Brien, T. D., and Johnson, K. H. (1987) Amyloid
fibrils in human insulinoma and islets of Langerhans of the diabetic
cat are derived from a neuropeptide-like protein also present in normal
islet cells, Proc. Natl. Acad. Sci. USA, 84,
3881-3885.
84.Masters, C. L., Multhaup, G., Simms, G.,
Pottgiesser, J., Martins, R. N., and Beyreuther, K. (1985) Neuronal
origin of a cerebral amyloid: neurofibrillary tangles of
Alzheimer’s disease contain the same protein as the amyloid of
plaque cores and blood vessels, EMBO J., 4,
2757-2763.
85.Oesch, B., Westaway, D., Walchli, M., McKinley,
M. P., Kent, S. B., Aebersold, R., Barry, R. A., Tempst, P., Teplow, D.
B., and Hood, L. E. (1985) A cellular gene encodes scrapie PrP 27-30
protein, Cell, 40, 735-746.
86.Vidal, R., Frangione, B., Rostagno, A., Mead, S.,
Revesz, T., Plant, G., and Ghiso, J. (1999) A stop-codon mutation in
the BRI gene associated with familial British dementia, Nature,
399, 776-781.
87.Glenner, G. G. (1980) Amyloid deposits and
amyloidosis. The β-fibrilloses (first of two parts), N. Engl.
J. Med., 302, 1283-1292.
88.Glenner, G. G., and Wong, C. W. (1984)
Alzheimer’s disease: initial report of the purification and
characterization of a novel cerebrovascular amyloid protein,
Biochem. Biophys. Res. Commun., 120, 885-890.
89.Gruys, E., and Snel, F. W. (1994) Animal models
for reactive amyloidosis, Baillieres Clin. Rheumatol., 8,
599-611.
90.Solomon, A., Weiss, D. T., Schell, M., Hrncic,
R., Murphy, C. L., Wall, J., McGavin, M. D., Pan, H. J., Kabalka, G.
W., and Paulus, M. J. (1999) Transgenic mouse model of AA amyloidosis,
Am. J. Pathol., 154, 1267-1272.
91.Liu, Y., Cui, D., Hoshii, Y., Kawano, H., Une,
Y., Gondo, T., and Ishihara, T. (2007) Induction of murine AA
amyloidosis by various homogeneous amyloid fibrils and amyloid-like
synthetic peptides, Scand. J. Immunol., 66, 495-500.
92.Johan, K., Westermark, G., Engstrom, U.,
Gustavsson, A., Hultman, P., and Westermark, P. (1998) Acceleration of
amyloid protein A amyloidosis by amyloid-like synthetic fibrils,
Proc. Natl. Acad. Sci. USA, 95, 2558-2563.
93.Lundmark, K., Westermark, G. T., Olsen, A., and
Westermark, P. (2005) Protein fibrils in nature can enhance amyloid
protein A amyloidosis in mice: cross-seeding as a disease mechanism,
Proc. Natl. Acad. Sci. USA, 102, 6098-6102.
94.Korenaga, T., Fu, X., Xing, Y., Matsusita, T.,
Kuramoto, K., Syumiya, S., Hasegawa, K., Naiki, H., Ueno, M., Ishihara,
T., Hosokawa, M., Mori, M., and Higuchi, K. (2004) Tissue distribution,
biochemical properties, and transmission of mouse type A AApoAII
amyloid fibrils, Am. J. Pathol., 164, 1597-1606.
95.Xing, Y., Nakamura, A., Chiba, T., Kogishi, K.,
Matsushita, T., Li, F., Guo, Z., Hosokawa, M., Mori, M., and Higuchi,
K. (2001) Transmission of mouse senile amyloidosis, Lab.
Invest., 81, 493-499.
96.Elliott-Bryant, R., and Cathcart, E. S. (1998)
Amyloid enhancing factor and dietary transmission in accelerated
amyloid A amyloidosis, Clin. Immunol. Immunopathol., 88,
65-69.
97.Yan, J., Fu, X., Ge, F., Zhang, B., Yao, J.,
Zhang, H., Qian, J., Tomozawa, H., Naiki, H., Sawashita, J., Mori, M.,
and Higuchi, K. (2007) Cross-seeding and cross-competition in mouse
apolipoprotein A-II amyloid fibrils and protein A amyloid fibrils,
Am. J. Pathol., 171, 172-180.
