REVIEW: Human Herpes Simplex Virus: Life Cycle and Development of Inhibitors
M. K. Kukhanova, A. N. Korovina*, and S. N. Kochetkov
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, 119991 Moscow, Russia; E-mail: kukhan86@hotmail.com; anna.korovina@gmail.com; kochet@eimb.ru* To whom correspondence should be addressed.
Received May 19, 2014
WHO reports that 90% of human population is infected by different types of herpesviruses, which develop latency or cause oral and genital herpes, conjunctivitis, eczema herpeticum, and other diseases. Herpesvirus almost always accompanies HIV-infection and complicates AIDS treatment. Herpes simplex virus type 1 is one of the most wide spread viruses from the Herpesviridae family. HSV virion, genome structure, replication mechanisms, antiherpes drug development strategies, including design of prodrugs, and mutations causing ACV-resistance in clinical HSV isolates are discussed in this review.
KEY WORDS: HSV, herpes simplex, life cycle, replication, drugs, mutations, resistanceDOI: 10.1134/S0006297914130124
Abbreviations: ACV, acyclovir; AraA, adenine arabinoside; BVDU, (E)-5-(2-bromovinyl)-2′-deoxyuridine (brivudin); CMV, cytomegalovirus; DAI, DNA-dependent activator of interferon regulatory factor; GCV, ganciclovir; HFC-1, host cell factor 1; HHV-6A, 6B, 7, 8, human herpes virus; HIV, human immunodeficiency virus; HpACV, acyclovir H-phosphonate; HSV-1, herpes simplex virus-1; IFI16, γ-interferon-inducible protein; IRF-3, interferon regulatory factor 3; LAT, latency associated transcript; ND-10, nuclear domain 10; PCV, penciclovir; PFA, phosphonoformic acid; PMEA, 9-(2-phosphonylmethoxyethyl)-adenine; RR, ribonucleotide reductase; VZV, varicella zoster virus.
According to the World Health Organization, viruses of the Herpesviridae
family infect 90% of the Earth’s population. Humans are the hosts
of at least nine unique herpes viruses. The most prevalent is herpes
simplex virus type 1 (HSV-1), which establishes latent infection but
reactivates causing cutaneous or genital herpes, conjunctivitis,
keratitis, encephalitis, or eczema herpeticum. HSV often coinfects
HIV-infected patients, complicating treatment of AIDS. HSV-1 might be
also involved in the pathogenesis of multiple sclerosis [1] and result in male infertility [2].
This review describes the structure, mechanism of replication, and search for new inhibitors of HSV-1.
HERPES SIMPLEX VIRUS TYPE 1: GENERAL DESCRIPTION, LIFE CYCLE, AND
REPLICATION
General description of Herpesviridae family. The Herpesviridae family includes more than 200 species that infect mammals, birds, reptiles, amphibians, fish, and bivalves. It is assumed that herpes viruses and tailed bacteriophages descend from a common ancestor in spite of differences in their morphology and hosts [3]. Conventionally, the Herpesviridae family involves viruses that share a common virion structure. The viral particle consists of a double-stranded DNA core surrounded by an icosahedral capsid consisting of 162 capsomeres surrounded by protein unstructured matrix called tegument, which, in turn, is surrounded by a lipid bilayer envelope with embedded branched glycoproteins.
Referring to these morphological characteristics, various viruses infecting different hosts are classified as herpes viruses.
To date, nine types of human herpes viruses have been identified: herpes simplex viruses types 1 and 2, varicella zoster virus, Epstein–Barr virus, cytomegalovirus, roseoloviruses HHV-6 (A and B) and HHV-7, and Kaposi sarcoma-associated herpes virus (HHV-8). Epstein–Barr virus and HHV-8 are carcinogenic [4, 5].
At the end of the 1970s, Herpesviridae was subdivided into three subfamilies.
1. The Alphaherpesvirinae subfamily includes lytic viruses with relatively short life cycle that infect different cell types, replicate rapidly, and establish latency mainly in sensory ganglia. Simplex virus (HSV-1 and -2), varicella zoster virus (VZV), and some avian viruses are referred to Alphaherpesvirinae. VZV provokes chickenpox in children and herpes zoster in adults.
2. The Betaherpesvirinae subfamily includes viruses with long life cycle and slow progression of infection of a limited number of hosts. Infected cells thrive and increase in size (cytomegaly). Latent infection localizes in secretory glands, lymphoreticular cells, kidneys, and other tissues. Human cytomegalovirus (CMV) and roseoloviruses (HHV-6) are Betaherpesvirinae.
3. The Gammaherpesvirinae subfamily includes Kaposi sarcoma-associated herpes virus (HHV-8), Epstein–Barr virus (EBV) that causes Burkitt’s lymphoma mainly in Central Africa residents, and infectious mononucleosis in USA and other countries [4].
Herpes viruses are highly complex. Their genomes encode many enzymes essential for nucleotide metabolism (thymidine kinase, thymidylate synthase, deoxyuridine triphosphatase, ribonucleotide reductase), DNA replication (DNA polymerase, helicase, primase), DNA reparation (uracil N-glycosylase, UL2), and posttranslational modifications (protein kinases). DNA synthesis and nucleocapsid assembly take place in the nucleus, and virion processing and maturation proceeds in the cytoplasm. The production of a new viral generation always results in host cell death.
