REVIEW: Role of Glutathione, Glutathione Transferase, and Glutaredoxin in Regulation of Redox-Dependent Processes
E. V. Kalinina*, N. N. Chernov, and M. D. Novichkova
Peoples’ Friendship University of Russia, ul. Miklukho-Maklaya 6, 117198 Moscow, Russia; E-mail: kevsan@orc.ru; elena.v.kalinina@gmail.com* To whom correspondence should be addressed.
Received July 17, 2014; Revision received August 10, 2014
Over the last decade fundamentally new features have been revealed for the participation of glutathione and glutathione-dependent enzymes (glutathione transferase and glutaredoxin) in cell proliferation, apoptosis, protein folding, and cell signaling. Reduced glutathione (GSH) plays an important role in maintaining cellular redox status by participating in thiol–disulfide exchange, which regulates a number of cell functions including gene expression and the activity of individual enzymes and enzyme systems. Maintaining optimum GSH/GSSG ratio is essential to cell viability. Decrease in the ratio can serve as an indicator of damage to the cell redox status and of changes in redox-dependent gene regulation. Disturbance of intracellular GSH balance is observed in a number of pathologies including cancer. Consequences of inappropriate GSH/GSSG ratio include significant changes in the mechanism of cellular redox-dependent signaling controlled both nonenzymatically and enzymatically with the participation of isoforms of glutathione transferase and glutaredoxin. This review summarizes recent data on the role of glutathione, glutathione transferase, and glutaredoxin in the regulation of cellular redox-dependent processes.
KEY WORDS: glutathione, glutathione transferase, glutaredoxin, redox regulationDOI: 10.1134/S0006297914130082
Abbreviations: AIF, apoptosis-inducing factor; AMPK, serine/threonine AMP-activated protein kinase; ARE, antioxidant responsive element; ASK1, apoptosis signal-regulating kinase-1; BSO, buthionine sulfoximine; ERK, extracellular signal-regulated kinase; γ-GCL, γ-glutamylcysteine ligase; GPx, glutathione peroxidase; Grx, glutaredoxin; GS, glutathione synthetase; GSH/GSSG, glutathione reduced/oxidized; GST, glutathione S-transferase; γ-GT, γ-glutamyltransferase; JNK, c-Jun N-terminal kinase; LPO, lipid peroxidation; MAPK, mitogen-activated protein kinase; mGSH, mitochondrial glutathione; nGSH, nuclear glutathione; OGC, 2-oxyglutarate carrier; PARP, poly(ADP-ribose)polymerase; Prx, peroxiredoxin; RNS, reactive nitrogen species; ROS, reactive oxygen species; Trx, thioredoxin; TrxR, thioredoxin reductase.
For living cells, control of metabolism and processes of cell
development is of great importance. This is provided to a large extent
by processes of thiol–disulfide exchange. Thiol groups of
cysteine residues are rather important for the functioning of enzymes
and processes underlying the system of the cell response to
environmental factors and the transmission of the information inside
the cell (cell signaling). The main mechanism of thiol-mediated redox
control in cell metabolism is attributed to the ability of the thiol
groups to change reversibly their redox state with subsequent
alterations in conformational, catalytic, or regulatory functions of a
protein. The basis for the redox homeostasis that maintains the redox
state of protein thiol groups is the ratio of the reduced (GSH) to
oxidized (GSSG) glutathione, a peptide that is present in most of cells
in millimolar concentrations [1, 2].
Reduced glutathione (GSH) is a tripeptide consisting of the amino acids L-glutamate, L-cysteine, and glycine. It is less susceptible to oxidation compared to cysteine, which makes it more suitable for maintaining the intracellular redox potential. The importance of GSH in the redox-dependent processes is determined by its involvement in the regulation of cell redox-dependent signaling and the activity of transcriptional factors. Besides, this peptide is also an intracellular antioxidant that scavenges free radicals, plays the role of the cosubstrate in the reaction of peroxide detoxification catalyzed by glutathione peroxidase (GPx) and glutathione transferase (GST), and reduces oxidized glutaredoxin (Grx), which is necessary for the reduction of disulfides [3-5]. Maintenance of the GSH/GSSG ratio at the optimal level is important for cell vitality. A decrease in the GSH content below the normal level may indicate the disturbance of cellular redox status and change in the redox-dependent regulation of genes. Alteration of intracellular GSH balance is observed in a number of pathologies including malignant tumors [6]. S-Glutathionylation of proteins is an important regulatory mechanism in biochemical processes that implies reversible modification of sulfhydryl groups of proteins by both nonenzymatic and enzymatic mechanisms with the participation of GST and Grx [7].
The combination of antioxidant properties with the ability to activate transcription of genes including those of the antioxidant enzymes and to inhibit redox-dependent activation of apoptosis suggests a significant impact of GST and Grx to the antioxidant defense system, which increases the cell resistance to oxidative stress [8-10]. One of the elements of the defense against oxidative stress is GST, which exhibits high activity towards the products of peroxide oxidation of DNA and lipids. Disulfides and mixed disulfides are substrates of Grx, which plays a significant role in thiol–disulfide exchange by regulating among other factors the activity of transcriptional factors and apoptosis. Isoforms of GST and Grx play a significant role in the regulation of cell signaling by protein–protein interactions with regulatory kinases controlling the cell response to stress, proliferation, and induction of apoptosis.
The present review summarizes recent data on the role of glutathione, glutathione transferase, and glutaredoxin in the regulation of redox-dependent processes in cells.
GLUTATHIONE. STRUCTURE AND REDOX-DEPENDENT FUNCTIONS
Structure and synthesis of glutathione. Glutathione (γ-glutamyl-cysteinylglycine) is one of the main intracellular low molecular weight thiol-containing compounds that is synthesized in almost all eukaryotic cells. Due to its structure and high intracellular concentration (1-10 mM; 10 mM in liver cells and malignant cells of various types), GSH acts as an antioxidant and also is involved in the maintenance of cell redox status, in the work of the detoxification system, in synthesis of eicosanoids, and in the regulation of numerous mechanisms of cell signaling including regulation of the cell cycle, gene expression, and apoptosis [6]. Glutathione is present in the cell mainly in the reduced form, while the content of GSSG does not exceed 1% of its total intracellular content. Approximately 85-90% of GSH is located in the cytoplasm, but some GSH after synthesis in the cytoplasm is found in the mitochondria, nucleus, peroxisomes, and endoplasmic reticulum [11]. The maintenance of optimal GSH/GSSG ratio in the cell is of importance for normal cell functioning and survival. Therefore, the system regulating this ratio must be strictly controlled. A decrease in GSH subjects the cell to the risk of oxidative damage. It has been shown that disturbance in the regulation of GSH level is observed in a wide range of pathologies such as cancer, neurodegenerative diseases, mucoviscidosis, and HIV infection [6].
Synthesis of GSH de novo includes two independent ATP-dependent stages that comprise a cycle of six enzymatic reactions called the γ-glutamyl cycle (Fig. 1). The first stage is the formation of the peptide bond between a cysteine and a glutamic acid catalyzed by γ-glutamylcysteine ligase (γ-GCL). This reaction is a rate-limiting step in GSH synthesis. The second stage yielding GSH is the reaction between the γ-glutamylcysteine and a glycine catalyzed by glutathione synthetase (GS) [3]. The enzyme that is capable of hydrolyzing the specific bond between the cysteine and glutamic acid residues in the GSH molecule, γ-glutamyl transferase (γ-GT), is localized on the outer side of the cytoplasmic membrane of certain cell types. γ-GT transfers the γ-glutamyl residue to an amino acid, this making possible the transport of the amino acid into the cell [3]. The dipeptide cysteinylglycine formed as a result of the action of γ-GT is cleaved by dipeptidase yielding cysteine and glycine, which become substrates for γ-GCL and GS, respectively. γ-Glutamyl cyclotransferase cleaves the bond in the dipeptide γ-Glu–amino acid yielding the free amino acid and 5-oxoproline. Under the action of oxoprolinase, the latter is converted into glutamic acid, which becomes a substrate for γ-GCL. Thus, extracellular glutathione can be cleaved yielding amino acids that are capable of penetrating inside the cell, where they can be used for the synthesis of a new GSH molecule. Most of the GSH in blood serum is provided by its synthesis in the liver, so disturbances in this process lead to systemic disorders of glutathione homeostasis in different organs [12].
Fig. 1. γ-Glutamyl cycle of glutathione synthesis. γ-GCL, γ-glutamylcysteine ligase; GS, glutathione synthetase; γ-GT, γ-glutamyl transferase.
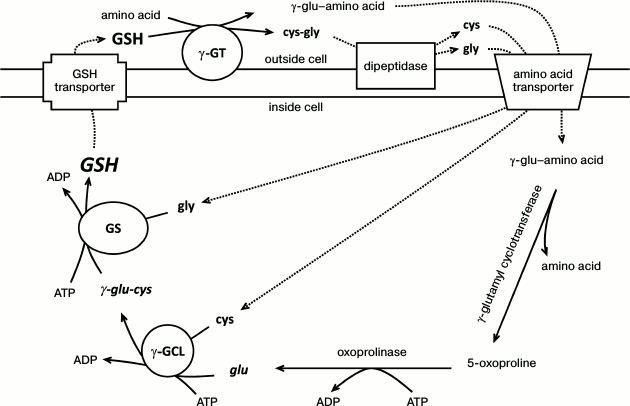
Recovery of the GSH content is provided not only by de novo synthesis, but also by the recycling of GSSG to GSH in the reaction catalyzed by glutathione reductase (GR) in the presence of NADPH(H+) as the cofactor [3].
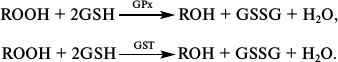
Role of glutathione in redox-dependent processes. The main functional element in the GSH molecule is the cysteine residue containing a reactive thiol group. Among the functions of glutathione, first should be mentioned its participation in the defense of the cell from products of oxidative stress. Hydrogen peroxide is reduced by glutathione peroxidase to H2O using GSH as the cosubstrate. Organic hydroperoxides can be reduced to corresponding alcohols in the reaction catalyzed by GPx as well as due to the peroxidase activity of Se-independent glutathione S-transferases that also use GSH as the cosubstrate:
Glutathione reduces oxidized glutaredoxin that is formed during the reduction of disulfides [5].
Glutathione is a low molecular weight antioxidant that can take part in the nonenzymatic antioxidant defense, playing a role as an efficient scavenger of free radicals [13, 14].
Oxidative stress leads to damage of carbohydrates, lipids, and nucleic acids, resulting in cell dysfunction and death. Oxidative stress and/or change in cellular redox status can affect the state of the nuclear chromatin and alter gene expression. Progression of oxidative stress results in single- or double-strand breaks in DNA molecules. Mitochondrial damage is followed by a decrease in the transmembrane potential, changes in the membrane permeability, and accelerated release of apoptotic factors, which leads to cell death [15]. Under physiological conditions, reactive oxygen and nitrogen species (ROS and RNS, respectively) are involved in processes of redox-signaling, which are fast, specific, and reversible reactions regulating the activity of proteins that are important for cell functioning. The processes involved in redox-signaling can occur in various cell compartments at certain times with participation of different redox pairs, such as GSH/GSSG or NADH(H+)/NAD+ [16]. Now special attention is paid to the GSH/GSSG ratio as the main marker of redox status and an important factor of signal transduction [17].
Many proteins contain functionally important cysteine residues that are subjected to posttranslational modifications including oxidation. Under physiological conditions, the free amino acid cysteine exhibits pK of 8.5, which excludes oxidative modification. Within a protein molecule, the cysteine can be activated, i.e. exists as the thiolate anion. This is due to numerous factors including hydrogen bonding, impact of neighboring amino acid residues, microenvironment of cysteine residues, and binding with substrate [18]. The cysteine residue of GSH can interact with residues of a protein yielding a disulfide bond (protein–SSG). This process called glutathionylation defends proteins from oxidative stress and makes a significant contribution to redox signaling and regulation of protein activities [19]. S-Glutathionylation can affect the ability of the protein to form disulfide bonds and to correct its folding, which influences the functional state of the protein. Also, S-glutathionylation protects sulfenic acid derivatives (Cys-SOH) formed during the oxidation of cysteine residues from oxidation to sulfinic acid (Cys-SO2H) and then further to sulfonic acid (Cys-SO3H), which cannot be reduced under physiological conditions [20].
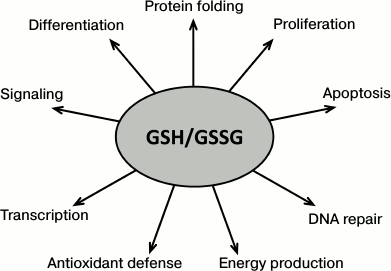
The reversibility of oxidation of cysteine residues is of great importance for the normal functioning of proteins and their ability to participate in signal transduction cascades. Glutathione is the main substrate for the reduction of oxidized cysteine residues. The state of the thiol–disulfide system is determined by the cellular redox status, which is characterized by the GSH/GSSG ratio. Under physiological conditions, GSH/GSSG is about 100 : 1, which minimizes the oxidative action of ROS/RNS. Disturbance of this ratio significantly affects in the context of the redox regulation the processes of signal transduction, control of gene expression, cell proliferation, differentiation, state of cell metabolism, and vital functions overall [18, 21] (Fig. 2). Overproduction of glutathionylated proteins indicates the progression of oxidative stress leading to cell death. Change in sulfhydryl homeostasis of the cell, especially the steady state of glutathionylation of specific regulatory proteins, modulates various pathways of signal transduction, shifting the cell state from survival to death. For example, the functioning of the cell actin is regulated by reversible S-glutathionylation, and disturbance in S-glutathionylation changes the structural organization of stress fibrils of the actin cytoskeleton [22]. Under normal conditions, modifications such as protein–SSG are transient and reversible. If the cysteine residue is essential, its S-glutathionylation can affect the functioning of the protein. For example, S-glutathionylation of subunits p65 and p50 of the transcriptional factor NF-κB inhibits their binding with DNA [23], while S-glutathionylation of the β-subunit of IκB kinase suppresses the activation of NF-κB [24].
Fig. 2. Role of glutathione in redox regulation of the main vital functions of cells.
Glutathione is synthesized only in the cytoplasm, and then transferred to the mitochondria, peroxisomes, endoplasmic reticulum, and nucleus. More than 70% of the total pool of GSH remains in the cytoplasm, while the nucleus and mitochondria are able to accumulate up to 10 and 30% of the total intracellular GSH content, respectively [25].
Recent investigations have shown that GSH is accumulated in the nucleus in the beginning of the G1 phase [26], so it could play an important role in the maintenance of the redox status of the nucleus during the cell cycle [27]. During mitosis, the nuclear membrane breaks down and appears again around the daughter DNA molecules packed into the chromosomes. During cell division, a high pool of GSH is maintained in close proximity to the chromatin, which is consistent with data on high redox potential of the dividing cell. The pool of nuclear glutathione (nGSH) is resistant to factors decreasing its content compared to changes observed for the total level of the cell and the mitochondrial GSH under the action of thiol-binding compounds N-ethylmaleimide and diethyl maleate, as well as the inhibitor of GSH synthesis buthionine sulfoximine (BSO). It has been found that cells with a high level of nGSH are more resistant to apoptosis under oxidative stress conditions. However, the role of nGSH in oxidative stress has been little studied [27].