98.Bales, K. R., Verina, T., Dodel, R. C., Du, Y.,
Altstiel, L., Bender, M., Hyslop, P., Johnstone, E. M., Little, S. P.,
Cummins, D. J., Piccardo, P., Ghetti, B., and Paul, S. M. (1997) Lack
of apolipoprotein E dramatically reduces amyloid β-peptide
deposition, Nat. Genet., 17, 263-264.
99.Wisniewski, T., Castano, E. M., Golabek, A.,
Vogel, T., and Frangione, B. (1994) Acceleration of Alzheimer’s
fibril formation by apolipoprotein E in vitro, Am. J.
Pathol., 145, 1030-1035.
100.Holtzman, D. M. (2004) In vivo effects
of ApoE and clusterin on amyloid-β metabolism and
neuropathology, J. Mol. Neurosci. MN, 23, 247-254.
101.Vidal, J., Verchere, C. B., Andrikopoulos, S.,
Wang, F., Hull, R. L., Cnop, M., Olin, K. L., LeBoeuf, R. C.,
O’Brien, K. D., Chait, A., and Kahn, S. E. (2003) The effect of
apolipoprotein E deficiency on islet amyloid deposition in human islet
amyloid polypeptide transgenic mice, Diabetologia, 46,
71-79.
102.Kindy, M. S., and Rader, D. J. (1998) Reduction
in amyloid A amyloid formation in apolipoprotein-E-deficient mice,
Am. J. Pathol., 152, 1387-1395.
103.Elliott-Bryant, R., and Cathcart, E. S. (1997)
Apolipoprotein E and apolipoprotein A-1 knock-out mice readily develop
amyloid A protein amyloidosis, Clin. Immunol. Immunopathol.,
85, 104-108.
104.Botto, M., Hawkins, P. N., Bickerstaff, M. C.,
Herbert, J., Bygrave, A. E., McBride, A., Hutchinson, W. L., Tennent,
G. A., Walport, M. J., and Pepys, M. B. (1997) Amyloid deposition is
delayed in mice with targeted deletion of the serum amyloid P component
gene, Nat. Med., 3, 855-859.
105.Togashi, S., Lim, S. K., Kawano, H., Ito, S.,
Ishihara, T., Okada, Y., Nakano, S., Kinoshita, T., Horie, K.,
Episkopou, V., Gottesman, M. E., Costantini, F., Shimada, K., and
Maeda, S. (1997) Serum amyloid P component enhances induction of murine
amyloidosis, Lab. Investig. J. Tech. Methods Pathol., 77,
525-531.
106.Castillo, G. M., Lukito, W., Wight, T. N., and
Snow, A. D. (1999) The sulfate moieties of glycosaminoglycans are
critical for the enhancement of β-amyloid protein fibril
formation, J. Neurochem., 72, 1681-1687.
107.Li, J.-P., Galvis, M. L. E., Gong, F., Zhang,
X., Zcharia, E., Metzger, S., Vlodavsky, I., Kisilevsky, R., and
Lindahl, U. (2005) In vivo fragmentation of heparan sulfate by
heparanase overexpression renders mice resistant to amyloid protein A
amyloidosis, Proc. Natl. Acad. Sci. USA, 102,
6473-6477.
108.Ailles, L., Kisilevsky, R., and Young, I. D.
(1993) Induction of perlecan gene expression precedes amyloid formation
during experimental murine AA amyloidogenesis, Lab. Investig. J.
Tech. Methods Pathol., 69, 443-448.
109.Elimova, E., Kisilevsky, R., Szarek, W. A., and
Ancsin, J. B. (2004) Amyloidogenesis recapitulated in cell culture: a
peptide inhibitor provides direct evidence for the role of heparan
sulfate and suggests a new treatment strategy, FASEB J. Off. Publ.
Fed. Am. Soc. Exp. Biol., 18, 1749-1751.
110.Lundmark, K., Westermark, G. T., Nystrom, S.,
Murphy, C. L., Solomon, A., and Westermark, P. (2002) Transmissibility
of systemic amyloidosis by a prion-like mechanism, Proc. Natl. Acad.
Sci. USA, 99, 6979-6984.
111.Solomon, I. H., Schepker, J. A., and Harris, D.
A. (2010) Prion neurotoxicity: insights from prion protein mutants,
Curr. Issues Mol. Biol., 12, 51-61.
112.Sandberg, M. K., Al-Doujaily, H., Sharps, B.,
Clarke, A. R., and Collinge, J. (2011) Prion propagation and toxicity
in vivo occur in two distinct mechanistic phases, Nature,
470, 540-542.