Under latent infection, cells bear a circular form of the viral genome, and only a small amount of RNA is transcribed. In a latent infection, the viral genome retains its ability to replicate and to provoke disease on reactivation. The mechanism of reactivation is not studied completely and can vary in different organisms. Different cell types maintain latency of distinct herpes viruses. For example, HSV-1 is detected only in neurons and ganglia that innervate liable to infection epithelium [6], while latent EBV is observed mainly in B-cells [7].
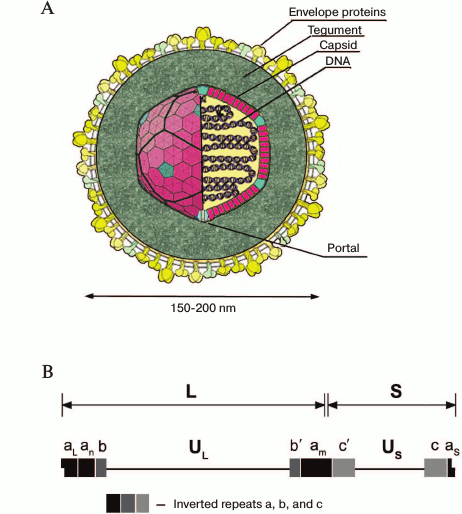
HSV-1 virion structure. HSV-1 virion structure is shown in Fig. 1A. Cryoelectron tomography has provided the most detailed data on the virion structure at resolution of 7 nm [9]. The virions are spherical particles 186 nm in diameter with glycoprotein spikes protruded from each virion, making their full diameter about 225 nm. The nucleocapsid occupies an eccentric position: on one virion side (the proximal pole), it is close to the envelope; on the other side (the distal pole), it is 30-35 nm apart from it. The tegument is an amorphous layer with some structured regions containing 7-nm width filaments apposed to the membrane.
The virion consists of 40 proteins of viral and cellular origin, 10 of which are glycosylated. Eleven proteins are located on the virion surface.
The core contains the linear double-stranded DNA genome wrapped as a toroid. A small fraction of the viral DNA appears to be circular. Host polyamines spermine and spermidine are found in the viral core, neutralizing the negative charges on the viral DNA and providing its proper packing. The virion contains 70,000 and 40,000 molecules of spermine and spermidine per virion, respectively. The polyamines are strongly bound to the DNA and cannot be exchanged with added radioactively labeled polyamines. By the degradation of the outer envelope using detergents and urea, spermidine, but not spermine, can be removed from the virion. Recently, polyamines and modified polyamines have been considered as possible regulators or inhibitors of some viral infections. Dextran-conjugated polyamines, in particular dextran-propan-1,3-diamine, inhibited HSV-1 growth in BS-C-1 cell line [10].
The tegument is comprised of 26 proteins, some of them participating in capsid transport to the nucleus and other organelles (UL36, UL37, ICP0) [11], viral DNA entry into the nucleus (VP1-2, UL36) [12], activation of early genes transcription (VP16, encoded be UL48 gene) [13], suppression of cellular protein biosynthesis, and mRNA degradation (VHS, UL41) [14].
The tegument contains RNA-binding proteins US11, UL47, and UL49 presumably bound to viral and cellular transcripts packaged in the virion.
The capsid has icosahedral configuration and is composed of 162 capsomeres (Fig. 1A) – 150 hexons and 12 pentons.
Fig. 1. A) HSV-1 virion structure; B) HSV-1 genome structure. The long component of the genome (UL) is flanked by inverted repeats designated as ab and ab′, the short one (US) is flanked by ac and ac′ sequences. The number of a sequence repeats at the UL–US junction and at the UL terminus is variable. The terminal aL and aS sequences are unique and asymmetric, and an and am are terminal a sequence repeats at n ≥ 0 and m ≥ 1. The structure of the a sequence (400-500 bp) is highly conserved, but it consists of a variable number of repeat elements. The terminal sequence of UL component (aL) is truncated and contains one 5′-overhanging nucleotide, and the aS sequence contains one 3′-overhanging nucleotide. Upon genome circularization, aL and aS sequences join. The figure is based on data presented in article [8].
Three types of capsids can be isolated from infected cells: A-capsids (procapsids) lack both scaffold proteins and viral DNA; B-capsids do not contain viral DNA but contain the protein scaffold for it; C-capsids contain the viral genome [15, 16].
Capsids of any type consist of four principal proteins: the major capsid protein UL19 (VP5), VP26 accessory protein (UL35), and also UL18 (VP23) and UL38 (VP19C) proteins, whose functions are not well studied. Six copies of the major capsid protein, VP5, form the hexons, and five copies form the pentons. Six copies of VP26 occupy the outer surfaces of the hexons formed by VP5. A single molecule of VP19C and two copies of VP23 form a triplex that binds surrounding capsomeres to form connections between them. In the center of every capsomere, there is a channel joining the virion outer surface and core. The channels in hexamers are 4 nm in diameter, and in pentamers they are slightly narrower, and in B-capsids these channels are completely closed. The capsid contains UL6 protein, which forms the portal on the vertex of one of the 12 capsid axes, through which the viral genome is presumably packed into the capsid [17], and VP24 (UL26) protease, breaking the scaffold during DNA packaging.