The mechanisms of transport and depositing of nGSH are still little studied. A significant amount of nGSH presumably comes from the parental nucleus to the daughter nuclei during telophase due to the high concentration of GSH in proximity to the replicating genetic material [27].
Nuclear pores allow various ions and small molecules to penetrate inside the nucleus. Similarly, GSH can diffuse into the nucleus [28]. At the same time, an ATP-dependent mechanism of GSH transport into the nucleus was demonstrated [29]. Currently, the role of protein Bcl-2 in transmembrane transport of GSH is being discussed. It has been found that the content of nGSH is significantly increased in tumor cells exhibiting overexpression of the bcl-2 gene [30]. It should be noted that the BH-3 domain of Bcl-2 protein can bind to GSH. The participation of Bcl-2 in the maintenance of GSH level in mitochondria has been reported [31]. These data together with the fact that the antiapoptotic protein Bcl-2 is involved in the formation of pores in the membrane indicate the ability of members of the Bcl-2 protein family to serve as mediators of GSH translocation into the nucleus [26, 30].
A number of works on plant cells have demonstrated that gene expression is sensitive to the accumulation of GSH in the nucleus and to its decrease in the cytoplasm [30]. Decrease in redox potential in the cytoplasm and its growth in the nucleus affects not only gene expression, but also the ability of proteins to bind with their targets in the nucleus. It was shown that in the beginning of G1 phase of the cell cycle in animal cells, activation of oxidative processes in the cytoplasm caused by epidermal growth factor resulted in the accumulation of ROS, which activated phosphorylation cascades and DNA replication and induced cell division [32]. The decrease in the cytoplasmic GSH level in G1 phase may cause accumulation of ROS. It is suggested that the changes in the nuclear redox status may act as the trigger element for the other components that are essential for transcription. Such a mechanism was shown for proteins NF-κB, AP-1, and p53 [33]. For example, for the interaction of NF-κB with a DNA molecule, the cysteine residue in the DNA-binding region of NF-κB must be reduced. Similar behavior was reported for such transcriptional factors as Fos, Jun, and Nrf2 [30].
Changes in the GSH content in the nucleus can modulate the structural organization of chromatin [28]. The extent of glutathionylation of nuclear proteins increases in the beginning of cell proliferation [26]. In the animal cell, while going from proliferation through differentiation to cell death, the cellular redox status changes towards more oxidized state. Thus, the GSH/GSSG ratio is a kind of switch from proliferation to differentiation and then to programmed cell death [34]. The nuclear GSH is related to the synthesis of DNA, presumably being the redox sensor for the beginning of DNA synthesis. GSH maintains the necessary organization of the nucleus at the expense of the optimal redox status for replication of DNA and maintenance of its intact structure. Also, it was found that nGSH affects the proteasomal degradation of nuclear proteins [26].
Se-dependent glutathione peroxidase activity was found in the nuclear fraction of Wistar rat hepatocytes [35]. The sperm nucleus-specific isoform of glutathione peroxidase GPx4/snGPx was shown to maintain the stability of chromatin structure in sperm [36]. The nuclear localization of this enzyme emphasizes the important role of glutathione in the regulation of the cell cycle and the chromosomal organization, since nuclear proteins, mainly histones and other chromatin-binding proteins, presumably should be maintained in the reduced state for optimal functioning [27].
The role of GSH in the repair of DNA damage also should be pointed out. Glutathione is not the most efficient protector of DNA from X-radiation, but it controls repair mechanisms of damaged DNA molecules [37]. An important component of the repair mechanism of DNA oxidative damages is poly(ADP-ribose)polymerase (PARP), which catalyzes the growth of the polymer chains from the ADP-ribose molecules on target proteins (particularly on histones). This process proceeds in virtually all eukaryotic cells in response to DNA damage [38]. The expression of genes and activity of PARP protein family members are related to nGSH level during the cell cycle. For plant cells, it was demonstrated that mRNA of PARP1 and PARP2 accumulate with the growth of the GSH pool in the nucleus [39]. Similar character of changes in polyribosylation activity was observed in NIH3T3 fibroblast cells: polyribosylation of histones in their nuclei grew during proliferation, when the nGSH level was maximal [26, 40].
It should be mentioned that many points concerning the mechanisms of GSH transport into the nucleus and the role of nGSH in various genetic and epigenetic processes remain unclear [26].
Investigation of the role of mitochondrial glutathione (mGSH) is of great interest. Functions of mitochondria are closely related to the maintenance of cellular redox balance. The mitochondria are the main consumers of the oxygen and the main source of ROS, which are mainly generated during the functioning of the electron transfer chain. Under physiological conditions, incomplete one-electron reduction of molecular oxygen results in the formation of superoxide anion radical O2·–, which gives rise to other ROS. The concentration of O2·– in the mitochondrial matrix under steady-state conditions exceeds 5-10-fold its concentration in the cytoplasm [41]. Besides, the action of various toxins and some pathological states affecting mitochondrial functions can increase the production of ROS. The presence of the antioxidant defense system in mitochondria prevents disturbances in mitochondrial functioning. The main component of this system is mGSH, which prevents or repairs damage occurring under normal aerobic metabolism.
The superoxide anion O2·– in mitochondria is inactivated by Mn-dependent superoxide dismutase converting O2·– to H2O2, which can be neutralized by GSH-dependent systems with the participation of glutathione transferase and glutathione peroxidase. The isoenzyme of glutathione peroxidase GPx1 that is most active towards H2O2 is localized mostly in the cytoplasm, but a small amount is also present in the mitochondrial matrix [42]. In mitochondria, GST catalyzes the formation of GS-conjugates and the reduction of organic hydroperoxides using GSH as the cosubstrate. In contrast to Se-containing GPx, GST does not interact with H2O2, but it efficiently reduces hydroperoxides of polyunsaturated (linoleic and arachidonic) fatty acids, phospholipids, mononucleotides, and DNA. The Se-independent GPx4 plays an important role in the detoxication of lipid hydroperoxides in mitochondria. Recently, it was shown that GPx4 prevents the development of cell apoptosis in the presence of apoptosis inducing factor (AIF) and supports the process of oxidative phosphorylation in intestinal epithelial cells [43]. Due to the ability to reduce hydroperoxides of cardiolipin, GPx4 takes part in regulation of the release of apoptogenic proteins from mitochondria [44]. In the mitochondria of human cells, peroxidase activity is exhibited by GST isoenzymes hGSTA4-4, hGSTA1, hGSTA2, and hGSTP1, among which isoform hGSTA4-4 is the most active [45]. The mitochondrial isoform of glutaredoxin is Grx2, which also occurs in the nucleus. Interestingly, the oxidized form of Grx2 can be reduced by both thioredoxin reductase (TrxR) and GSH, this providing functional activity of Grx2 in the oxidized microenvironment that is common for mitochondria. Grx2 in mitochondria plays a significant role in the interaction of the GSH pool with protein thiol groups, in the antioxidant defense system, and in redox-dependent signaling [45-47]. The thioredoxin-dependent system in mitochondria is represented by thioredoxin 2 (Trx2) and thioredoxin reductase (TrxR2), and among the isoforms of peroxiredoxin the most important is Prx3. The systems mGSH/GPx and Prx3/Trx2 that defend against H2O2 are interrelated. For example, it was shown that decrease in mGSH resulted in oxidation of Trx2 [48]. Thus, the role of the redox-cycle of mGSH in the maintenance of the efficient antioxidant system and homeostasis of hydrogen peroxide in mitochondria is evident.
The content of mGSH in mitochondria is approximately the same as in the cytoplasm (10-14 mM) [42]. Glutathione is not synthesized in mitochondria, but it is transported from outside. GSH easily penetrates through the porin channels of the outer mitochondrial membrane. Since under physiological conditions GSH exists as an anion, it is unable to diffuse into the matrix through the inner mitochondrial membrane that has a high negative transmembrane potential. GSH is transferred into the mitochondrial matrix by transporters in the inner mitochondrial membrane working against the electrochemical gradient. In mitochondria of liver and kidneys, this role is played by carriers of 2-oxoglutarate (OGC) and dicarboxylates (DIC), which transfer GSH into the mitochondrial matrix by the antiport mechanism in exchange with 2-oxoglutarate and inorganic phosphate, respectively [42]. Since these transporters provide liver mitochondria with only 45-50% of their GSH, some additional mechanism must exist.
GSSG does not come out from mitochondria to the cytoplasm, being instead reduced by glutathione reductase yielding GSH. This process depends on the presence of a sufficient amount of NAPDH(H+). It should be noted that the accumulation of GSSG affects the glutathionylation of mitochondrial proteins, thus changing their functioning. For example, the activity of NADH-ubiquinone reductase (complex I of the mitochondrial respiratory chain) depends on the GSH/GSSG ratio [49]. In an experimental model with mitochondrial membranes from rat heart, it was found that the addition of GSSG after the action of Grx2 resulted in the glutathionylation of complex I. In contrast, the addition of GSH and Grx2 caused its deglutathionylation [50]. For mitochondria from bovine heart, the sites of glutathionylation in complex I were found to be cysteine residues Cys531 and Cys704 of the 75-kDa NDUSF1 subunit [51]. It should be pointed out that the role of S-glutathionylation is of importance for the defense of NADH-ubiquinone reductase from the irreversible oxidation and for the control of the ROS production by the mitochondria in response to the changes in the local redox environment. In this case, the important condition is the simultaneous S-glutathionylation of the complex I and the α-ketoglutarate dehydrogenase complex, since the latter supplies NADH(H+) for the oxidation by the complex I, i.e. both protein complexes contribute to the generation of ROS by the mitochondria [52]. Under conditions of oxidative stress, the glutathionylation of these two enzyme complexes decreases their activities and production of ROS [53, 54]. After return of the content of O2·– and H2O2 to the normal level, the α-ketoglutarate dehydrogenase complex and NADH dehydrogenase are deglutathionylated by Grx2, and oxidative phosphorylation is restored.
Role of glutathione in redox-dependent regulation of apoptosis. Numerous works have considered the protective role of GSH in the mechanism of apoptosis. According to a contemporary concept, decrease in GSH level below a crucial value results in the appearance of a signal for apoptosis, which is initiated by the activation of the death receptor or by mitochondrial apoptotic signaling. In contrast, an increase in GSH level provides defense of cells from Fas-induced apoptosis [55]. Numerous data indicate the crucial role of GSH in cell defense from various apoptotic stimuli, since disturbances of redox homeostasis of the cell caused by GSH oxidation or GSH export facilitate the development of apoptosis [56, 57].
In a number of works with various cell types, it has been shown that disturbance of the GSH/GSSG balance caused by accumulation of GSSG caused by an oxidant precedes the induction of mitochondrial apoptotic signal [58-61]. The restoration of the GSH/GSSG ratio to the normal level after the action of the oxidant does not prevent the development of apoptosis, suggesting that it is induced in the early stage of the disbalance between GSH and GSSG. The use of the thiol antioxidant N-acetyl cysteine before the action of compounds resulting in the oxidative (tert-butyl hydroperoxide) or carbonyl (methylglyoxal) stress prevents the induction of apoptosis. These data are consistent with other reports and indicate that the signal for apoptotic death is triggered in the very beginning of the decrease in GSH/GSSG ratio [58, 59, 61, 62]. After the action of oxidants, N-acetyl cysteine cannot prevent the development of apoptosis.
Induction of apoptosis under oxidative stress is caused by the activation of mitogen-activated protein kinases (MAPKs) [63]. There are three classes of MAPKs: ERK (extracellular signal-regulated kinase), JNK (c-Jun N-terminal kinase), and p38 [63]. The signal transduction cascade includes consecutive phosphorylation steps resulting in the activation of specific MAPKKK (MAP3K, kinase of MAPK kinase) that activates MAPK kinase (MAP2K) that in turn activates MAPK [64]. Stress-induced apoptosis is related to the activation of JNK and p38 MAPK, and this can be triggered by the kinase cascade involving apoptosis signal-regulating kinase 1 (ASK1), MAPK kinase 4/7 (MEK4/7), and JNK, or through the cascade of ASK1, MAPK kinase-3/6 (MEK 3/6), and p38 [63, 65].
The role of GSH in redox-dependent regulation of MAPK-induced apoptotic signaling has been little studied to date. Since GSH is a key factor in the maintenance of intracellular redox homeostasis and plays an important role in antioxidant defense of the cell, it might be a modulator of MAPK-dependent signaling pathways. Actually, in some cell models it was shown that the disbalance of GSH/GSSG activated the MAPK-signaling pathway and facilitated apoptosis. For example, the induction of ROS formation by aloe-emodin resulted in disbalance of the GSH/GSSG ratio and redox-dependent activation of the GSTP1/JNK-signaling pathway in hepatoma cells [66].
Other works report that the inhibition of the de novo GSH synthesis by BSO promotes the redox-activation of MAPK and the apoptotic signaling pathway. The treatment of breast cancer cells with the antitumor agent aplidine after treatment with BSO resulted in the activation of the JNK- and p38-dependent signal pathways and in the development of apoptosis [67]. Apoptosis of HepG2 cells treated with BSO was induced by andrographolide through the activation of the ASK1/MEK4/JNK signal cascade [68]. The addition of thiols (N-acetyl cysteine and GSH) prevented the activation of MAPK induced by the toxins, which suggests the participation of GSH in the functioning of MAPK and in the cell response to stress [67].
While investigating the role of GSSG in the initiation of apoptosis, it was found that extracellular GSSG is able to activate selectively the MAP kinase cascade ASK1/MEK3/6/p38 through the mechanism of GSSG-induced thiol–disulfide exchange on the cellular membrane and the formation of mixed protein disulfides [69]. Such a redox stress can in turn result in the breakdown of the Trx1/ASK1 complex and in the activation of the p38-dependent pathway of apoptosis [69]. The fact that each of these events can be prevented by GSH is consistent with the idea of the protective role of glutathione [69]. In this connection, it should be noted that in SH-SY5Y neuroblastoma cells that are resistant to GSSG-induced apoptosis, apoptosis was activated after preliminary treatment with BSO, which was accompanied by increased ROS production and activation of JNK [70]. Such data point to the assumption that drop in GSH concentration below a certain critical level is a necessary condition for the activating effect of GSSG on the MAPK-signaling pathway and induction of apoptosis.
GSH regulates the redox state of Trx1 and the Trx1-dependent ASK1 signal cascade inducing apoptosis. For example, the action of agents oxidizing GSH (diamide and dithio-nitrobenzoate) on stomach adenocarcinoma cells initiated the mitochondrial pathway of apoptosis [71]. In this case, redox activation of the Trx1/ASK1/p38 signal cascade was triggered by increase in GSSG content. The resistance of the cells towards systems producing H2O2 and ROS (paraquat and xanthine/xanthine oxidase) correlated with the Nrf2-dependent increase in GSH content and with protein S-glutathionylation [71]. Another response was observed in SH-SY5Y neuroblastoma cells, where H2O2 activated the Trx1/p38/p53 cascade and cellular apoptosis, while diamide activated the ERK-signaling pathway, Nrf2-dependent increase in GSH content, and expression of the heme oxygenase-1 gene, which assisted cell survival.
Concerning the regulation of GSH-dependent posttranslational modifications of cysteine residues of the proteins involved in the MAPK-signaling pathways, it can be noted that under the oxidative stress caused by menadione (2-methyl-1,4-naphthoquinone), S-glutathionylation of Cys1238 in the ATP-binding domain of MEKK1 inhibits the activity of the kinase [73]. However, a question is still open concerning the specific relation between the oxidative stress and S-glutathionylation in terms of the activation/inactivation of specific MAPK-dependent signal pathways and induction of apoptosis.