113.Dovidchenko, N. V., Finkelstein, A. V., and
Galzitskaya, O. V. (2014) How to determine the size of folding nuclei
of protofibrils from the concentration dependence of the rate and
lag-time of aggregation. I. Modeling the amyloid protofibril formation,
J. Phys. Chem. B, 118, 1189-1197.
114.Chayen, N. E. (2005) Methods for separating
nucleation and growth in protein crystallization, Prog. Biophys.
Mol. Biol., 88, 329-337.
115.Naiki, H., Hashimoto, N., Suzuki, S., Kimura,
H., Nakakuki, K., and Gejyo, F. (1997) Establishment of a kinetic model
of dialysis-related amyloid fibril extension in vitro,
Amyloid, 4, 223-232.
116.Serio, T. R., Cashikar, A. G., Kowal, A. S.,
Sawicki, G. J., Moslehi, J. J., Serpell, L., Arnsdorf, M. F., and
Lindquist, S. L. (2000) Nucleated conformational conversion and the
replication of conformational information by a prion determinant,
Science, 289, 1317-1321.
117.Uversky, V. N., Li, J., Souillac, P., Millett,
I. S., Doniach, S., Jakes, R., Goedert, M., and Fink, A. L. (2002)
Biophysical properties of the synucleins and their propensities to
fibrillate: inhibition of α-synuclein assembly by β- and
γ-synucleins, J. Biol. Chem., 277, 11970-11978.
118.Pedersen, J. S., Christensen, G., and Otzen, D.
E. (2004) Modulation of S6 fibrillation by unfolding rates and
gatekeeper residues, J. Mol. Biol., 341, 575-588.
119.Fezoui, Y., and Teplow, D. B. (2002) Kinetic
studies of amyloid β-protein fibril assembly. Differential effects
of α-helix stabilization, J. Biol. Chem., 277,
36948-36954.
120.Tanaka, M., Chien, P., Yonekura, K., and
Weissman, J. S. (2005) Mechanism of cross-species prion transmission:
an infectious conformation compatible with two highly divergent yeast
prion proteins, Cell, 121, 49-62.
121.Wright, C. F., Teichmann, S. A., Clarke, J.,
and Dobson, C. M. (2005) The importance of sequence diversity in the
aggregation and evolution of proteins, Nature, 438,
878-881.
122.Krebs, M. R. H., Morozova-Roche, L. A., Daniel,
K., Robinson, C. V., and Dobson, C. M. (2004) Observation of sequence
specificity in the seeding of protein amyloid fibrils, Protein Sci.
Publ. Protein Soc., 13, 1933-1938.
123.Harper, J. D., Lieber, C. M., and Lansbury, P.
T., Jr. (1997) Atomic force microscopic imaging of seeded fibril
formation and fibril branching by the Alzheimer’s disease
amyloid-β protein, Chem. Biol., 4, 951-959.
124.Harper, J. D., Wong, S. S., Lieber, C. M., and
Lansbury, P. T. (1997) Observation of metastable Abeta amyloid
protofibrils by atomic force microscopy, Chem. Biol., 4,
119-125.
125.Walsh, D. M., Lomakin, A., Benedek, G. B.,
Condron, M. M., and Teplow, D. B. (1997) Amyloid β-protein
fibrillogenesis. Detection of a protofibrillar intermediate, J.
Biol. Chem., 272, 22364-22372.
126.Walsh, D. M., Hartley, D. M., Kusumoto, Y.,
Fezoui, Y., Condron, M. M., Lomakin, A., Benedek, G. B., Selkoe, D. J.,
and Teplow, D. B. (1999) Amyloid β-protein fibrillogenesis.
Structure and biological activity of protofibrillar intermediates,
J. Biol.Chem., 274, 25945-25952.
127.Conway, K. A., Harper, J. D., and Lansbury, P.
T., Jr. (2000) Fibrils formed in vitro from α-synuclein
and two mutant forms linked to Parkinson’s disease are typical
amyloid, Biochemistry, 39, 2552-2563.
128.Kayed, R., Sokolov, Y., Edmonds, B., McIntire,
T. M., Milton, S. C., Hall, J. E., and Glabe, C. G. (2004)
Permeabilization of lipid bilayers is a common conformation-dependent
activity of soluble amyloid oligomers in protein misfolding diseases,
J. Biol. Chem., 279, 46363-46366.
129.Ionescu-Zanetti, C., Khurana, R., Gillespie, J.