The outer envelope of the virion consists of lipid bilayer and 11 glycoproteins (gB, gC, gD, gE, gG, gH, gI, gJ, gK), membrane gL, and gM (Fig. 1A) [18], and at least two unglycosylated membrane proteins (UL20 and US9). The lipid bilayer is formed by cell membrane during virus egress by exocytosis. The function of glycoproteins in virus entry into the cell is now studied extensively.
HSV-1 genome structure. The HSV-1 genome (GenBank accession number X14112) (Fig. 1B) was estimated to be a 152,261-bp linear double-stranded GC-rich (G+C, 68%) DNA sequence [19]. The ends of the DNA are possibly held together or in close proximity inasmuch as a small fraction of the packaged DNA appears to be circular. The linear DNA circularizes in the absence of protein biosynthesis after entering from the nucleus of infected cells.
The HSV-1 genome can be considered as consisting of two unique units, long (UL) and short (US), separated by a set of inverted repeats. The repeats bracketing the UL component are designated ab and ab′, whereas those of the US component are ac and ac′ (Fig. 1B).
Due to the presence of inverted repeats, the UL and US units of the genome can be inverted relative to one another to yield four linear isomers. However, it was shown that neither the presence of internal repeats nor orientation of the genome components affect viral viability in Vero cells [20].
The HSV-1 genome encodes around 90 transcriptional units, and at least 84 encode proteins. With some minor exceptions, each viral transcript encodes a single protein and does not contain any introns. Several transcripts appear not to encode open reading frames (ORFs). Those best known are the latency-associated transcripts (LATs) [21] and oriS encoded regulatory microRNAs [22].
During the course of the HSV-1 infection, different genes are expressed, one gradually turning on and regulating another. According to this cascade fashion, the viral genes are classified into at least three general classes: α or immediate early, β or early, and γ or late genes [23]. Immediate early genes α are mapped near the termini of UL and US. The α0 and α4 genes map within the inverted repeats of UL and US, respectively [24].
Life cycle, gene expression, and replication of HSV-1. The viral life cycle can be divided into the following major steps: entry into the host cell, expression of viral genes, replication, virion assembly, and egress of the new generation of viral particles (Fig. 2A). In permissive cell lines, this cycle takes about 18-20 h.
Fig. 2. A) HSV-1 life cycle: 1) virion attachment and entry into the cell; 2) transport to the nucleus; 3) viral gene expression: immediate early (a), early (b), and late (c); 4) viral DNA replication; 5) nucleocapsid assembly; 6) capsid maturation; 7) primary envelope formation; 8) egress. B) Overall structure of HSV-1 DNA polymerase in ribbon diagram. The figure was composed by the PDB Viewer program and is based on data represented in article [53].
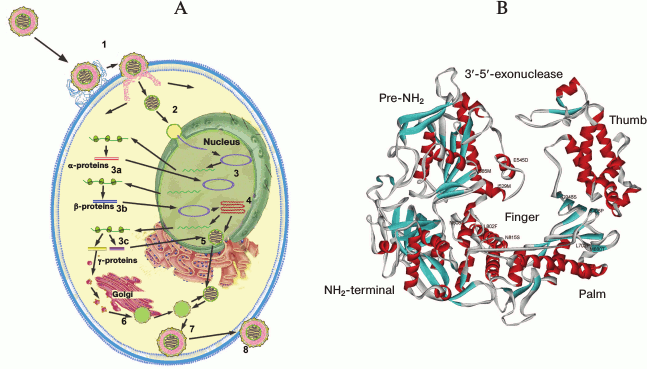
To date, two HSV-1 entry pathways have been proposed (Fig. 2A). The main mechanism assumes the fusion of the viral envelope with the plasma membrane and further transport of the viral capsid to the nucleus. The essential stage of this process is interaction of surface glycoproteins of the virus with specific cell surface receptors. The additional pathway by which the virus enters the cell is endocytosis of the enveloped virion followed by fusion of the envelope with intracellular vesicles [25].
Attachment of the virion to the cell surface is mediated by viral glycoproteins C (gC) and B (gB), which interact with cell surface glycosaminoglycans, in particular heparan sulfate [26].
The interaction between four glycoproteins, gD, gB, and the heterodimer gH/gL, is required for viral entry into the host cell by fusion of the viral outer envelope with the plasma membrane [27, 28]. Glycoprotein gD can bind to the receptors of three types: nectin-1 and nectin-2, herpes virus entry mediator (HVEM), and 3-O-sulfated heparan sulfate (3-O-S-HS). The last is produced by 3-O-sulfotransferases 2-7 (3-OST) [25] making them attractive therapeutic targets for the development of antiherpetic drugs [29].
In addition to binding of gD to cellular receptors, it triggers membrane fusion by interaction with the gB and gH/gL complex. The exact mechanism and participants of this process are poorly understood, but it is known that the N-terminal region of gD interacts with cellular receptors causing the release of its C-terminal domain, which activates gB and gH/gL complex thereby triggering membrane fusion. When gD is not bound to the ligand, the C-terminal domain is blocked [27]. An interesting additional function of gD is suppression of apoptosis in the HSV-infected cell [30]. Interaction between gB and paired immunoglobulin-like type 2 receptor α (PILRα) is necessary for viral entry into the cell. Upon addition of antibodies to these receptors, infection in cell culture is suppressed [31].