The presented data show that GSH plays an important role in the redox regulation of MAPK-dependent pathways of signal transduction. However, the difference between the induction of different signal pathways that are responsible for cell death or survival is likely determined by not only the cellular content of GSH, but also by the cell type [71, 72].
In recent years, special attention has been given to investigation of the mitochondrion as the cell organelle involved in the activation of apoptosis. The GSH/GSSG ratio is considered to be the main redox system maintaining redox homeostasis of the mitochondrial matrix and defending mitochondrial proteins and DNA from the action of ROS. Using various cell models, it has been shown that selective decrease in the mGSH content resulted in a drop in the activity of the respiratory chain complexes, growth in the production of ROS, decrease in transmembrane potential (ΔΨ), and the release of apoptogenic factors from mitochondria. For example, in diabetic cardiomyocytes, stress-induced oxidation of mitochondrial, but not cytoplasmic GSH, resulted in a decrease in the ΔΨ value and the activation of caspase-9 and caspase-3 [74]. In human B-cell lymphoma cells, ROS-induced decrease in mGSH level initiated apoptosis that was accompanied by drop in ΔΨ value, release of cytochrome c, and activation of caspase-3 [75].
A direct relation between the decrease in the mGSH content and the activation of apoptosis has been demonstrated for different cell types. In hepatocytes, decrease in mGSH content was a necessary condition for TNF-α-induced apoptosis, which was preceded by tBid/Bax-initiated permeabilization of the mitochondrial membrane, release of cytochrome c, assembly of apoptosomes, and activation of caspase-3 [76]. In large intestine cells, the oxidation of mGSH was the main factor in the development of menadione-induced mitochondrial dysfunction and cytochrome c-dependent activation of apoptosis [77].
The precise mechanism of mitochondrial dysfunction caused by decrease in mGSH content is not completely clear. However, it was found that cisplatin-induced apoptosis is related to disbalance in the mGSH/GSSG ratio, decrease in NADPH(H+) content, and oxidative damage of cardiolipin and aconitase, which disturbs the functioning of mitochondria and activates caspase-3 [78, 79]. Later works demonstrated that a sharp drop in the mGSH content induced the generation of ROS/RNS, leading to apoptosis in HL-60 and Raji cells. In this case, apoptosis was caused by the breakdown of complex I of the respiratory chain due to the destabilization of the Fe-S cluster of the NDUGS3 subunit of the complex, resulting in inhibition of respiration and drop in ΔΨ value [80]. Of note, in hepatocytes a slight decrease in mGSH content caused by moderate hypoxia did not lead to apoptosis. This fact demonstrates that to induce apoptosis, the content of mGSH must drop to a certain critical level [25].
A drop in mGSH content may control the permeability of the mitochondrial membrane. In early works, decrease in mGSH is attributed to changes in mitochondrial permeability that is caused by redox modulation of adenine nucleotide translocase and subsequent release of apoptogenic factors such as cytochrome c and AIF from the mitochondria to the cytoplasm [81, 82]. The later investigations showed that change in redox balance of mGSH is a crucial factor in the control of mitochondrial membrane permeability [83]. Drop in mGSH/GSSG ratio from 300 : 1 to 20 : 1 leads to the opening of the anion channel in the inner mitochondrial membrane and the mitochondrial pore. If the mGSH/GSSG ratio lies in the region from 150 : 1 to 100 : 1, instability of the ΔΨ value is observed. In the case of pronounced oxidation, when the ratio is less than 50 : 1, irreversible depolarization of the mitochondrial membrane takes place, accompanied by opening channels and breakdown of the mitochondria [83]. The accelerated transport of glutathione into mitochondria suppressed the menadione-induced growth of mGSSG level, preventing the decrease in the ATP content, drop in ΔΨ value, release of cytochrome c into the cytoplasm, and activation of caspase-3 and caspase-9 [77].
ROLE OF GLUTATHIONE S-TRANSFERASE IN REDOX-DEPENDENT
PROCESSES
A significant role in redox-dependent processes cellular belongs to glutathione S-transferase (EC 2.5.1.18). This enzyme is represented by a superfamily of isoenzymes catalyzing the conjugation of glutathione with a wide range of nonpolar compounds of exogenous and endogenous origin containing electrophilic atoms of carbon, sulfur, nitrogen, and phosphorous, which assists in the defense of the cell against possible toxic action of these compounds [84-87]. Isoenzymes of GST have now been found in most living organisms including aerobic bacteria, yeast, plants, insects, and vertebrates. The GST superfamily includes three subfamilies of isoenzymes: cytosolic, mitochondrial, and microsomal.
In mammals, GST is present in virtually all organs and tissues, but the highest content of the enzyme is found in the liver. The cytosolic isoenzymes of GST account for approximately 90% of the total GST activity in the cell. Based the amino acid sequence homology, mammalian cytosolic GST isoenzymes are grouped into seven classes (α, µ, π, θ, ζ, ω, and σ) that comprise 17 isoenzymes [84, 85]. In humans and rodents, the cytosolic isoenzymes within the same class exhibit more than 40% homology (sometimes more than 90%), while the homology between the enzymes in different classes is less than 25%. Special attention in the contemporary classification is given to the primary structure of the conserved N-terminus of the polypeptide chain containing catalytic residues of tyrosine, cysteine, or serine [85, 86]. In species other than mammals, GST isoenzymes of β, δ, ε, φ, λ, τ, and ν classes have been found [85, 88].
Microsomal GSTs are integral membrane proteins that are now called membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG) [89, 90]. Isoenzymes of the MAPEG microsomal subfamily are divided into four subgroups (I-IV), the amino acid sequence homology between the subgroups constituting less than 20%. In humans, six isoenzymes have been found that belong to subgroups I, II, and IV [90]. Similarly to the cytosolic and mitochondrial GST isoenzymes, the microsomal isoenzymes catalyze conjugation of GSH with electrophilic compounds, but they also participate in reactions of isomerization of unsaturated compounds and biosynthesis of leukotrienes and prostaglandins [85].
A mitochondrial isoenzyme of human GST is GSTK1-1, which belongs to κ-class [89]. The same isoenzyme was observed in human peroxisomes [12]. GSTK1-1 of rodents and humans is active towards a number of traditional GST substrates, in particular 1-chloro-2,4-dinitrobenzol. In Caenorhabditis elegans, GST is involved in the metabolism of lipids [91].
Cytosolic and mitochondrial isoforms of GST are homo- or heterodimers, and the subunits within heterodimers belong to the same class. Although the cytosolic GSTs are dimers, representatives of the microsomal subfamily can be mono-, di-, and trimers, and multienzyme complexes also occur [85, 92]. Each subunit is composed of two domains linked with a small irregular region. The N-terminal domain is the GSH-binding site (G-site). It exhibits topology that is similar to thioredoxin and contains four β-sheets (β1, β2, β3, and β4), three of which are antiparallel, and three α-helices. These elements of the secondary structure are composed into the sequences βαβαββα. The C-terminal domain is the cosubstrate-binding site (H-site), a completely α-helical region composed of either five or six α-helices (α4-8 or α4-9). In contrast to other classes, the isoenzymes of the α, θ, and ω classes have an additional α9 helix. The isoenzymes of the μ-class have a unique μ-loop at the C-terminus, while the isoenzymes of the ω-class contain an additional 19 amino acid sequence at the N-terminus. The θ-isoenzymes have a large loop between the α4- and α5-helices [84, 93]. The differences in the structures of the representatives of various GST isoenzymes provide their wide substrate specificity and diversity of functions.
Detailed comparative analysis of the amino acid sequence and structure of cytosolic GST isoenzymes considering the presence of certain amino acid residues in their active site allowed their division into two subgroups: Y-GST, isoenzymes using a tyrosine residue for the activation of GSH (α, µ, π, and σ classes), and S/C-GST, isoenzymes binding GSH through a serine (φ, τ, θ, and ζ classes) or a cysteine (β and ω classes) residue [93]. In GST of both subgroups and in mitochondrial GST, these amino acid residues essential for GSH activation are located in the so-called catalytic loop that is next to the first β-sheet in the thioredoxin-like domain. As mentioned above, the structure of the H-site exhibits significant variability in representatives of different classes.
While forming the dimeric structure, domain I of one subunit and domain II of the second interact with each other by the lock-and-key principle. Certain aromatic amino acid residues of the loop between the α3-helix and β2-sheet of the first monomer play a role as the key. They are located in the hydrophobic “lock” formed by the cavity between the α4- and α5-helices of the second monomer.
The main function of GST is its participation in the antioxidant system through its ability to reduce organic hydroperoxides to alcohols using GSH as the cosubstrate [85]:
![]()
Via Se-independent glutathione-peroxidase activity, GST reduces hydroperoxides of polyunsaturated higher fatty acids, phospholipids, and cholesterol [94, 95]. Among the substrates of GSTA4-4 are products of lipid peroxide oxidation acrolein and 4-hydroxynonenal (4-HNE) [96]. Conjugation of these compounds with glutathione protects proteins and DNA against covalent modification. As a result of oxidative stress, nucleotides can be oxidized to propenals and hydroperoxides, which are substrates of GSTP1-1. Oxidation of catecholamines also results in the formation of compounds (aminochrome, dopachrome, noradrenochrome, and adrenochrome) that are the substrates of GST isoforms. Conjugation of such compounds with GSH contributes to the cellular antioxidant defense system, since they contain a quinone structure producing O2·–, and, consequently, promoting oxidative stress [85]. The cytosolic GSTM2-2 was shown to detoxify o-quinones of dopamine, which can protect dopaminergic systems of the brain against degenerative processes [97]. GSTP1-1 mediates defense against oxidative stress, recovering the peroxidase activity of the oxidized peroxiredoxin Prx6 [98].
Of special importance is the role of GST in the regulation of cell signaling due to the protein–protein interactions with kinases that are activated by oxidative stress. Under physiological conditions, some GSTP1-1 is bound to kinase JNK1, resulting in its inactivation, which regulates the level of active JNK1. Under conditions increasing ROS content, which is observed, for example, under the action of a number of antitumor drugs, the complex between GSTP1-1 and JNK1 dissociates, and GSTP1-1 associates into oligomers. The release of JNK1 induces a cascade of processes, starting from the phosphorylation of Jun-c and resulting in apoptosis. The enhanced expression of the GSTP1-1 gene observed in some tumors can significantly inhibit JNK1, and consequently, suppress the signal pathway leading to apoptosis, which contributes into the formation of drug resistance of the tumor cells [8, 99]. A similar interaction of GSTP1-1 with TRAF2 (factor 2 bound to the TNF-α receptor) blocks the action of kinases JNK1, p38, and ASK1 in the case of the signal cascade induced by TNF-α. The dissociation of the complex between GSTP1-1 and TRAF2 activates proliferation under gentle oxidative stress, while a prolonged and strong oxidative stress leads to apoptosis [100]. It should be noted that the catalytic activity of GSTP1-1 does not change during protein–protein interaction, suggesting that the active sites of the enzyme are not involved in this process [86].
The isoenzyme GSTA1-1 also participates in the regulation of apoptotic signal pathways through protein–protein interactions with JNK1. Enhanced expression of the GSTA1-1 gene significantly decreases the number of cells subjected to apoptosis due to inhibition of JNK1-dependent phosphorylation of Jun-c and the activation of caspase-3 [101]. GSTM1-1 exhibits regulatory functions that are similar to those of GSTP1-1. The complex between GSTM1-1 and ASK1 is important for maintenance of the normal level of phosphorylation of p38. Under stress conditions of heat shock or increased ROS level, the complex dissociates, and the GSTM1-1 associates into oligomers, while ASK1 is activated [102]. Since ASK1 is a kinase of MAPK kinase activating the JNK1- and p38-dependent signal pathways, the dissociation of this complex results in cytokine- and stress-induced apoptosis [103].
As discussed above, GST was shown to be involved in the process of S-glutathionylation. Originally, it was considered that growth in ROS production leads to S-glutathionylation to prevent irreversible oxidation of protein cysteine residues and disturbance of protein functions [34]. Later it was found that S-glutathionylation plays an important role in the mechanisms of the cell signaling, changing the sensitivity of cysteine residues towards redox modification. The list of proteins whose structure and functions are modulated by S-glutathionylation is large: proteins involved in metabolism, proteins forming the cytoskeleton and ion channels, signal proteins (kinases and phosphatases), transcription factors, ras-proteins, and heat shock proteins [104].
The process of S-glutathionylation can proceed both non-enzymatically and with participation of enzymes, one of which is GSTP1-1. The ability of GSTP1-1 for S-glutathionylation is based on the catalytic activity of the enzyme. Under oxidative stress, GSTP1-1 is auto-S-glutathionylated at residues Cys47 and Cys101, each of which affects the catalytic activity of the enzyme and its ability to bind target proteins. Besides, specific S-glutathionylation causes oligomerization of GSTP1-1, which presumably has significant consequences for other components of the cellular stress response. S-Glutathionylation of the GSTP1-1 monomer decreases the number of α-helical regions, i.e. alters the secondary structure of the enzyme, which subsequently leads to a change in the tertiary and quaternary structures [105], affecting the ability of the GSTP1-1 to interact with proteins. An example of such regulation is the complex between GSTP1-1 and JNK1. The S-glutathionylation of GSTP1-1 at residues Cys47 and/or Cys101 results in the dissociation of the complex between GSTP1-1 and JNK1, activation of JNK1, and aggregation of GSTP1-1 [105].
The ability of homodimers of GSTP1-1 (sometimes GSTM1-1) to dissociate and form heterodimers with other monomeric proteins underlies its ability to provide these proteins with glutathione [106, 107]. The cytosolic isoforms of GST are catalytically active in the dimeric form, the surface of the dimer being the site of the noncatalytic binding of ligands. A number of works report that the isoforms of mammalian GSTP1-1 and GSTM1-1 in the monomeric form can interact with ASK1, JNK1, or with peroxiredoxin 6 (Prx6) [98, 100, 102]. Investigations of the GSTP1-1 molecule have shown that the structural features of its C-terminus promote the dissociation of the homodimer into monomers. At the same time, the Trx-like domain at the N-terminus promotes the formation of heterodimers between GSTP1-1 monomers with other proteins, especially with those containing a Trx-like domain [108].
The recovery of the peroxidase activity of Prx6 can be an example of the protein–protein interaction of GSTP1-1 with simultaneous reduction of the protein by glutathione. The Prx6 molecule has one catalytically active cysteine residue, Cys47, at the N-terminus. The oxidation of this residue yields a sulfenic acid derivative, this inactivating the peroxidase activity of Prx6 towards H2O2 and hydroperoxides of phospholipids. It has been found that the isoenzyme GSTP1-1 in complex with GSH forms a heterodimer with Prx6 and reduces the Cys47 residue. The binding of GSH induces conformational changes allowing the formation of the heterodimer between GSTP1-1 and Prx6 [109]. Then Prx6 is S-glutathionylated at the oxidized Cys47 residue with subsequent disulfide bonding between Cys47 of Prx6 and Cys47 of GSTP1-1, and then the disulfide bond is reduced by GSH.