R., Petrick, J. S., Trabachino, L. C., Minert, L. J., Carter, S. A.,
and Fink, A. L. (1999) Monitoring the assembly of Ig light-chain
amyloid fibrils by atomic force microscopy, Proc. Natl. Acad. Sci.
USA, 96, 13175-13179.
130.Quintas, A., Vaz, D. C., Cardoso, I., Saraiva,
M. J., and Brito, R. M. (2001) Tetramer dissociation and monomer
partial unfolding precedes protofibril formation in amyloidogenic
transthyretin variants, J. Biol. Chem., 276,
27207-27213.
131.Gosal, W. S., Morten, I. J., Hewitt, E. W.,
Smith, D. A., Thomson, N. H., and Radford, S. E. (2005) Competing
pathways determine fibril morphology in the self-assembly of
β2-microglobulin into amyloid, J. Mol. Biol., 351,
850-864.
132.Malisauskas, M., Zamotin, V., Jass, J., Noppe,
W., Dobson, C. M., and Morozova-Roche, L. A. (2003) Amyloid
protofilaments from the calcium-binding protein equine lysozyme:
formation of ring and linear structures depends on pH and metal ion
concentration, J. Mol. Biol., 330, 879-890.
133.Plakoutsi, G., Taddei, N., Stefani, M., and
Chiti, F. (2004) Aggregation of the acylphosphatase from Sulfolobus
solfataricus: the folded and partially unfolded states can both be
precursors for amyloid formation, J. Biol. Chem., 279,
14111-14119.
134.Bader, R., Bamford, R., Zurdo, J., Luisi, B.
F., and Dobson, C. M. (2006) Probing the mechanism of amyloidogenesis
through a tandem repeat of the PI3-SH3 domain suggests a generic model
for protein aggregation and fibril formation, J. Mol. Biol.,
356, 189-208.
135.Morozova-Roche, L. A., Zamotin, V.,
Malisauskas, M., Ohman, A., Chertkova, R., Lavrikova, M. A., Kostanyan,
I. A., Dolgikh, D. A., and Kirpichnikov, M. P. (2004) Fibrillation of
carrier protein albebetin and its biologically active constructs.
Multiple oligomeric intermediates and pathways, Biochemistry,
43, 9610-9619.
136.Petty, S. A., Adalsteinsson, T., and Decatur,
S. M. (2005) Correlations among morphology, β-sheet stability, and
molecular structure in prion peptide aggregates, Biochemistry,
44, 4720-4726.
137.Petty, S. A., and Decatur, S. M. (2005)
Inter-sheet rearrangement of polypeptides during nucleation of
β-sheet aggregates, Proc. Natl. Acad. Sci. USA, 102,
14272-14277.
138.Bitan, G., Kirkitadze, M. D., Lomakin, A.,
Vollers, S. S., Benedek, G. B., and Teplow, D. B. (2003) Amyloid
β-protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize
through distinct pathways, Proc. Natl. Acad. Sci. USA,
100, 330-335.
139.Roher, A. E., Chaney, M. O., Kuo, Y. M.,
Webster, S. D., Stine, W. B., Haverkamp, L. J., Woods, A. S., Cotter,
R. J., Tuohy, J. M., Krafft, G. A., Bonnell, B. S., and Emmerling, M.
R. (1996) Morphology and toxicity of Abeta-(1-42) dimer derived from
neuritic and vascular amyloid deposits of Alzheimer’s disease,
J. Biol. Chem., 271, 20631-20635.
140.Podlisny, M. B., Ostaszewski, B. L., Squazzo,
S. L., Koo, E. H., Rydell, R. E., Teplow, D. B., and Selkoe, D. J.
(1995) Aggregation of secreted amyloid β-protein into sodium
dodecyl sulfate-stable oligomers in cell culture, J. Biol.
Chem., 270, 9564-9570.
141.Walsh, D. M., Tseng, B. P., Rydel, R. E.,
Podlisny, M. B., and Selkoe, D. J. (2000) The oligomerization of
amyloid β-protein begins intracellularly in cells derived from
human brain, Biochemistry, 39, 10831-10839.
142.Modler, A. J., Gast, K., Lutsch, G., and
Damaschun, G. (2003) Assembly of amyloid protofibrils via critical
oligomers – a novel pathway of amyloid formation, J. Mol.
Biol., 325, 135-148.