After entering of the viral particle, which is the capsid coated with the tegument, it is transported to the nuclear pores with subsequent transfer into the nucleus (Fig. 2A). HSV-1 travels through the cells for rather long distances, especially in neurons. Using indirect immunofluorescence microscopy, it was shown that the viral capsid is transported to the nucleus along a network of microtubules. The transport is powered by the cytoplasmic motor protein dynein [32]. In a cell-free system, capsids coated with the inner tegument exposing US3, UL36, UL37, ICP0, UL14, UL16, and UL21 proteins recruited motor proteins associated with microtubules (dynein, dynactin, kinesin-1, and kinesin-2). The most likely candidates to play a role of a linking chain between the motor proteins and the capsids are UL36 and UL37 proteins. The capsids, which are not coated with the tegument or are coated with tegument that contains other proteins, do not bind the motor proteins. Presumably, when the outer envelope of the virion is fused with the cell membrane, the outer tegument proteins remain bound to the membrane. So, the proteins of the inner tegument are exposed to the capsid surface and bind to the motor proteins [11].
On the surface of the nuclear membrane, the capsid is associated with nuclear pore complex [33]. The inner tegument protein UL36 (VP1/2) bearing a nuclear localization signal [34] and nucleoporins Nup358 and Nup214, which bind the capsid indirectly or directly, appeared to be key participants in this process. The capsid binds to the nuclear pore complex in such a way that its unique “portal”-containing vertex sits just above the nuclear pore. Presumably, all these interactions are necessary for transport of the viral DNA by the nuclear import pathway mediated by importin β [35].
Transcription and replication of the viral genome (Fig. 2A) as well as the assembly of progeny capsids take place within the nucleus. The infection comes with reorganization of the nucleus causing an increase of its size, disruption of nucleolus [36] and nuclear domain-10 (ND-10) [37], and chromatin condensation and subsequent destruction of the latter and the nuclear lamina [38] in the late steps of infection. Key cellular processes – transcription [39], splicing of the cellular RNA [40], protein biosynthesis [41], and cellular response to infection [42] – are also blocked. All these steps increase the efficiency of viral replication and transcription.
The viral mRNA is synthesized by the host cell RNA-polymerase II with the participation of viral factors in all steps in infection. Viral proteins regulate sequential transcriptional cascades (α, β, and γ genes; Fig. 2A) and a series of posttranslational modifications.
For the transcription of immediate early α genes, the presence of the tegument protein VP16 is important [43]. Unlike other viral genes, all α genes contain several copies of the consensus sequence: 5′-GyATGnTAATGArATTCyTTGnGGG-3′, where y is a pyrimidine base, r is a purine base, n is any base [43]. The cellular transcription factor Oct-1 binds to this sequence. VP16 protein interacts with this transcription factor and together with HCFC1 protein forms a complex that activates transcription of α genes.
An intriguing feature of VP16 is its ability to regulate methylation and demethylation of histone H3 that binds with non-nucleosomal viral DNA at the α, β, and γ gene promoters during infection. During infection, VP16 triggers a cascade of viral gene expression by directly or indirectly activating the viral α gene promoters and removal of histone H3. H3 histone binding to the α gene promoters is most likely the result of the cellular response to foreign DNA detected by the cell in the nucleus in order to inactivate it [44].
Six genes (ICP0, ICP4, ICP22, ICP27, ICP47, and US1.5) are ascribed to the group of immediate early genes; five of them (ICP0, ICP4, ICP22, ICP27, and US1.5) activate transcription of β genes at least in several types of cells. Immediate early proteins accomplish multiple functions and perform dramatic reorganization of cellular processes in the interests of the virus. For instance, ICP0 protein contains the E3-domain possessing ubiquitin ligase activity towards a wide range of substrates. Through direct or indirect interaction or by substrate phosphorylation, the triggering of proteasomal degradation of some of the proteins participating in cellular defense against the viral infection can occur. So, in a primary culture of fibroblasts the target of ICP0-mediated ubiquitination resulting in proteasomal degradation is interferon-inducible protein 16 (IFI16) localized in the nucleus. This DNA sensor triggers the cascade of the innate immune response signaling IRF-3 activation [45]. ICP0 similar to VP16 protein mentioned above can activate viral chromatin condensation and decondensation [46].
At the same time, in a confrontation between the virus and the cell the latter also has some tools for suppression of infection. For example, a DNA-dependent activator of interferon-regulatory factor (DAI) – the cytosolic DNA sensor, in addition to membrane-associated Toll-like receptor 9, recognizes the pathogen’s DNA [47] and inhibits HSV-1 early gene expression via repression of ICP0 promoter activation [48]. ICP22 protein functions as a repressor in a number of cellular and viral promoters. Using immunoprecipitation, this protein was shown to form a complex with transcription elongation factor b (P-TEFb), like viral transcriptional activator VP16, and to block its binding to viral promoters [49].