GST takes part in the regulation of serine/threonine AMP-activated protein kinase (AMPK), which controls the energy balance of the cell [110]. A diversity of AMPK functions are involved in the control of various metabolic pathways and physiological processes such as proliferation and cell motility. AMPK is activated by ROS and RNS through AMP-dependent and AMP-independent mechanisms and can be involved in cellular redox regulation [111]. In vitro studies demonstrated that, under conditions close to physiological, mammalian isoenzymes GSTM1-1 and GSTP1-1 promoted S-glutathionylation of AMPK at the same cysteine residues that were glutathionylated during the nonenzymatic H2O2-dependent process, which also increased the kinase activity [111-113]. The interaction with AMPK activates GSTM1-1 and GSTP1-1, which in turn results in the S-glutathionylation and activation of AMPK. These data illustrate well the role of AMPK as an important element in redox-dependent signal transduction [114-116]. The activated AMPK activates the transcriptional factor FOXO3 that affects such processes as cell proliferation, gluconeogenesis, and defense against oxidative stress through the activation of the PI3K/AKT signal pathway [117]. The contribution of FOXO3 to antioxidant defense is accounted for by the enhanced expression of Mn-superoxide dismutase, catalase, thioredoxin [118, 119], metallothioneins [120], mitochondrial uncoupling protein UCP2 [121], γ-glutamylcysteine synthetase [118], glutathione peroxidase [119], and GSTM1-1 [120]. The GST-mediated S-glutathionylation and activation of AMPK can be considered as an additional mechanism of regulation of AMPK as a redox sensor of energetic stress and antioxidant defense [111].
As mentioned above, the main products of lipid peroxidation are 4-hydroxynonenals (4-HNEs), which form adducts with proteins and nucleic acids. 4-HNEs are involved in the MAPK-dependent signal pathways of cellular stress response, particularly by facilitation of the phosphorylation of JNK and p38, which results in their activation [122]. A dose-dependent regulation of cellular signal pathways by 4-HNEs has been demonstrated: at concentrations above 10 μM, 4-HNEs exerted cytotoxic effect, while at concentrations below 10 μM (physiological range) 4-HNEs modulated cell growth, i.e. affected cell proliferation [123]. Besides, 4-HNEs inhibit expression of cyclins D1, D2, and A, and consequently the activity of the cyclin-dependent kinases 4/6 (Cdk4/6) and Cdk2 [124], as well as increase the expression of p21waf1, which inhibits the functioning of some cyclin-dependent complexes [125]. Thus, 4-HNEs can simultaneously affect the expression of different genes involved in the control of cell proliferation. Undoubtedly, the intracellular content of 4-HNEs must be regulated to protect the cell from damage and/or to control stress-dependent signal pathways. Most 4-HNE is metabolized by GST, yielding conjugates with GSH with their subsequent detoxication. The most specific isoform towards 4-HNE is GSTA4-4 [126-128].
It has been found that under conditions of oxidative stress, the phosphorylation of GSTA4 subunits in the cytoplasm increases, this facilitating their binding to the Hsp70 protein, fast dimerization, and subsequent translocation into mitochondria. If the subunits are not hyperphosphorylated, they do not exhibit high affinity to Hsp70. In this case, the formed dimers remain in the cytoplasm [129]. Thus, the oxidative stress-activated import to mitochondria of the GSTA4-4 isoform exhibiting high specificity towards 4-HNEs protects mitochondria from oxidative stress and modulates signal pathways that are affected by 4-hydroxynonenals [127-129]. It should be noted that TNFα, IL-6, and epidermal growth factor enhance the GSTA4-4 content in mitochondria in vivo [130]. The decrease in the GSTA4-4 level results in the growth of ROS production and disturbs the mitochondrial functions, which promotes the development of the insulin resistance and type 2 diabetes [131]. In whole, the data of different studies show that the concentration of 4-HNEs in the cell is important for the activation of the cell cycle and signal cascades regulating cell differentiation, proliferation, and apoptosis, the level of 4-HNEs being strongly dependent on the activity of GSTA4-4 both in the cytoplasm and in the mitochondria.
There are a number of specific features of the impact of the GSTP1-1 isoform on the redox-dependent pathways regulating cell signaling and metabolism. It is supposed that some of numerous changes occurring in regulatory proteins observed under acute or chronic cocaine injections could be related to the S-glutathionylation catalyzed by GSTP1-1. For example, actin, JNK, and AMP-dependent protein kinase are regulated through S-glutathionylation under the action of cocaine [22, 105, 132-135]. Presumably, it is the enhanced S-glutathionylation that results in neuroadaptation under cocaine-induced oxidative stress [135]. GSTP1-1 was shown to directly inhibit the cyclin-dependent kinase Cdk5, interacting with its regulatory p25/p35 subunit [136]. The stimulation of Cdk5 results in the generation of ROS, which leads to cell death due to a feedback mechanism. Under neurotoxic conditions, the introduction of the GSTP1-1 gene provides successful neuroprotection due to the ability of GSTP1-1 to modulate the Cdk5-dependent signaling, which protects the cell from oxidative stress and prevents neurodegeneration [137].
GSTP1-1 was shown to prevent the origin and progression of Parkinson’s disease, suppressing the activation of Jun-c [140]. GSTP1-1 gene knockout mice were more sensitive to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, which led to the early degeneration of dopaminergic neurons and corpus striatum fibers.
The expression level of GST isoforms differs in normal and tumor tissues. High expression of the GSTP1-1 gene often correlates with drug resistance that is observed in tumor tissues of the ovaries, lungs, mammary glands, large intestine, and in some oncological diseases of blood [8]. The ability of GSTP1-1 as the inhibitor of JNK1, ASK1, and TRAF2 to regulate kinase signal pathways determining the cell fate can provide the resistance of tumor cells to antitumor drugs including those with prooxidant action [138, 139]. Transfection of the GSTA1-1 gene into H69 cells (small-cell lung cancer) leads to resistance towards doxorubicin, exhibiting prooxidant action [101]. Overexpression of the GSTA1-1 gene protected the cells from the decrease in GSH level caused by doxorubicin, decelerating lipid peroxide oxidation. Besides, overexpression of the GSTA1-1 gene significantly decreased the number of apoptotic cells due to the inhibition of JNK1-dependent phosphorylation of both Jun-c and caspase-3 [101].
The mechanisms regulating the work of genes of GST isoforms are still not completely understood. It was shown that exogenous or endogenous compounds of different structure are inducers of GST. Some of them activate the transcriptions of GST genes, acting on the antioxidant-responsive (ARE), xenobiotic-responsive (XRE), or glucocorticoid-responsive (GRE) elements of the promoter region [141, 142]. The presence of ARE in the promoter region is characteristic for genes whose products are involved in the defense of the cell from oxidative stress or xenobiotics. GST isoforms are also encoded by genes that are often called ARE genes, and the corresponding proteins are called ARE proteins. Expression of ARE genes is controlled by transcription factor Nrf2. Normally, Nrf2 is located in the cytoplasm in a complex with Keap1 protein, which provides ubiquitinylation of Nrf2 and its proteasomal degradation [85]. The mechanism that was supposed to be responsible for the activation of Nrf2 involves oxidation of cysteine residues of Keap1 under oxidative stress, resulting in the dissociation of the Keap1–Nrf2 complex and translocation of Nrf2 to the nucleus, where it forms a dimeric complex with small Maf protein. This complex activates expression of genes whose products are involved in cell defense [143]. However, some data suggest that the idea of direct dissociation of the Keap1–Nrf2 complex is incorrect, since the affinity of this interaction is rather high. It is suggested that stress conditions do not affect the affinity of the complex, but rather decrease the ability of Keap1 to ubiquitinylate Nrf2, which finally allows the transcription factor to be accumulated in the nucleus and stimulate the expression of ARE genes [144].
GLUTAREDOXIN. ROLE IN REDOX-DEPENDENT CONTROL
Glutaredoxin (EC 1.20.4.1) is one of the most important enzymes involved in processes of disulfide reduction and deglutathionylation. Isoenzymes of Grx are thermoresistant and low molecular weight proteins (10-16 kDa) functioning as GSH-dependent oxidoreductases. According to their structure, they belong to the thioredoxin superfamily, and together with Trx they play an important role in redox-dependent processes in cells. The active site sequence Cys-X-X-Cys/Ser is located in the N-terminal region of Grx, while the conserved glutathione-binding domain is at the C-terminal part of the molecule. Isoenzymes of Grx are found in virtually all living organisms except for some types of bacteria and archaea [145]. The family of Grx isoenzymes is classified in terms of the presence of a cysteine residue in the second position of the active site sequence. Isoenzymes with the sequence Cys-Cys-X-Cys/Ser in the active site are called Grx of C–C type. Originally, it was suggested that the main functions of Grx are reduction of disulfide bonds and deglutathionylation of proteins. However, later it was found that certain Grx isoenzymes rather serve as transfer proteins for iron–sulfur clusters [FeS] using GSH as a ligand [146]. The oxidized form of Grx formed after the reduction of protein disulfides and glutathionylated thiols is reduced by GSH (Fig. 3). However, some Grx isoenzymes are reduced by ferredoxin- or NADPH-dependent thioredoxin reductase, for example Grx4 of E. coli and human Grx2 [46]. Depending on the active site structure, Grx isoenzymes can be dithiol or monothiol (active site sequences Cys-X-X-Cys and Cys-X-X-Ser, respectively) [147]. The binding of Fe-S clusters can lead to the formation of dimers and tetramers. In these interactions, alternative protein–protein contact sites are possible in mono- and dithiol Grx isoenzymes, providing for the existence of both mono- and multidomain forms of Grx [148].
Fig. 3. Scheme of reactions catalyzed by the glutaredoxin-dependent system. The oxidized form of Grx formed after the reduction of protein disulfides and glutathionylated thiols is reduced by GSH. The oxidized GSH is reduced by glutathione reductase using NADPH(H+) as the coenzyme.
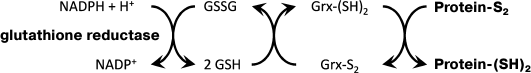
Concerning the classification of Grx isoenzymes, it should be noted that since bacteria, yeast, and mammals have a limited number of these proteins, their classification into mono- and dithiol isoenzymes is sufficient. For photosynthesizing organisms containing a wide range of Grx isoenzymes, a new classification is used [149] according to which the isoenzymes are divided into six classes based the homology of their amino acid sequences. Dithiol Grx isoenzymes belong to class I, monothiol Grx isoenzymes occur in both classes I and II, and glutaredoxins of the C–C type belong to III class. Grx isoenzymes of classes I and II are found in virtually all organisms. Isoenzymes of class III are present in higher plants, where they control the functional activity of plants, for example, flowering [5]. Isoenzymes of class IV are found in photosynthesizing eukaryotes. Glutaredoxins of class V occur in cyanobacteria and proteobacteria, while Grx of class VI are present only in cyanobacteria [150].
Grx isoenzymes use two catalytic mechanisms: monothiol and dithiol. The monothiol mechanism is characteristic for the reactions of deglutathionylation (Fig. 4a). In this case, only the catalytic cysteine residue (the first of two active-site cysteines at the N-terminus) participates in the catalysis. The reduction of a glutathionylated substrate starts from nucleophilic attack of the thiol group of the Grx CysA residue. The substrate is released with the formation of the intermediate glutathionylated product Grx-SSG. Further, glutaredoxin is regenerated by GSH, yielding Grx(SH)2 and GSSG. The monothiol mechanism is used by both monothiol and dithiol Grx isoenzymes [5]. The dithiol mechanism, besides the catalytic cysteine residue, requires another cysteine residue (so-called recycling cysteine) that can be either the second cysteine residue of the Grx active site (CysB) or a cysteine residue apart from the active site (CysC). If the substrate is deglutathionylated, the first stage proceeds by the monothiol mechanism, but the glutathionylated intermediate product Grx-SSG then releases GSH yielding the intramolecular disulfide bond Grx(S2) between the catalytic cysteine residue and one of the recycling cysteines. Further, the disulfide bond is reduced using two GSH molecules or by thioredoxin reductase. If the substrate of Grx requires the reduction of an intra- or intermolecular disulfide bond (Fig. 4b), the CysA residue of Grx forms a transient disulfide bond with one of the substrate cysteines, and then the reduced substrate is released, while the disulfide bond is formed between the CysA and CysB or CysC residues of Grx. Finally, the disulfide bond in Grx(S2) is reduced by two molecules of GSH or by thioredoxin reductase [151]. All dithiol isoforms of Grx investigated so far are capable of functioning by the monothiol mechanism, but not all have been tested for the ability for dithiol catalysis. However, all dithiol Grx isoforms that have the ability for dithiol catalysis can also function using the monothiol mechanism.
Four Grx isoenzymes have been found in mammals: Grx1, Grx2, Grx3 (also known as protein interacting cousin of Trx (PICOT)), and Grx5 [152]. The dithiol isoenzyme Grx1 is localized mainly in the cytoplasm, but also can be translocated into the nucleus, secreted from the cell, and localized in the intermembrane space of mitochondria [147, 153]. The dithiol Grx2 was originally found in mitochondria, but later it was found in the cytoplasm and nucleus of testes and in a number of tumor cells [154]. The monothiol isoenzyme Grx3 is a multidomain protein that is present in the nucleus as well as in the cytoplasm, while the monothiol Grx5 is found in mitochondria [152].
Fig. 4. Scheme of the thiol–disulfide exchange with participation of glutaredoxin (Grx). a) Grx catalyzes deglutathionylation of a protein by the monothiol mechanism through the formation of the mixed disulfide with Grx (1) that is reduced by GSH (2). Under conditions of decreased GSH/GSSG ratio, Grx can catalyze S-glutathionylation of proteins (3). b) Grx reduces disulfide bonds in proteins by the dithiol mechanism through the formation of an intermediate complex between Grx and the substrate (1, 2). Oxidized Grx is reduced by two GSH molecules (3, 4).
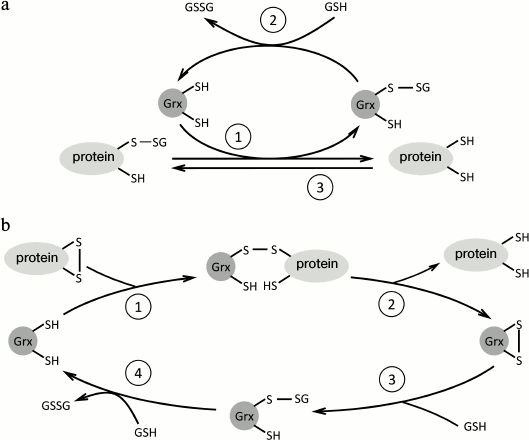
Although the amino acid sequences of Grx1 and Grx2 exhibit only 34% homology, these isoenzymes use the same catalytic mechanism [155, 156]. Grx1 is approximately 10-fold more active than Grx2, but the content of Grx2 in the intermembrane space of mitochondria is higher than that of Grx1, which presumably compensates the difference in their catalytic activity [73]. Oxidized Grx1 is reduced only by GSH, while Grx2 can be reactivated using either glutathione or thioredoxin reductase, which suggests that this protein exhibits properties of both Grx and Trx [46] and implies the connection between the metabolic pathways controlled by glutathione and thioredoxin in mitochondria. Besides, the possibility of reduction of Grx2 by thioredoxin reductase allows Grx2 to function over a wide range of GSH/GSSG values and rather strong oxidative stress in mitochondria [50].