The main function of the α gene-encoded proteins is activation of β gene expression. Proteins and enzymes encoded by the β genes are involved in viral genome replication (e.g. HSV DNA polymerase, UL30), regulation of nucleotide metabolism (e.g. thymidine kinase, UL23), suppression of early α genes, and activation of late γ genes. Regulation of β and γ gene expression is more diverse; that is why the start of initiation, duration, and level of expression of these genes do not coincide, in contrast to α gene expression regulation.
Being a result of low translation initiation efficiency, the level of expression of the key replication protein DNA polymerase is below as compared to other β genes, for instance, thymidine kinase. Upstream (+55) and downstream from the translation initiation site, the transcript of this gene contains sequences forming stable hairpins that might prevent access of cellular initiation factors. The level of expression of DNA polymerase reaches its maximum only 4 h after infection [50].
After initiation, viral DNA synthesis switches from a Θ replication mechanism to a rolling-circle mechanism [51], the latter producing concatemeric molecules that are cleaved during the process of nucleocapsid assembly.
The first step in replication of HSV DNA is the unwinding of the double helix by UL9 and/or ICP8 (UL29) proteins in the AT-rich regions of the oriL or oriS origins of replication. The latter are present in one copy in UL of the genome, and in two copies in US of the genome, respectively. ICP8 binds ssDNA fragments, and UL9 binds specifically to oriS and unwinds it. Then helicase–primase complex composed of UL5, UL8, and UL52 proteins is loaded. Its helicase activity efficiently catalyzes unwinding of dsDNA only if single-stranded overhang of greater than six nucleotides is available. The primase function can also be accomplished by the primase subunit of the cellular DNA polymerase α.
The leading and lagging DNA strands are synthesized by viral DNA polymerase (UL30) complexed with processivity factor UL42. The latter protein differs from proliferating cell nuclear antigen (PCNA) – it binds DNA as a monomer and thus does not form a toroidal structure [52].
In addition to seven viral proteins, a few cellular proteins appeared to participate in the replication. These are DNA ligase, topoisomerase II, and various components of the DNA repair and homologous recombination systems [54]. Moreover, the cellular chaperone Hsp90 was found to be essential for the viral replication; its inhibition impairs the latter and results in viral DNA polymerase mislocalization to the cytoplasm and its proteasome-dependent degradation [55].
Some viral proteins participate in nucleotide metabolism, e.g. thymidine kinase (UL23), ribonucleotide reductase (UL39, UL40), deoxyuridine triphosphatase (UL50), uracil N-glycosylase (UL2), and alkaline nuclease (UL12). These enzymes are essential for viral DNA synthesis and repair because the production of the corresponding host cell enzymes is suppressed.
After the viral DNA replication initiation, the levels of expression of late γ genes, especially encoding capsid proteins, increase providing the assembly of progeny virions. The capsid assembly and viral genome packaging occur in the nucleus (Fig. 2A) followed by nucleocapsid egress from the nucleus via nuclear pore or by budding through the nuclear membrane. With the participation of UL36 and UL37 proteins, the capsid is transported from the nucleus to the cytoplasm [56], where the virion maturation and outer shell formation occurs. The release of the virion from the cell by exocytosis accomplishes the envelope formation (Fig. 2A). As well as during virus entry into the cell, the egress of virions is associated with microtubule-based transport and with the UL37 interaction with molecular motor dystonin. Using live-cell imaging, dystonin depletion was shown to result in striking reduction in capsid movement in the cytoplasm during egress [57].
HSV-infected cells produce not only infective virions but also non-infectious light particles (L-particles), which are devoid of viral capsids and genomes. They presumably facilitate the infection by delivering additional tegument proteins to the host cell. Clathrin-like coats are probably associated with virion and L-particle envelopment in virion assembly sites [58].
A functional screening assay using small interfering RNAs (siRNA) has shown that at least 15 host proteins are implicated in pathways that are most likely relevant for HSV-1 viability and the viral propagation. Among them intracellular transport (ARF1, HSPA8, RAB2A, RAB5A, RAB6A, RAB10, RAB11A) and cytoskeleton (KRT10) components as well as proteins involved in gene expression (DDX3X, HSPA8, EIF4H), signal transduction (CD59, MIF, YWHAG, YWHAZ), and apoptosis (MIF, YWHAZ) have been identified. The incorporation of most of these proteins within mature viral particles seems to be necessary for the optimal course of the next round of infection, and the depletion of one of the above proteins results in poorer viral replication without any significant effect on the cell viability [59]. One more cellular protein, the small GTPase Rab27a, colocalizes with viral glycoproteins gH and gD in the trans-Golgi network (TGN) and probably takes part in viral egress from oligodendroglial cells. The viral titer of Rab27a-silenced infected cells is significantly decreased [1].
An interesting property of HSV-1 is its ability to establish a latent infection. After primary infection, HSV-1 either replicates productively in epithelial cells or enters sensory neuron axons and moves to the neuronal cell nucleus. There, the viral DNA remains circular and does not possess any lytic gene expression; however, latency associated transcripts (LATs) are expressed and then spliced to give some mRNAs. Both the transcriptionally active and silent regions of latent HSV DNA have a nucleosomal structure similar to that of cellular chromatin [60]. Recent views on the functions of LATs are conflicting, but their suggested major function is generation of miRNAs and siRNAs that downregulate ICP0 and other lytic gene expression. HSV-1 latency, reactivation, and recurrent diseases are studied in rabbit and mouse eye models [6].