Grx1 and monomeric Grx2 can catalyze both deglutathionylation and the reverse reaction of S-glutathionylation. The direction of the reaction depends on the relative concentrations of the participants protein-SSG, protein-SH, GSH, and GSSG. The redox potential of the GSH/GSSG pair is most important for determining the cellular redox potential. The value of the GSH/GSSG redox potential substantially depends on the functional state of the cell. During cell proliferation, this value is approximately –240 mV, during cell differentiation it reaches –200 mV, and apoptosis results in further growth of this value to –170 mV [157]. It has been ascertained that Grx acts as the GSH-dependent reductase at –240 mV, while at –170 mV it acts as the GSSG-dependent oxidase [158]. Under conditions when GSH/GSSG value is decreased, i.e. under the action of oxidizing factors, Grx can catalyze the S-glutathionylation reaction, while under weakening oxidative stress, Grx catalyzes deglutathionylation [159, 160]. Grx facilitates S-glutathionylation of proteins via the reaction of the disulfide bond with the radical GS• yielding the intermediate anion radical Grx-SSG•–, which then gives the mixed disulfide PSSG [159]. The reversion of S-glutathionylation depends on the extent and duration of the initiating stress, the removal of which usually results in deglutathionylation. The half-reduction time of the glutathionylated bonds is 2-3 h [105]. Evidently, Grx contributes to the control of signal transduction, regulating the processes of glutathionylation and deglutathionylation.
The thiol groups of the active site of some Grx isoenzymes can form complexes with iron–sulfur clusters. These enzymes include a limited number of dithiol Grx isoenzymes of humans, plants, and trypanosomes and virtually all monothiol Grx isoenzymes [161-163]. Most such complexes were found in mitochondria. The cluster [2Fe2S]2+ is located between two monomers of Grx, forming coordination bonds with two active site cysteine residues at the N-termini and with two noncovalently bound GSH molecules. The GSH comes from the free GSH pool, indicating the important role of GSH in stabilizing Fe-S clusters [164]. Since the cofactor [2Fe2S]2+ within the holo-Grx complex interacts with the cysteine residues involved in the catalysis, such complex is catalytically inactive [161]. The degradation of the cluster and dissociation of the holo-complex restore the activity of Grx. Slow degradation of the complex under aerobic conditions is efficiently prevented by GSH. In contrast, GSSG facilitates degradation of the cluster and the activation of Grx [161]. Two GSH molecules in the complex successfully screen the iron atoms from the environment. Thus, the iron of the [2Fe2S]2+ cluster has no possibility to interact with oxidants that require direct molecular interaction, particularly H2O2. It has been shown that release of Grx monomers is caused by O2·– [165]. Presumably, the breakdown of the cluster in response to the action of the oxidant is related to the formation of GSSG. It is suggested that the human Grx2/Fe-S complex is a kind of the redox sensor: under high GSH/GSSG values, Grx is bound to the complex in the inactive state, while changes in the cellular redox status result in the release of the catalytically active Grx [161, 166]. Besides, Grx2 is resistant to oxidative inactivation [167] and successfully functions as an alternative reducing system of Trx1 in the cytosol and Trx2 in mitochondria under the action of inhibitors of thioredoxin reductase. Overexpression of the Grx2 gene in mitochondria protects Trx2 against oxidation, this significantly decreasing the development of apoptosis caused by the production of ROS in mitochondria [167].
Recent studies have shown the importance of the yeast cytosolic multidomain monothiol isoenzymes Grx3 and Grx4 in the intracellular distribution of iron [168]. Simultaneous decrease in Grx3 and Grx4 decelerates all iron-dependent processes in the cytoplasm, mitochondria, and nucleus, which is caused by iron deficiency in the organelles and insufficiency of its incorporation into proteins despite sufficient amount of iron in the cytoplasm. The ability of Grx to bind Fe-S complexes is necessary for bioavailability of iron in the cell [168].
For better understanding the functions of various human Grx isoenzymes, their specificity towards different disulfides must be investigated considering different cellular localization. Besides, their particular contribution to the processes of iron transport and maintenance of Fe-S clusters are also of importance. In this connection, recent investigation of Grx5 seems to be interesting. It was demonstrated that the gene of Grx5 had high expression level in bone tissue and played an antiapoptotic regulatory role in osteoblasts [169]. However, it remains unclear, what precisely affected the development of the oxidative stress-induced apoptosis while changing the level of Grx5 (overexpression or knockout of the gene): alterations of the thiol–disulfide homeostasis or homeostasis of the iron–sulfur clusters.
Thiol–disulfide exchange influences not only the substrate structure, but also the structure of Grx. Comparison of the oxidized and reduced forms of Grx1 from E. coli and T4 bacteriophage demonstrated that the structures of the two forms of the isoenzymes are very similar, although there are some differences [170, 171]. In the presence of the mixed disulfide bond with GSH, the Grx1 of E. coli exhibits properties of the oxidized protein [172]. The structural changes involve the region of the active site, increasing the flexibility of this region in the reduced enzyme form. Besides, in the oxidized Grx, the surface of the molecule involved in protein–protein interactions is masked. Therefore, the affinity of Grx to the substrate decreases as soon as the substrate is reduced, resulting in the dissociation of the Grx–substrate complex.
The specific regulation of protein activities through glutathionylation/deglutathionylation processes are important for many aspects of cell functioning including the regulation of apoptotic signal cascades. It was found that apoptosis induced by TNF-α and FasL is highly sensitive to S-glutathionylation. Thus, in epithelial lung cells, the Fas receptor is glutathionylated on Cys294 during degradation of Grx1 involving caspase-8 and/or caspase-3, this resulting in the acceleration of apoptosis [173]. The results indicate the existence of feedback between caspase-3 and Grx, since Grx activates procaspase-3, which causes degradation of Grx. At the same time, in vitro studies demonstrated inactivation of caspase-3 by glutathionylation [174]. For better understanding of the interactions between Grx and caspase-3, it is necessary to determine the specific cysteine residues of caspase-3 that are glutathionylated/deglutathionylated. This is of importance for ascertaining the relation between the redox status of caspase-3 and the mechanism of activation of apoptosis. It is assumed that the deglutathionylation catalyzed by Grx can play a crucial role in the mechanism of the redox regulation of the processes from the proliferation to apoptosis that is specific for different cell types [175].
It should be noted that Grx is capable of protein–protein interactions. For example, Grx participates in the regulation of ASK1 protein kinase activating JNK1- and p38-dependent signal pathways of apoptosis [103]. Using different cell lines, it was shown that ASK1 is activated by ROS, in particular by H2O2, due to the breakdown of the complex with Grx1. The reduced Grx1 binds to the C-terminal domain of ASK1, resulting in the inactivation of the kinase. In contrast, oxidation of Grx1 leads to the dissociation of the complex, activation of ASK1, and induction of apoptosis [176]. This dissociation is prevented by catalase or N-acetylcysteine. The decrease in GSH content using BSO inhibits the binding of Grx1 to ASK1. Presumably, GSH is necessary for the reduction of the intermolecular disulfide bonds between the adjacent cysteine residues in the Grx1 molecule, which enables the protein to bind ASK1 [176]. These data suggest that Grx1 can be considered as a redox-sensitive factor involved in the regulation of signal cascades of JNK1 and p38 MAP-kinases.
Glutathionylation in cells of humans and other mammals participates in the regulation of a number of key proteins and processes in response to redox signals. More than 200 mammalian proteins are known to be involved in thiol–disulfide exchange. For example, S-glutathionylation has been shown to inhibit phosphofructokinase, carboanhydrase III, nuclear factor NF1, glyceraldehyde-3-phosphate dehydrogenase, protein tyrosine phosphatase 1B, protein kinase Cα, creatine kinase, actin, protein phosphatase 2A, protein kinase A, tyrosine hydroxylase, complex I of the mitochondrial respiratory chain, NR-κB transcription factor, and IκB kinase (IKK). In contrast, such proteins as microsomal S-glutathione transferase, phosphatase of carbonic anhydrase III, HIV-1 protease, matrix metalloproteinase, HRAS GTPase, sarcoplasmic calcium ATPase, and complex II of the mitochondrial respiratory chain are activated by S-glutathionylation. The progression of oxidative stress and change in the functioning of Grx disturb the regulation of S-glutathionylation, which may facilitate a number of pathophysiological changes observed in diabetes, disorders of the lungs and heart, oncological diseases, and different neurodegenerative processes. For example, disturbance of the glutathionylation of cytoskeletal elements promotes pathological changes in heart and skeletal muscles in ischemia and in neurons in Friedreich’s ataxia [177]. Glutathionylation of actin prevents its polymerization, so the redox-dependent reversible glutathionylation of actin regulates the cytoskeleton structure, which is of special importance for the functioning of such cells as thrombocytes, in which actin is the main protein [178]. Besides, glutathionylation of actin was shown to be necessary for the dissociation of actin–myosin complexes during cell adhesion [22].
In Alzheimer’s disease, change in metabolism is partially associated with decrease in the activity of α-ketoglutarate dehydrogenase. The activity of this enzyme decreased with glutathionylation under conditions of oxidative stress, which may take place in brain cells in Alzheimer’s disease [179]. Besides, the selective glutathionylation of protein p53 in brain cells was found in Alzheimer’s disease, which also may facilitate the progression of oxidative stress [180].
In patients with type 2 diabetes, glutathionylated hemoglobin was found, and its content correlated with the development of microangiopathy [181]. At the same time, in a rat diabetes model an increased expression of Grx1 gene was revealed, this facilitating the translocation of NF-κB into the nucleus and activation of cell adhesion molecules ICAM-1. Both these processes make a significant contribution to the development of retinopathy. The disturbance in their regulation, in which glutathionylation plays an important role, is observed under the activation of Grx1 [182].
The crystalline lens contains high concentrations of GSH (6 mM) that plays a role of an antioxidant, maintaining the transparency of the lens [183]. During the progression of cataract, the GSH/GSSG ratio decreases and the lens proteins undergo structural changes resulting in the unfolding of the protein globules and exposure of the buried cysteine residues, which increases disulfide bonding and S-glutathionylation [183]. Presumably, the maintenance of GSH level prevents or decelerates the progression of cataract. In the rat diabetes model, N-acetyl-cysteine and glutathione ethyl ester that are easily converted into GSH in vivo successfully suppressed the development of the cataract in early stages [184].
Glutathionylation of transcription factor p53 significantly decreased its ability to bind with the DNA molecule. Consequently, glutathionylation inhibits p53 that suppresses the development of malignant tumors, which may influence oncogenesis [185]. It is supposed that inactivation of p53 through its glutathionylation provides the mechanism for cell adaptation that suppresses the development of the apoptotic response in the early stage of oxidative stress and allows the cell to avoid immediate death [185].
Of note is that age is a risk factor for many diseases, since different damages accumulate with age, while repair systems slow their activity. With age, mitochondrial functions can also be affected by negative changes facilitating ROS production, which is observed simultaneously with decrease in the activity of antioxidant enzymes. Such disorders in redox-regulation affect the S-glutathionylation of proteins, which makes the cell more sensitive to apoptosis and promotes the development of pathologies [175].
CONCLUSION
Summarizing the data we have described, we conclude that an important role in the system of the antioxidant defense and the redox-dependent regulation belongs to GSH and redox-dependent enzymes. For the last decade, new details concerning the participation of glutathione-dependent enzymes (glutathione transferase and glutaredoxin) in the processes of proliferation, apoptosis, protein folding, and cell signaling have been revealed. Reduced glutathione (GSH) is an important intracellular antioxidant that plays a special role in the maintenance of the cellular redox status due to participation in thiol–disulfide exchange, providing the regulation of a number of cellular functions from gene expression to the activity of separate enzymes and enzyme systems. The maintenance of the optimal GSH/GSSG level is of importance for cell viability. Decrease in GSH content below the normal level can be the indicator of the disturbances of cellular redox status and the alteration of redox-dependent gene regulation. Disturbance of the intracellular GSH balance is observed in a number of pathologies including malignant tumors. This significantly alters the mechanism of cellular redox signaling that is controlled both nonenzymatically and enzymatically with the participation of isoenzymes of glutathione transferase and glutaredoxin.
REFERENCES
1.Nagy, P. (2013) Kinetics and mechanisms of
thiol-disulfide exchange covering direct substitution and thiol
oxidation-mediated pathways, Antioxid. Redox Signal., 18,
1623-1641.
2.Janssen-Heininger, Y. M., Nolin, J. D., Hoffman, S.
M., van der Velden, J. L., Tully, J. E., Lahue, K. G., Abdalla, S. T.,
Chapman, D. G., Reynaert, N. L., van der Vliet, A., and Anathy, V.
(2013) Emerging mechanisms of glutathione-dependent chemistry in
biology and disease, J. Cell. Biochem., 114,
1962-1968.
3.Lu, S. C. (2013) Glutathione synthesis, Biochim.
Biophys. Acta, 1830, 3143-3153.
4.Franco, R., and Cidlowski, J. A. (2009) Apoptosis
and glutathione: beyond an antioxidant, Cell Death Different.,
16, 1303-1314.
5.Deponte, M. (2013) Glutathione catalysis and the
reaction mechanisms of glutathione-dependent enzymes, Biochim.
Biophys. Acta, 1830, 3217-3266.
6.Townsend, D. M., Tew, K. D., and Tapiero, H. (2003)
The importance of glutathione in human disease, Biomed.
Pharmacother., 57, 145-155.
7.Grek, C. L., Zhang, J., Manevich, Y., Danyelle, M.,
Townsend, D. M., and Tew, K. D. (2013) Causes and consequences of
cysteine S-glutathionylation, J. Biol. Chem., 288,
26497-26504.
8.Board, P. G., and Menon, D. (2013) Glutathione
transferases, regulators of cellular metabolism and physiology,
Biochim. Biophys. Acta, 1830, 3267-3288.
9.Allen, E. M., and Mieyal, J. J. (2012)
Protein-thiol oxidation and cell death: regulatory role of
glutaredoxins, Antioxid. Redox Signal., 17,
1748-1763.
10.Lillig, C. H., and Berndt, C. (2013)
Glutaredoxins in thiol/disulfide exchange, Antioxid. Redox
Signal., 18, 1654-1665.
11.Green, R. M., Graham, M., O’Donovan, M. R.,
Chipman, J. K., and Hodges, N. J. (2006) Subcellular
compartmentalization of glutathione: correlations with parameters of
oxidative stress related to genotoxicity, Mutagenesis,
21, 383-390.
12.Ookhtens, M., and Kaplowitz, N. (1998) Role of
the liver in interorgan homeostasis of glutathione and cyst(e)ine,
Sem. Liver Dis., 18, 313-329.
13.Galano, A., and Alvarez-Idaboy, J. R. (2011)
Glutathione: mechanism and kinetics of its non-enzymatic defense action
against free radicals, RSC Adv., 1, 1763-1771.
14.Winterbourn, C. C. (1993) Superoxide as an
intracellular radical sink, Free Rad. Biol. Med., 14,
85-90.
15.Ortega, A. L., Mena, S., and Estrela, J. M.
(2011) Glutathione in cancer cell death, Cancers, 3,
1285-1310.
16.Go, Y.-M., and Jones, D. P. (2008) Redox
compartmentalization in eukaryotic cells, Biochim. Biophys.
Acta, 1780, 1273-1290.
17.Jones, D. P. (2006) Redefining oxidative stress,
Antioxid. Redox Signal., 8, 1865-1879.
18.Cai, Z., and Yan, L. J. (2013) Protein oxidative
modifications: beneficial roles in disease and health, J. Biochem.
Pharmacol. Res., 1, 15-26.
19.Jacob, C., Battaglia, E., Burkholz, T., Peng, D.,
Bagrel, D., and Montenarh, M. (2012) Control of oxidative
posttranslational cysteine modifications: from intricate chemistry to
widespread biological and medical applications, Chem. Res.
Toxicol., 25, 588-604.
20.Rhee, S. G., Jeong, W., Chang, T.-S., and Woo, H.
A. (2007) Sulfiredoxin, the cysteine sulfinic acid reductase specific
to 2-Cys peroxiredoxin: its discovery, mechanism of action, and
biological significance, Kidney Int. Suppl., 72,
S3-S8.
21.Fitzpatrick, A. M., Jones, D. P., and Brown, L.
A. (2012) Glutathione redox control of asthma: from molecular
mechanisms to therapeutic opportunities, Antioxid. Redox
Signal., 17, 375-408.
22.Fiaschi, T., Cozzi, G., Raugei, G., Formigli, L.,
Ramponi, G., and Chiarugi, P. (2006) Redox regulation of beta-actin
during integrin-mediated cell adhesion, J. Biol. Chem.,
281, 22983-22991.
23.Qanungo, S., Starke, D. W., Pai, H. V., Mieyal,
J. J., and Nieminen, A. L. (2007) Glutathione supplementation
potentiates hypoxic apoptosis by S-glutathionylation of
p65-NFκΒ, J. Biol. Chem., 282,
18427-18436.
24.Reynaert, N. L., van der Vliet, A., Guala, A. S.,
McGovern, T., Hristova, M., Pantano, C., Heintz, N. H., Heim, J., Ho,
Y. S., Matthews, D. E., Wouters, E. F., and Janssen-Heininger, Y. M.
(2006) Dynamic redox control of NF-κB through
glutaredoxin-regulated S-glutathionylation of inhibitory κB
kinase β, Proc. Natl. Acad. Sci. USA, 103,
13086-13091.
25.Lluis, J. M., Morales, A., Blasco, C., Colell,
A., Mari, M., Garcia-Ruiz, C., and Fernandez-Checa, J. C. (2005)
Critical role of mitochondrial glutathione in the survival of
hepatocytes during hypoxia, J. Biol. Chem., 280,
3224-3232.
26.Markovic, J., Borras, C., Ortega, A., Sastre, J.,
Vina, J., and Pallardo, F. V. (2007) Glutathione is recruited into the
nucleus in early phases of cell proliferation, J. Biol. Chem.,
282, 20416-20424.
27.Garcia-Gimenez, J. L., Markovic, J., Dasi, F.,
Queval, G., Schnaubelt, D., Foyer, C. H., and Pallardo, F. V. (2013)
Nuclear glutathione, Biochim. Biophys. Acta, 1830,
3304-3316.
28.Bellomo, G., Palladini, G., and Vairetti, M.
(1997) Intranuclear distribution, function and fate of glutathione and
glutathione-S-conjugate in living rat hepatocytes studied by
fluorescence microscopy, Microsc. Res. Techn., 36,
243-252.
29.Ho, Y. F., and Guenthner, T. M. (1994) Uptake and
biosynthesis of glutathione by isolated hepatic nuclei,
Toxicologist, 14, 178.
30.Voehringer, D. W., McConkey, D. J., McDonnell, T.
J., Brisbay, S., and Meyn, R. E. (1998) Bcl-2 expression causes
redistribution of glutathione to the nucleus, Proc. Natl. Acad. Sci.
USA, 95, 2956-2960.
31.Zimmermann, A. K., Loucks, F. A., Schroeder, E.
K., Bouchard, R. J., Tyler, K. L., and Linseman, D. A. (2007)
Glutathione binding to the Bcl-2 homology-3 domain groove: a molecular
basis for Bcl-2 antioxidant function at mitochondria, J. Biol.
Chem., 282, 29296-29304.
32.Carpenter, G., and Cohen, S. (1990) Epidermal
growth factor, J. Biol. Chem., 265, 7709-7712.
33.Jang, J. H., and Surh, Y. J. (2003) Potentiation
of cellular antioxidant capacity by Bcl-2: implications for its
antiapoptotic function, Biochem. Pharmacol., 66,
1371-1379.
34.Schafer, F. Q., and Buettner, G. R. (2001) Redox
environment of the cell as viewed through the redox state of the
glutathione disulfide/glutathione couple, Free Rad. Biol. Med.,
30, 1191-1212.
35.Soboll, S., Grundel, S., Harris, J.,
Kolb-Bachofen, V., Ketterer, B., and Sies, H. (1995) The content of
glutathione and glutathione S-transferases and the glutathione
peroxidase activity in rat liver nuclei determined by a non-aqueous
technique of cell fractionation, Biochem. J., 311 (Pt.
3), 889-894.
36.Conrad, M., Moreno, S. G., Sinowatz, F., Ursini,
F., Kolle, S., Roveri, A., Brielmeier, M., Wurst, W., Maiorino, M., and
Bornkamm, G. W. (2005) The nuclear form of phospholipid hydroperoxide
glutathione peroxidase is a protein thiol peroxidase contributing to
sperm chromatin stability, Mol. Cell. Biol., 25,
7637-7644.
37.Pujari, G., Berni, A., Palitti, F., and
Chatterjee, A. (2009) Influence of glutathione levels on
radiation-induced chromosomal DNA damage and repair in human peripheral
lymphocytes, Mutat. Res., 675, 23-28.
38.Berger, F., Ramirez-Hernandez, M. H., and
Ziegler, M. (2004) The new life of a centenarian: signaling functions
of NAD(P), Trends Biochem. Sci., 29, 111-118.
39.Pellny, T. K., Locato, V., Vivancos, P. D.,
Markovic, J., De Gara, L., Pallardo, F. V., and Foyer, C. H. (2009)
Pyridine nucleotide cycling and control of intracellular redox state in
relation to poly(ADP-ribose) polymerase activity and nuclear
localization of glutathione during exponential growth of
Arabidopsis cells in culture, Mol. Plant,
2, 442-456.
40.Garcia-Gimenez, J. L., Ledesma, A. M., Esmoris,
I., Roma-Mateo, C., Sanz, P., Vina, J., and Pallardo, F. V. (2012)
Histone carbonylation occurs in proliferating cells, Free Rad. Biol.
Med., 52, 1453-1464.
41.Cadenas, E., and Davies, K. J. (2000)
Mitochondrial free radical generation, oxidative stress, and aging,
Free Rad. Biol. Med., 29, 222-230.
42.Mari, M., Morales, A., Colell, A., Garcia-Ruiz,
C., and Fernandez-Checa, J. C. (2009) Mitochondrial glutathione, a key
survival antioxidant, Antioxid. Redox Signal., 11,
2685-2700.
43.Cole-Ezea, P., Swan, D., Shanley, D., and
Hesketh, J. (2012) Glutathione peroxidase 4 has a major role in
protecting mitochondria from oxidative damage and maintaining oxidative
phosphorylation complexes in gut epithelial cells, Free Rad. Biol.
Med., 53, 488-497.
44.Liang, H., Ran, Q., Jang, Y. C., Holstein, D.,
Lechleiter, J., McDonald-Marsh, T., Musatov, A., Song, W., Van Remmen,
H., and Richardson, A. (2009) Glutathione peroxidase 4 differentially
regulates the release of apoptogenic proteins from mitochondria,
Free Rad. Biol. Med., 47, 312-320.
45.Mari, M., Morales, A., Colell, A., Garcia-Ruiz,
C., Kaplowitz, N., and Fernandez-Checa, J. C. (2013) Mitochondrial
glutathione: features, regulation and role in disease, Biochim.
Biophys. Acta, 1830, 3317-3328.
46.Johansson, C., Lillig, C. H., and Holmgren, A.
(2004) Human mitochondrial glutaredoxin reduces S-glutathionylated
proteins with high affinity accepting electrons from either glutathione
or thioredoxin reductase, J. Biol. Chem., 279,
7537-7543.
47.Knoops, B., Goemaere, J., Van der Eecken, V., and
Declercq, J. P. (2011) Peroxiredoxin 5: structure, mechanism, and
function of the mammalian atypical 2-Cys peroxiredoxin, Antioxid.
Redox Signal., 15, 817-829.
48.Zhang, H., Go, Y. M., and Jones, D. P. (2007)
Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in
parallel with mitochondrial GSH system in protection against oxidative
stress, Arch. Biochem. Biophys., 465, 119-126.
49.Taylor, E. R., Hurrell, F., Shannon, R. J., Lin,
T. K., Hirst, J., and Murphy, M. P. (2003) Reversible glutathionylation
of complex I increases mitochondrial superoxide formation, J. Biol.
Chem., 278, 19603-19610.
50.Beer, S. M., Taylor, E. R., Brown, S. E., Dahm,
C. C., Costa, N. J., Runswick, M. J., and Murphy, M. P. (2004)
Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation
of mitochondrial membrane thiol proteins: implications for
mitochondrial redox regulation and antioxidant defense, J. Biol.
Chem., 279, 47939-47951.
51.Hurd, T. R., Requejo, R., Filipovska, A., Brown,
S., Prime, T. A., Robinson, A. J., Fearnley, I. M., and Murphy, M. P.
(2008) Complex I within oxidatively stressed bovine heart mitochondria
is glutathionylated on Cys531 and Cys704 of the 75-kDa subunit:
potential role of Cys residues in decreasing oxidative damage, J.
Biol. Chem., 283, 24801-24815.
52.Mailloux, R. J., Jin, X., and Willmore, W. G.
(2013) Redox regulation of mitochondrial function with emphasis on
cysteine oxidation reactions, Redox Biol., 2,
123-139.
53.Tretter, L., and Adam-Vizi, V. (2000) Inhibition
of Krebs cycle enzymes by hydrogen peroxide: a key role of
α-ketoglutarate dehydrogenase in limiting NADH production under
oxidative stress, J. Neurosci., 20, 8972-8979.
54.Gibson, G. E., Park, L. C., Sheu, K. F., Blass,
J. P., and Calingasan, N. Y. (2000) The alpha-ketoglutarate
dehydrogenase complex in neurodegeneration, Neurochem. Int.,
36, 97-112.
55.Cazanave, S., Berson, A., Haouzi, D., Vadrot, N.,
Fau, D., Grodet, A., Letteron, P., Feldmann, G., El-Benna, J.,
Fromenty, B., Robin, M. A., and Pessayre, D. (2007) High hepatic
glutathione stores alleviate Fas-induced apoptosis in mice, J.
Hepatol., 46, 858-868.
56.Aoyama, K., Watabe, M., and Nakaki, T. (2012)
Modulation of neuronal glutathione synthesis by EAAC1 and its
interacting protein GTRAP3-18, Amino Acids, 42,
163-169.
57.Thompson, J. A., and Franklin, C. C. (2009)
Enhanced glutathione biosynthetic capacity promotes resistance to
As3+-induced apoptosis, Toxicol. Lett., 193,
33-40.
58.Pias, E. K., and Aw, T. Y. (2002) Early redox
imbalance mediates hydroperoxide-induced apoptosis in mitotic competent
undifferentiated PC-12 cells, Cell Death Different., 9,
1007-1016.
59.Pias, E. K., and Aw, T. Y. (2002) Apoptosis in
mitotic competent undifferentiated cells is induced by cellular redox
imbalance independent of reactive oxygen species production, FASEB
J., 16, 781-790.
60.Wang, T. G., Gotoh, Y., Jennings, M. H., Rhoads,
C. A., and Aw, T. Y. (2000) Lipid hydroperoxide-induced apoptosis in
human colonic CaCo-2 cells is associated with an early loss of cellular
redox balance, FASEB J., 14, 1567-1576.
61.Ekshyyan, O., and Aw, T. Y. (2005) Decreased
susceptibility of differentiated PC12 cells to oxidative challenge:
relationship to cellular redox and expression of apoptotic protease
activator factor-1, Cell Death Different., 12,
1066-1077.
62.Okouchi, M., Okayama, N., and Aw, T. Y. (2005)
Differential susceptibility of naive and differentiated PC-12 cells to
methylglyoxal-induced apoptosis: influence of cellular redox, Curr.
Neurovasc. Res., 2, 13-22.
63.Runchel, C., Matsuzawa, A., and Ichijo, H. (2011)
Mitogen-activated protein kinases in mammalian oxidative stress
responses, Antioxid. Redox Signal., 15, 205-218.
64.Kyriakis, J. M., and Avruch, J. (2001) Mammalian
mitogen-activated protein kinase signal transduction pathways activated
by stress and inflammation, Physiol. Rev., 81,
807-869.
65.Circu, M. L., and Aw, T. Y. (2010) Reactive
oxygen species, cellular redox systems, and apoptosis, Free Rad.
Biol. Med., 48, 749-762.
66.Lu, G. D., Shen, H. M., Chung, M. C., and Ong, C.
N. (2007) Critical role of oxidative stress and sustained JNK
activation in aloe-emodin-mediated apoptotic cell death in human
hepatoma cells, Carcinogenesis, 28, 1937-1945.
67.Cuadrado, A., Garcia-Fernandez, L. F., Gonzalez,
L., Suarez, Y., Losada, A., Alcaide, V., Martinez, T., Fernandez-Sousa,
J. M., Sanchez-Puelles, J. M., and Munoz, A. (2003) Aplidin induces
apoptosis in human cancer cells via glutathione depletion and sustained
activation of the epidermal growth factor receptor, Src, JNK, and p38
MAPK, J. Biol. Chem., 278, 241-250.
68.Ji, L., Shen, K., Jiang, P., Morahan, G., and
Wang, Z. (2011) Critical roles of cellular glutathione homeostasis and
jnk activation in andrographolide-mediated apoptotic cell death
in human hepatoma cells, Mol. Carcinogen., 50,
580-591.
69.Filomeni, G., Rotilio, G., and Ciriolo, M. R.
(2003) Glutathione disulfide induces apoptosis in U937 cells by a
redox-mediated p38 MAP kinase pathway, FASEB J., 17,
64-66.
70.Filomeni, G., Aquilano, K., Civitareale, P.,
Rotilio, G., and Ciriolo, M. R. (2005) Activation of c-Jun-N-terminal
kinase is required for apoptosis triggered by glutathione disulfide in
neuroblastoma cells, Free Rad. Biol. Med., 39,
345-354.
71.Piccirillo, S., Filomeni, G., Brune, B., Rotilio,
G., and Ciriolo, M. R. (2009) Redox mechanisms involved in the
selective activation of Nrf2-mediated resistance versus p53-dependent
apoptosis in adenocarcinoma cells, J. Biol. Chem., 284,
27721-27733.
72.Filomeni, G., Piccirillo, S., Rotilio, G., and
Ciriolo, M. R. (2012) p38(MAPK) and ERK1/2 dictate cell death/survival
response to different pro-oxidant stimuli via p53 and Nrf2 in
neuroblastoma cells SH-SY5Y, Biochem. Pharmacol., 83,
1349-1357.
73.Cross, J. V., and Templeton, D. J. (2004)
Oxidative stress inhibits MEKK1 by site-specific glutathionylation in
the ATP-binding domain, Biochem. J., 381, 675-683.
74.Ghosh, S., Pulinilkunnil, T., Yuen, G.,
Kewalramani, G., An, D., Qi, D., Abrahani, A., and Rodrigues, B. (2005)
Cardiomyocyte apoptosis induced by short-term diabetes requires
mitochondrial GSH depletion, Am. J. Physiol. Heart Circ.