HSV-1 DNA polymerase. HSV-1 DNA polymerase is the key enzyme in viral DNA replication. It belongs to the family B polymerases that includes human polymerases α, δ, and ε. HSV-1 DNA polymerase is a 136-kDa protein being at least 300 amino acid residues longer than other B polymerases and exhibiting 16-50% sequence homology.
DNA polymerase associates with the DNA-binding accessory protein UL42 (65 kDa), which binds to the C-terminal region of the enzyme and acts as a processivity factor [61, 62]. The DNA polymerase region, which binds UL42, contains a C-terminal nuclear localization signal (NLS) corresponding to the RRMLHR motif (amino acid residues 1224-1229) [63]. In addition to polymerase activity, HSV polymerase has 3′-5′-exonuclease activity [64], presumably 5′-3′-exonuclease activity, and RNase H activity [65] required for removing RNA primers during synthesis of Okazaki fragments. Unexpectedly, HSV-1 DNA polymerase exhibits apurinic/apyrimidinic (AP) and 5′-deoxyribose phosphate (dRP) lyase activities typical for repair polymerase [66]. AP activity of HSV-1 DNA polymerase in conjunction with the viral uracil N-glycosylase (UL2) is involved in a viral DNA repair system similar to cellular base excision repair (BER). The functional consequence of interaction between UL30 and UL2 is replication block of uracil-containing templates upstream from template uracil due to an AP-site [67].
HSV-1 polymerase is comprised of six structure domains. In addition to domains that usually perform the DNA polymerase activity, i.e. palm, fingers, and thumb domains, it has a pre-NH2 domain, an NH2-domain, and a 3′-5′-exonuclease domain containing regions exo I, exo II (region IV), and exo III (δ-C region) [53] (Fig. 2B). Regions III and VI belong to the fingers, regions I, II, and VII are located in the palm subdomain, and the thumb subdomain contains the conservative region V. Located in palm subdomain residues D717, D888, and F718 are involved in metal ion coordination required for polymerase catalysis. The ribose of the incoming nucleotide interacts with the strictly conservative Y722 residue, providing a “steric gating” effect against incorporation of ribonucleotides into the growing DNA chain. The side chain of N815 stacks against the base of the incoming nucleotide and stabilizes it. The R785, R789, and K811 residues from the fingers domain interact with the phosphate groups of the incoming nucleotide and are important for the positioning of the phosphate moiety to the 3′-OH of the primer. The KKKY (938-941) motif and residues Y818, Y884, and D886 serve for sensing mismatches in newly synthesized DNA duplex [53].
It was found that the pre-NH2 domain, namely the conservative motif FYNPYL (amino acid residues 44-49) in the herpes virus, is required for efficient HSV-1 replication. Mutant viruses containing DNA polymerases without the extreme N-terminal 52 residues exhibited 5-7-fold decreases in viral yield, and virus with DNA polymerase without 141 residue failed to replicate. Mutant enzymes lacking the N-terminal 52 residues and containing six alanine residues instead of the FYNPYL motif displayed basal polymerase activity similar to that of wild-type HSV-1 polymerase in vitro. According to a suggested scenario, this HSV-1 polymerase region interacts with an unknown factor that recruits the polymerase to the replication fork. The possible candidates are helicase–primase complex component UL8, single-stranded DNA binding protein ICP8, alkaline nuclease UL12, and chaperone Asf1b [68].
INHIBITORS OF HERPES VIRUS REPLICATION: CLINICAL DRUGS AND
LABORATORY DEVELOPMENTS
As mentioned in the first section of this review, human herpetic infection is widespread and is a central problem in both Russia and abroad [69]. Among HIV-infected patients who are coinfected by the herpes virus, about 6-10% have virus strains resistant to available antiherpetic drugs.
Most modern drugs for the treatment of herpetic infections are based on the use of modified nucleosides or their prodrugs [70]1. The action of drugs is directed mostly to the suppression of activity of the main replication enzyme of the virus – the DNA polymerase. It should be noted that the drugs do not save the patient from the recurrent character of the disease, and their prolonged administration can cause the emergence of resistant virus strains. These circumstances make the search for new antiherpetic drugs and their new targets a high priority. Below we present data on the application of clinically approved antiherpetic drugs as well as the search for new substances with high efficiency and low toxicity suppressing the replication of human HSV-1.
1A prodrug is a medication that does not manifest any antiviral action itself, but forms an inhibitor of the virus after penetration into the infected cell due to chemical or enzymatic activation.
Clinically approved antiherpetic drugs. The first nucleoside antiherpetic drug, 5-iodo-2′-deoxyuridine, was created in the end of 1960s by W. Prusoff [71] and was widely used in clinical practice to treat herpetic keratitis. This was the first antiviral drug based on a nucleoside analog, and this became the basis for the development of antiviral therapy. In the next two decades, trifluorothymidine, vidarabine, and brivudin, which are also nucleoside analogs, were approved to therapy. However, these drugs showed low selectivity and rather high toxicity and were used only for topical administration. The second generation of antiherpetic drugs was created based on acyclic nucleosides (acyclovir, valacyclovir, ganciclovir, penciclovir, and famciclovir) that suppress infections caused by HSV-1, HSV-2, VZV, and CMV [69, 70]. The chemical structures of these compounds are presented in Fig. 3.
Fig. 3. Chemical structures of antiherpetic drugs used in clinics: a) acyclovir; b) valacyclovir; c) ganciclovir; d) penciclovir; e) famciclovir; f) foscarnet.
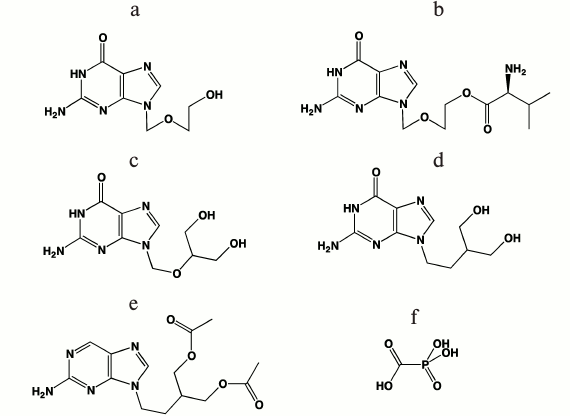
Acyclovir and valacyclovir. Acyclovir (ACV, earlier referred to as acycloguanosine; Fig. 3a) was first proposed as an antiherpetic drug in 1977 [72]. A new period in the development of antiherpetic chemotherapy started with its discovery. To date, acyclovir remains the gold standard in the search for antiherpetic drugs [73], and the author of its development, Gertrude Elion, was awarded the Nobel Prize in Physiology and Medicine in 1988 [74].
Acyclovir turned out to be an effective and low-toxicity drug. The mechanism of action of ACV is based on its phosphorylation by viral thymidine kinase with the formation of corresponding monophosphate (ACVMP). The next two phosphorylation steps are catalyzed by cellular kinases with the formation of triphosphate (ACVTP), which acts as a substrate of viral DNA polymerase, is incorporated in the viral DNA chain, and terminates its synthesis [72].
Disadvantages of acyclovir are poor oral bioavailability (10-30%), limited solubility in water, and short half-life of the drug in the bloodstream. Therefore, rather high doses and frequent drug administration are necessary to support the proper concentration of acyclovir in the patients’ blood that, in turn, increases toxicity. To improve solubility and increase bioavailability, several prodrugs of acyclovir were prepared, and glycine and L-alanine esters were the first among the acyclovir prodrugs [70]. However, the drugs had rather high toxicity during clinic investigations.
The L-valine ester of acyclovir – valacyclovir – turned out to be an effective and safe drug (Fig. 3b). The increased oral bioavailability of valacyclovir can be attributed to rapid intestinal absorption via the human intestinal peptide transporter hPEPT1, followed by rapid conversion to ACV by ester hydrolysis in the small intestine [70].
Ganciclovir. Ganciclovir (GCV) – 9-(1,3-dihydroxy-2-propoxymethyl)guanine – is an acyclic guanosine analog (Fig. 3c) that is structurally related to ACV [75]. The drug appeared to be active towards CMV, HSV-1, HSV-2, VZV, and EBV; however, it is used in clinic only for the treatment of CMV infections. As with ACV, the oral bioavailability of ganciclovir is limited and, to overcome this problem the prodrug of ganciclovir, the valine ester of ganciclovir, was synthesized [76].
Penciclovir and famciclovir. Penciclovir (PCV, Fig. 3d) is an acyclic guanosine analog with a structure that is similar to that of ACV and GCV but without the oxygen atom in the acyclic “sugar” moiety and with an OH group in the position equivalent to that of the 3′-OH group in the natural deoxynucleoside. PCV was synthesized for the first time at Beecham Pharmaceuticals Laboratories. The drug was less active than acyclovir against HSV-1 in experiments with infected cells; however, it efficiently suppressed VZV replication in animals and is widely used for treatment of HSV-1 induced skin lesions. In contrast to acyclovir, the drug also inhibited the replication of an HSV-1 strain encoding a mutant DNA polymerase [77]. Like ACV, PCV is converted to the monophosphate by the viral thymidine kinase. The initial step of PCV phosphorylation to PCVMP is more efficient than the phosphorylation of ACV, but the PCVTP formed in infected cells is less active than ACVTP as a substrate of HSV DNA polymerase. Its oral bioavailability was even lower than that of ACV. To improve these limitations, the PCV prodrug famciclovir, the diacetyl derivative of penciclovir, was synthesized (Fig. 3e). Famciclovir is converted to penciclovir in two steps in vivo: removal of the two acetyl groups by esterase and oxidation of the purine by aldehyde oxidase. It should be noted that famciclovir is inactive in cell cultures since oxidation of penciclovir does not occur there; however, being administered orally, it was found to be even more effective than acyclovir in eliminating the virus from its target sites. The more important advantage of famciclovir is its ability to prevent latent infection of HSV-1. When treating with famciclovir, significantly less latent virus was detected in mice ganglia as compared to valacyclovir [78]. The reason of preventing recurrences of infectious virus remains to be elucidated.