Physiol., 289, H768-H776.
75.Armstrong, J. S., Steinauer, K. K., Hornung, B.,
Irish, J. M., Lecane, P., Birrell, G. W., Peehl, D. M., and Knox, S. J.
(2002) Role of glutathione depletion and reactive oxygen species
generation in apoptotic signaling in a human B lymphoma cell line,
Cell Death Different., 9, 252-263.
76.Mari, M., Colell, A., Morales, A., Caballero, F.,
Moles, A., Fernandez, A., Terrones, O., Basanez, G., Antonsson, B.,
Garcia-Ruiz, C., and Fernandez-Checa, J. C. (2008) Mechanism of
mitochondrial glutathione-dependent hepatocellular susceptibility to
TNF despite NF-κB activation, Gastroenterology,
134, 1507-1520.
77.Circu, M. L., Rodriguez, C., Maloney, R., Moyer,
M. P., and Aw, T. Y. (2008) Contribution of mitochondrial GSH transport
to matrix GSH status and colonic epithelial cell apoptosis, Free
Rad. Biol. Med., 44, 768-778.
78.Martins, N. M., Santos, N. A., Curti, C.,
Bianchi, M. L., and Santos, A. C. (2008) Cisplatin induces
mitochondrial oxidative stress with resultant energetic metabolism
impairment, membrane rigidification and apoptosis in rat liver, J.
Appl. Toxicol., 28, 337-344.
79.Santos, N. A., Catao, C. S., Martins, N. M.,
Curti, C., Bianchi, M. L., and Santos, A. C. (2007) Cisplatin-induced
nephrotoxicity is associated with oxidative stress, redox state
unbalance, impairment of energetic metabolism and apoptosis in rat
kidney mitochondria, Arch. Toxicol., 81, 495-504.
80.Chen, G., Chen, Z., Hu, Y., and Huang, P. (2011)
Inhibition of mitochondrial respiration and rapid depletion of
mitochondrial glutathione by beta-phenethyl isothiocyanate: mechanisms
for anti-leukemia activity, Antioxid. Redox Signal., 15,
2911-2921.
81.Chernyak, B. V., and Bernardi, P. (1996) The
mitochondrial permeability transition pore is modulated by oxidative
agents through both pyridine nucleotides and glutathione at two
separate sites, Europ. J. Biochem., 238, 623-630.
82.Costantini, P., Chernyak, B. V., Petronilli, V.,
and Bernardi, P. (1996) Modulation of the mitochondrial permeability
transition pore by pyridine nucleotides and dithiol oxidation at two
separate sites, J. Biol. Chem., 271, 6746-6751.
83.Aon, M. A., Cortassa, S., Maack, C., and
O’Rourke, B. (2007) Sequential opening of mitochondrial ion
channels as a function of glutathione redox thiol status, J. Biol.
Chem., 282, 21889-21900.
84.Wu, B., and Dong, D. (2012) Human cytosolic
glutathione transferases: structure, function, and drug discovery,
Trends Pharmacol. Sci., 33, 656-668.
85.Hayes, J. D., Flanagan, J. U., and Jowsey, I. R.
(2005) Glutathione transferases, Ann. Rev. Pharmacol. Toxicol.,
45, 51-88.
86.Tew, K. D., and Townsend, D. M. (2012)
Glutathione-S-transferases as determinants of cell survival and death,
Antioxid. Redox Signal., 17, 1728-1737.
87.Kulinskii, V. I. (1999) Detoxification of
xenobiotics, Soros Ed. J., 1, 8-12.
88.Krajewski, M. P., Kanawati, B., Fekete, A.,
Kowalski, N., Schmitt-Kopplin, P., and Grill, E. (2013) Analysis of
Arabidopsis glutathione-transferases in yeast,
Phytochemistry, 91, 198-207.
89.Ladner, J. E., Parsons, J. F., Rife, C. L.,
Gilliland, G. L., and Armstrong, R. N. (2004) Parallel evolutionary
pathways for glutathione transferases: structure and mechanism of the
mitochondrial class kappa enzyme rGSTK1-1, Biochemistry,
43, 352-361.
90.Jakobsson, P.-J., Morgenstern, R., Mancini, J.,
Ford-Hutchinson, A., and Persson, B. (1999) Common structural features
of MAPEG – a widespread superfamily of membrane associated
proteins with highly divergent functions in eicosanoid and glutathione
metabolism, Protein Sci., 8, 689-692.
91.Morel, F., and Aninat, C. (2011) The glutathione
transferase kappa family, Drug Metab. Rev., 43,
281-291.
92.Mandal, A. K., Skoch, J., Bacshai, B. J., Hyman,
B. T., Christmas, P., Miller, D., Yamin, T. T., Xu, S., Wisniewski, D.,
Evans, J. F., and Soberman, R. J. (2004) The membrane organization of
leukotriene synthesis, Proc. Natl. Acad. Sci. USA, 101,
6587-6592.
93.Oakley, A. (2011) Glutathione transferases: a
structural perspective, Drug Metab. Rev., 43,
138-151.
94.Prabhu, K. S., Reddy, P. V., Jones, E. C., Liken,
A. D., and Reddy, C. C. (2004) Characterization of a class alpha
glutathione S-transferase with glutathione peroxidase activity in human
liver microsomes, Arch. Biochem. Biophys., 424,
72-80.
95.Hiratsuka, A., Yamane, H., Yamazaki, S., Ozawa,
N., and Watabe, T. (1997) Subunit Ya-specific glutathione peroxidase
activity toward cholesterol 7-hydroperoxides of glutathione
S-transferases in cytosols from rat liver and skin, J. Biol.
Chem., 272, 4763-4769.
96.Hubatsch, I., Ridderstrom, M., and Mannervik, B.
(1998) Human glutathione transferase A4-4: an alpha class enzyme with
high catalytic efficiency in the conjugation of 4-hydroxynonenal and
other genotoxic products of lipid peroxidation, Biochem. J.,
330, 175-179.
97.Dagnino-Subiabre, A., Cassels, B. K., Baez, S.,
Johansson, A. S., Mannervik, B., and Segura-Aguilar, J. (2000)
Glutathione transferase M2-2 catalyzes conjugation of dopamine and dopa
o-quinones, Biochem. Biophys. Res. Commun., 274,
32-36.
98.Manevich, Y., Feinstein, S., and Fisher, A. B.
(2004) Activation of the antioxidant enzyme 1-Cys peroxiredoxin
requires glutathionylation mediated by heterodimerization with pi GST,
Proc. Natl. Acad. Sci. USA, 101, 3780-3785.
99.Adler, V., Yin, Z., Fuchs, S. Y., Benezra, M.,
Rosario, L., Tew, K. D., Pincus, M. R., Sardana, M., Henderson, C. J.,
Wolf, C. R., Davis, R. J., and Ronai, Z. (1999) Regulation of JNK
signaling by GSTp, EMBO J., 18, 1321-1334.
100.Wu, Y., Fan, Y., Xue, B., Luo, L., Shen, J.,
Zhang, S., Jiang, Y., and Yin, Z. (2006) Human glutathione
S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1
signals, Oncogene, 25, 5787-5800.
101.Sharma, A., Patrick, B., Li, J., Sharma, R.,
Jeyabal, P. V., Reddy, P. M., Awasthi, S., and Awasthi, Y. C. (2006)
Glutathione S-transferases as antioxidant enzymes: small cell lung
cancer (H69) cells transfected with hGSTA1 resist doxorubicin-induced
apoptosis, Arch. Biochem. Biophys., 452, 165-173.
102.Dorion, S., Lambert, H., and Landry, J. (2002)
Activation of the p38 signaling pathway by heat shock involves the
dissociation of glutathione S-transferase Mu from Ask1, J. Biol.
Chem., 277, 30792-30797.
103.Ichijo, H., Nishida, E., Irie, K., ten Dijke,
P., Saitoh, M., Moriguchi, T., Takagi, M., Matsumoto, K., Miyazono, K.,
and Gotoh, Y. (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK
that activates SAPK/JNK and p38 signaling pathways, Science,
275, 90-94.
104.Lindahl, M., Mata-Cabana, A., and Kieselbach,
T. (2011) The disulfide proteome and other reactive cysteine proteomes:
analysis and functional significance, Antioxid. Redox Signal.,
14, 2581-2642.
105.Townsend, D. M., Manevich, Y., He, L.,
Hutchens, S., Pazoles, C. J., and Tew, K. D. (2009) Novel role for
glutathione S-transferase pi. Regulator of protein S-glutathionylation
following oxidative and nitrosative stress, J. Biol. Chem.,
284, 436-445.
106.Ralat, L. A., Misquitta, S. A., Manevich, Y.,
Fisher, A. B., and Colman, R. F. (2008) Characterization of the complex
of glutathione S-transferase pi and 1-cysteine peroxiredoxin, Arch.
Biochem. Biophys., 474, 109-118.
107.Pettigrew, N. E., and Colman, R. F. (2001)
Heterodimers of glutathione S-transferase can form between isoenzyme,
Arch. Biochem. Biophys., 396, 225-230.
108.Tew, K. D., Manevich, Y., Grek, C., Xiong, Y.,
Uys, J., and Townsend, D. M. (2011) The role of glutathione
S-transferase P in signaling pathways and S-glutathionylation in
cancer, Free Rad. Biol. Med., 51, 299-313.
109.Ralat, L. A., Manevich, Y., Fisher, A. B., and
Colman, R. F. (2006) Direct evidence for the formation of a complex
between 1-cysteine peroxiredoxin and glutathione S-transferase pi with
activity changes in both enzymes, Biochemistry, 45,
360-372.
110.Hardie, D. G., Ross, F. A., and Hawley, S. A.
(2012) AMPK: a nutrient and energy sensor that maintains energy
homeostasis, Nature Rev. Mol. Cell Biol., 13,
251-262.
111.Klaus, A., Zorman, S., Berthier, A., Polge, C.,
Ramirez, S., Michelland, S., Seve, M., Vertommen, D., Rider, M.,
Lentze, N., Auerbach, D., and Schlattner, U. (2013) Glutathione
S-transferases interact with AMP-activated protein kinase: evidence for
S-glutathionylation and activation in vitro, PLoS One,
8, e62497.
112.Oakhill, J. S., Chen, Z. P., Scott, J. W.,
Steel, R., Castelli, L. A., Ling, N., Macaulay, S. L., and Kemp, B. E.
(2010) β-Subunit myristoylation is the gatekeeper for initiating
metabolic stress sensing by AMP-activated protein kinase (AMPK),
Proc. Natl. Acad. Sci. USA, 107, 19237-19241.
113.Zmijewski, J. W., Banerjee, S., Bae, H.,
Friggeri, A., Lazarowski, E. R., and Abraham, E. (2010) Exposure to
hydrogen peroxide induces oxidation and activation of AMP-activated
protein kinase, J. Biol. Chem., 285, 33154-33164.
114.Hawley, S. A., Ross, F. A., Chevtzoff, C.,
Green, K. A., Evans, A., Fogarty, S., Towler, M. C., Brown, L. J.,
Ogunbayo, O. A., Evans, A. M., and Hardie, D. G. (2010) Use of cells
expressing gamma subunit variants to identify diverse mechanisms of
AMPK activation, Cell Metab., 11, 554-565.
115.Zou, M. H., Kirkpatrick, S. S., Davis, B. J.,
Nelson, J. S., Wiles, W. G., Schlattner, U., Neumann, D., Brownlee, M.,
Freeman, M. B., and Goldman, M. H. (2004) Activation of the
AMP-activated protein kinase by the anti-diabetic drug metformin in
vivo. Role of mitochondrial reactive nitrogen species, J. Biol.
Chem., 279, 43940-43951.
116.Xie, Z., Dong, Y., Zhang, M., Cui, M. Z.,
Cohen, R. A., Riek, U., Neumann, D., Schlattner, U., and Zou, M. H.
(2006) Activation of protein kinase C zeta by peroxynitrite regulates
LKB1-dependent AMP-activated protein kinase in cultured endothelial
cells, J. Biol. Chem., 281, 6366-6375.
117.Tikhanovich, I., Cox, J., and Weinman, S. A.
(2013) Forkhead box class O transcription factors in liver function and
disease, J. Gastroenterol. Hepatol., 28, Suppl. 1,
125-131.
118.Colombo, S. L., and Moncada, S. (2009)
ΑΜPΚα1 regulates the antioxidant status of
vascular endothelial cells, Biochem. J., 421,
163-169.
119.Wang, S., Dale, G. L., Song, P., Viollet, B.,
and Zou, M. H. (2010) ΑΜPΚα1 deletion shortens
erythrocyte life span in mice: role of oxidative stress, J. Biol.
Chem., 285, 19976-19985.
120.Gree, E. L., Oskoui, P. R., Banko, M. R.,
Maniar, J. M., Gygi, M. P., Gygi, S. P., and Brunet, A. (2007) The
energy sensor AMP-activated protein kinase directly regulates the
mammalian FOXO3 transcription factor, J. Biol. Chem.,
282, 30107-30119.
121.Xie, Z., Zhang, J., Wu, J., Viollet, B., and
Zou, M. H. (2008) Upregulation of mitochondrial uncoupling protein-2 by
the AMP-activated protein kinase in endothelial cells attenuates
oxidative stress in diabetes, Diabetes, 57,
3222-3230.
122.Usatyuk, P. V., and Natarajan, V. (2004) Role
of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced
actin remodeling and barrier function in endothelial cells, J. Biol.
Chem., 279, 11789-11797.
123.Zarkovic, N., Ilic, Z., Jurin, M., Schaur, R.
J., Puhl, H., and Esterbauer, H. (1993) Stimulation of HeLa cell growth
by physiological concentrations of 4-hydroxynonenal, Cell Biochem.
Function, 11, 279-286.
124.Pizzimenti, S., Barrera, G., Dianzani, M. U.,
and Brusselbach, S. (1999) Inhibition of D1, D2, and A-cyclin
expression in HL-60 cells by the lipid peroxidation product
4-hydroxynonenal, Free Rad. Biol. Med., 26,
1578-1586.
125.Barrera, G., Pizzimenti, S., Laurora, S.,
Moroni, E., Giglioni, B., and Dianzani, M. U. (2002) 4-Hydroxynonenal
affects pRb/E2F pathway in HL-60 human leukemic cells, Biochem.
Biophys. Res. Commun., 295, 267-275.
126.He, N. G., Singhal, S. S., Srivastava, S. K.,
Zimniak, P., Awasthi, Y. C., and Awasthi, S. (1996) Transfection of a
4-hydroxynonenal metabolizing glutathione S-transferase isozyme, mouse
GSTA4-4, confers doxorubicin resistance to Chinese hamster ovary cells,
Arch. Biochem. Biophys., 333, 214-220.
127.Gallagher, E. P., Gardner, J. L., and Barber,
D. S. (2006) Several glutathione S-transferase isozymes that protect
against oxidative injury are expressed in human liver mitochondria,
Biochem. Pharmacol., 71, 1619-1628.
128.Gallagher, E. P., and Gardner, J. L. (2002)
Comparative expression of two alpha class glutathione S-transferases in
human adult and prenatal liver tissues, Biochem. Pharmacol.,
63, 2025-2036.
129.Raza, H. (2011) Dual localization of
glutathione S-transferase in the cytosol and mitochondria: implications
in oxidative stress, toxicity and disease, FEBS J., 278,
4243-4251.
130.Desmots, F., Rissel, M., Gilot, D.,
Lagadic-Gossmann, D., Morel, F., Guguen-Guillouzo, C., Guillouzo, A.,
and Loyer, P. (2002) Pro-inflammatory cytokines tumor necrosis factor
alpha and interleukin-6 and survival factor epidermal growth factor
positively regulate the murine GSTA4 enzyme in hepatocytes, J. Biol.
Chem., 277, 17892-17900.
131.Curtis, J. M., Grimsrud, P. A., Wright, W. S.,
Xu, X., Foncea, R. E., Graham, D. W., Brestoff, J. R., Wiczer, B. M.,
Ilkayeva, O., Cianflone, K., Muoio, D. E., Arriaga, E. A., and
Bernlohr, D. A. (2010) Downregulation of adipose glutathione
S-transferase A4 leads to increased protein carbonylation, oxidative
stress, and mitochondrial dysfunction, Diabetes, 59,
1132-1142.
132.Hyman, S. E., Malenka, R. C., and Nestler, E.
J. (2006) Neural mechanisms of addiction: the role of reward-related
learning and memory, Ann. Rev. Neurosci., 29,
565-598.
133.Kalivas, P. W., and O’Brien, C. (2008)
Drug addiction as a pathology of staged neuroplasticity,
Neuropsychopharmacology, 33, 166-180.
134.Klatt, P., and Lamas, S. (2002) c-Jun
regulation by S-glutathionylation, Methods Enzymol., 348,
157-174.
135.Humphries, K. M., Deal, M. S., and Taylor, S.
S. (2005) Enhanced dephosphorylation of cAMP-dependent protein kinase
by oxidation and thiol modification, J. Biol. Chem., 280,
2750-2758.
136.Uys, J. D., Knackstedt, L., Hurt, P., Tew, K.
D., Manevich, Y., Hutchens, S., Townsend, D. M., and Kalivas, P. W.
(2011) Cocaine-induced adaptations in cellular redox balance
contributes to enduring behavioral plasticity,
Neuropsychopharmacolgy, 36, 2551-2560.
137.Sun, K. H., Chang, K. H., Clawson, S., Ghosh,
S., Mirzaei, H., Regnier, F., and Shah, K. (2011)
Glutathione-S-transferase P1 is a critical regulator of Cdk5 kinase
activity, J. Neurochem., 118, 902-914.
138.Castro-Caldas, M., Carvalho, A. N., Rodrigues,
E., Henderson, C., Wolf, C. R., and Gama, M. J. (2012) Glutathione
S-transferase pi mediates MPTP-induced c-Jun N-terminal kinase
activation in the nigrostriatal pathway, Mol. Neurobiol.,
45, 466-477.
139.McIlwain, C. C., Townsend, D. M., and Tew, K.
D. (2006) Glutathione S-transferase polymorphisms: cancer incidence and
therapy, Oncogene, 25, 1639-1648.
140.Davis, Jr. W., Ronai, Z., and Tew, K. D. (2001)
Cellular thiols and reactive oxygen species in drug-induced apoptosis,
J. Pharmacol. Exp. Therap., 296, 1-6.
141.Hayes, J. D., and Pulford, D. J. (1995) The
glutathione S-transferase supergene family: regulation of GST and the
contribution of the isoenzymes to cancer chemoprotection and drug
resistance, Crit. Rev. Biochem. Mol. Biol., 30,
445-600.
142.Higgins, L. G., and Hayes, J. D. (2011)
Mechanisms of induction of cytosolic and microsomal glutathione
transferase (GST) genes by xenobiotics and pro-inflammatory agents,
Drug Metab. Rev., 43, 92-137.
143.Pool-Zobel, B., Veeriah, S., and Bohmer, F. D.
(2005) Modulation of xenobiotic metabolizing enzymes by
anticarcinogens – focus on glutathione S-transferases and
their role as targets of dietary chemoprevention in colorectal
carcinogenesis, Mutat. Res., 591, 74-92.
144.McMahon, M., Thomas, N., Itoh, K., Yamamoto,
M., and Hayes, J. D. (2006) Dimerization of substrate adaptors can
facilitate cullin-mediated ubiquitylation of proteins by a
“tethering” mechanism: a two-site interaction model for the
Nrf2–Keap1 complex, J. Biol. Chem., 281,
24756-24768.
145.Alves, R., Vilaprinyo, E., Sorribas, A., and
Herrero, E. (2009) Evolution based on domain combinations: the case of
glutaredoxins, BMC Evol. Biol., 9, artical 66.
146.Rouhier, N. (2010) Plant glutaredoxins: pivotal
players in redox biology and iron-sulfur center assembly, New
Phytol., 186, 365-372.
147.Lillig, C. H., Berndt, C., and Holmgren, A.
(2008) Glutaredoxin systems, Biochim. Biophys. Acta,
1780, 1304-1317.
148.Johansson, C., Roos, A. K., Montano, S. J.,
Sengupta, R., Filippakopoulos, P., Guo, K., von Delft, F., Holmgren,
A., Oppermann, U., and Kavanagh, K. L. (2011) The crystal structure of
human GLRX5: iron-sulfur cluster coordination, tetrameric assembly and
monomer activity, Biochem. J., 433, 303-311.
149.Couturier, J., Jacquot, J. P., and Rouhier, N.
(2009) Evolution and diversity of glutaredoxins in photosynthetic
organisms, Cell. Mol. Life Sci., 66, 2539-2557.
150.Benyamina, S. M., Baldacci-Cresp, F.,
Couturier, J., Chibani, K., Hopkins, J., Bekki, A., de Lajudie, P.,
Rouhier, N., Jacquot, J. P., Alloing, G., Puppo, A., and Frendo, P.
(2013) Two Sinorhizobium meliloti glutaredoxins regulate iron
metabolism and symbiotic bacteroid differentiation, Environ.
Microbiol., 15, 795-810.
151.Xing, S., Lauri, A., and Zachgo, S. (2006)
Redox regulation and flower development: a novel function for
glutaredoxins, Plant Biol., 8, 547-555.
152.Hanschmann, E. M., Godoy, J. R., Berndt, C.,
Hudemann, C., and Lillig, C. H. (2013) Thioredoxins, glutaredoxins, and
peroxiredoxins – molecular mechanisms and health
significance: from cofactors to antioxidants to redox signaling,
Antioxid. Redox Signal., 19, 1539-1605.
153.Lundberg, M., Fernandes, A. P., Kumar, S., and
Holmgren, A. (2004) Cellular and plasma levels of human glutaredoxin 1
and 2 detected by sensitive ELISA systems, Biochem. Biophys. Res.
Commun., 319, 801-809.
154.Lonn, M. E., Hudemann, C., Berndt, C.,
Cherkasov, V., Capani, F., Holmgren, A., and Lillig, C. H. (2008)
Expression pattern of human glutaredoxin 2 isoforms: identification and
characterization of two testis/cancer cell-specific isoforms,
Antioxid. Redox Signal., 10, 547-557.
155.Gallogly, M. M., Starke, D. W., and Mieyal, J.
J. (2009) Mechanistic and kinetic details of catalysis of
thiol-disulfide exchange by glutaredoxins and potential mechanisms of
regulation, Antioxid. Redox Signal., 11, 1059-1081.
156.Stroher, E., and Millar, A. H. (2012) The
biological roles of glutaredoxins, Biochem. J., 446,
333-348.
157.Watson, W. H., Chen, Y., and Jones, D. P.
(2003) Redox state of glutathione and thioredoxin in differentiation
and apoptosis, Biofactors, 17, 307-314.
158.Aslund, F., Berndt, K. D., and Holmgren, A.
(1997) Redox potentials of glutaredoxins and other thiol-disulfide
oxidoreductases of the thioredoxin superfamily determined by direct
protein–protein redox equilibria, J. Biol. Chem.,
272, 30780-30786.
159.Starke, D. W., Chock, P. B., and Mieyal, J. J.
(2003) Glutathione-thiyl radical scavenging and transferase properties
of human glutaredoxin (thioltransferase). Potential role in redox
signal transduction, J. Biol. Chem., 278,
14607-14613.
160.Ruoppolo, M., Lundstrom-Ljung, J., Talamo, F.,
Pucci, P., and Marino, G. (1997) Effect of glutaredoxin and protein
disulfide isomerase on the glutathione-dependent folding of
ribonuclease A, Biochemistry, 36, 12259-12267.
161.Lillig, C. H., Berndt, C., Vergnolle, O., Lonn,
M. E., Hudemann, C., Bill, E., and Holmgren, A. (2005) Characterization
of human glutaredoxin 2 as iron-sulfur protein: a possible role as
redox sensor, Proc. Natl. Acad. Sci. USA, 102,
8168-8173.
162.Riondet, C., Desouris, J. P., Montoya, J. G.,
Chartier, Y., Meyer, Y., and Reichheld, J.-P. (2012) A
dicotyledon-specific glutaredoxin GRXC1 family with dimer-dependent
redox regulation is functionally redundant with GRXC2, Plant Cell
Environ., 35, 360-373.
163.Feng, Y. G., Zhong, N., Rouhier, N., Hase, T.,
Kusunoki, M., Jacquot, J. P., Jin, C. W., and Xia, B. (2006) Structural
insight into poplar glutaredoxin C1 with a bridging iron-sulfur cluster
at the active site, Biochemistry, 45, 7998-8008.
164.Berndt, C., Hudemann, C., Hanschmann, E.-M.,
Axelsson, R., Holmgren, A., and Lillig, C. H. (2007) How does
iron-sulfur cluster coordination regulate the activity of human
glutaredoxin 2? Antioxid. Redox Signal., 9, 151-157.
165.Mitra, S., and Elliott, S. J. (2009) Oxidative
disassembly of the [2Fe-2S] cluster of human Grx2 and redox regulation
in the mitochondria, Biochemistry, 48, 3813-3815.
166.Johansson, C., Kavanagh, K. L., Gileadi, O.,
and Oppermann, U. (2007) Reversible sequestration of active site
cysteines in a 2Fe-2S-bridged dimer provides a mechanism for
glutaredoxin 2 regulation in human mitochondria, J. Biol. Chem.,
282, 3077-3082.
167.Zhang, H., Du, Y., Zhang, X., Lu, J., and
Holmgren, A. (2014) Glutaredoxin 2 reduces both thioredoxin 2 and
thioredoxin 1 and protects cells from apoptosis induced by auranofin
and 4-hydroxynonenal, Antioxid. Redox Signal., in press.
168.Muhlenhoff, U., Molik, S., Godoy, J. R.,
Uzarska, M. A., Richter, N., Seubert, A., Zhang, Y., Stubbe, J.,
Pierrel, F., Herrero, E., Lillig, C. H., and Lill, R. (2010) Cytosolic
monothiol glutaredoxins function in intracellular iron sensing and
trafficking via their bound iron-sulfur cluster, Cell Metab.,
12, 373-385.
169.Linares, G. R., Xing, W., Govoni, K. E., Chen,
S. T., and Mohan, S. (2009) Glutaredoxin 5 regulates osteoblast
apoptosis by protecting against oxidative stress, Bone,
44, 795-804.
170.Xia, T. H., Bushweller, J. H., Sodano, P.,
Billeter, M., Bjornberg, O., Holmgren, A., and Wuthrich, K. (1992) NMR
structure of oxidized Escherichia coli glutaredoxin: comparison
with reduced E. coli glutaredoxin and functionally related
proteins, Protein Sci., 1, 310-321.
171.Wang, Y., Amegbey, G., and Wishart, D. S.
(2004) Solution structures of reduced and oxidized bacteriophage T4
glutaredoxin, J. Biomol. NMR, 29, 85-90.
172.Bushweller, J. H., Billeter, M., Holmgren, A.,
and Wuthrich, K. (1994) The nuclear magnetic resonance solution
structure of the mixed disulfide between Escherichia coli
glutaredoxin (C14S) and glutathione, J. Mol. Biol., 235,
1585-1597.
173.Anathy, V., Aesif, S. W., Guala, A. S.,
Havermans, M., Reynaert, N. L., Ho, Y. S., Budd, R. C., and
Janssen-Heininger, Y. M. (2009) Redox amplification of apoptosis by
caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of
Fas, J. Cell Biol., 184, 241-252.
174.Huang, Z., Pinto, J. T., Deng, H., and Richie,
J. P., Jr. (2008) Inhibition of caspase-3 activity and activation by
protein glutathionylation, Biochem. Pharmacol., 75,
2234-2244.
175.Allen, E. M., and Mieyal, J. J. (2012)
Protein-thiol oxidation and cell death: regulatory role of
glutaredoxins, Antioxid. Redox Signal., 17,
1748-1763.
176.Song, J. J., Rhee, J. G., Suntharalingam, M.,
Walsh, S. A., Spitz, D. R., and Lee, Y. J. (2002) Role of glutaredoxin
in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative
stress mediated by H2O2, J. Biol. Chem.,
277, 46566-46575.
177.Sparaco, M., Gaeta, L. M., Santorelli, F. M.,
Passarelli, C., Tozzi, G., Bertini, E., Simonati, A., Scaravilli, F.,
Taroni, F., Duyckaerts, C., Feleppa, M., and Piemonte, F. (2009)
Friedreich’s ataxia: oxidative stress and cytoskeletal
abnormalities, J. Neurol. Sci., 287, 111-118.
178.Johansson, M., and Lundberg, M. (2007)
Glutathionylation of beta-actin via a cysteinyl sulfenic acid
intermediary, BMC Biochem., 8, 26.
179.Shi, Q., Xu, H., Kleinman, W. A., and Gibson,
G. E. (2008) Novel functions of the α-ketoglutarate dehydrogenase
complex may mediate diverse oxidant-induced changes in mitochondrial
enzymes associated with Alzheimer’s disease, Biochim.
Biophys. Acta, 1782, 229-238.
180.Di Domenico, F., Cenini, G., Sultana, R.,
Perluigi, M., Uberti, D., Memo, M., and Butterfield, D. A. (2009)
Glutathionylation of the pro-apoptotic protein p53 in Alzheimer’s
disease brain: implications for AD pathogenesis, Neurochem.
Res., 34, 727-733.
181.Sampathkumar, R., Balasubramanyam, M.,
Sudarslal, S., Rema, M., Mohan, V., and Balaram, P. (2005) Increased
glutathionylated hemoglobin (HbSSG) in type 2 diabetes subjects with
microangiopathy, Clin. Biochem., 38, 892-899.
182.Shelton, M. D., Kern, T. S., and Mieyal, J. J.
(2007) Glutaredoxin regulates nuclear factor κB and intercellular
adhesion molecule in Muller cells: model of diabetic retinopathy, J.
Biol. Chem., 282, 12467-12474.
183.Craghill, J., Cronshaw, A. D., and Harding, J.
J. (2004) The identification of a reaction site of glutathione mixed
disulphide formation on γS-crystallin in human lens, Biochem.
J., 379, 595-600.
184.Zhang, S., Chai, F. Y., Yan, H., Guo, Y., and
Harding, J. J. (2008) Effects of N-acetylcysteine and glutathione ethyl
ester drops on streptozotocin-induced diabetic cataract in rats,
Mol. Vision, 14, 862-870.
185.Velu, C. S., Niture, S. K., Doneanu, C. E.,
Pattabiraman, N., and Srivenugopal, K. S. (2007) Human p53 is inhibited
by glutathionylation of cysteines present in the proximal DNA-binding
domain during oxidative stress, Biochemistry, 46,
7765-7780.