Foscarnet. Foscarnet (PFA) (Fig. 3f) is a pyrophosphate analog and a nonnucleoside inhibitor of HSV DNA polymerase. Foscarnet is a noncompetitive inhibitor with respect to nucleotide substrates; it binds to the enzyme active site and prevents the binding of incoming nucleotide [79]. PFA is not widely used in clinic because of high toxicity compared to acyclovir and is used only if the treatment with acyclovir and other nucleoside drugs is ineffective, e.g. in patients that have acquired resistance to them [80].
Search for new antiherpetic drugs. A series of interesting new nucleoside analogs having antiherpetic activity in cell cultures and laboratory animals has been described. Among such compounds worth mentioning are guanine derivatives: acyclic H2G (Fig. 4a), carbocyclic cyclobutane (lobucavir) (Fig. 4b) and cyclopropane (A-5021) (Fig. 4c) analogs. These compounds did not pass clinical trials and are not approved as drugs because of increased toxicity [81]; however, they can be used as the basis for the development of new drugs.
Fig. 4. Chemical structures of antiherpetic drugs: a) H2G; b) lobucavir; c) A-5021; d) HpACV; e) (Z)-9-[3-(phosphonomethoxyprop)-1-en-1-yl]adenine; f) (E)-9-[3-(phosphonomethoxyprop)-1-en-1-yl]adenine.
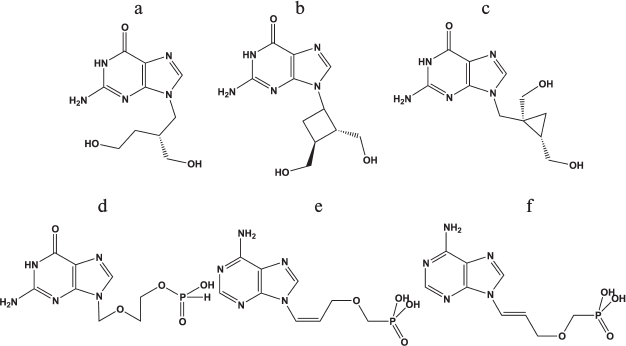
Nucleoside phosphonate derivatives. To date, three phosphonate derivatives of acyclic nucleosides acting directly without the primary phosphorylation stage for activation in the cell are used in clinics. These are cidofovir, a cytidine analog applied for treatment of CMV infection, and adefovir and tenofovir, adenine analogs that are employed in therapy of hepatitis B and HIV, respectively. Acyclic unsaturated phosphonate analogs, (Z)- and (E)-isomers of 9-[3-(phosphonometoxyprop)-1-en-1-yl]adenine [75], and a phosphonate analog of acyclovir (HpACV) (Fig. 4d) [82] were synthesized and tested as HSV-1 inhibitors.
An interesting property of the acyclic unsaturated phosphonate analogs is their ability to inhibit both replication of HSV-1 and human immunodeficiency virus in cells. In this case, the concentration of Z-isomer (Fig. 4e) that suppresses the development of viruses by 50% (IC50) was substantially lower than that of E-isomer (Fig. 4f). Toxicity of both compounds was lower than that of the known anti-HIV drug [2-(6-amino-9H-purin-9-yl)ethoxymethyl]phosphonic acid (PMEA). Both isomers also inhibit thymidine kinase-deficient HSV-1 strains resistant to acyclovir, since no first phosphorylation stage is required for their activation. The synthesized diphosphates acted as substrates of both HIV reverse transcriptase and HSV-1 DNA polymerase, were incorporated into 3′-end of the primer–template, and terminated further elongation [83]. These compounds were not substrates of the cellular DNA polymerase α, consistent with data on their low toxicity found in cell culture experiments. These compounds simultaneously suppress both HIV and HSV, a rare achievement.
Properties of H-phosphonate acyclovir (HpACV, Fig. 4d) are interesting. This drug suppressed HSV replication in cell culture and lowered the probability of lethal outcome of HSV-infected laboratory animals [84]. It was noted that like acyclovir, HpACV and interferon α act synergistically [85]. An unusual feature of HpACV is the suppression of acyclovir-resistant virus strains deficient in thymidine kinase; its concentration was only twice higher than that in the case of acyclovir-sensitive strains. However, the inhibiting concentration of acyclovir increased 500-fold in similar experiments. The resistance of strains to HpACV appears more slowly than to ACV and at higher concentrations (100-800 μg/ml against 2.5-100 μg/ml, respectively). Thus, we assume that ACV metabolism differs substantially from that of HpACV. Indeed, in contrast to ACV, which should be phosphorylated by thymidine kinase upon uptake into the cell, HpACV in Vero cells is mainly converted into acyclovir monophosphate (ACVMP), and only the small fraction is hydrolyzed to ACV [84].
Derivatives of triazolopyrimidines. Derivatives of 1,2,3-triazolo[1,5-α]pyrimidine are the base for the synthesis of many physiologically active compounds.
Compound (a) in Fig. 5 was described as an inhibitor of a PTEN-deficient cancer cell line [86], while compound (c) manifested antiherpetic activity [87], and ribosylated compound (b) was active against rhinoviruses [88]. Derivatives of triazolopyrimidines were studied as HSV-1 inhibitors in Vero cells [87]. The chemical structures of these compounds are shown in Figs. 5c-5g.
Fig. 5. Chemical structures of derivatives of triazolopyrimidines.
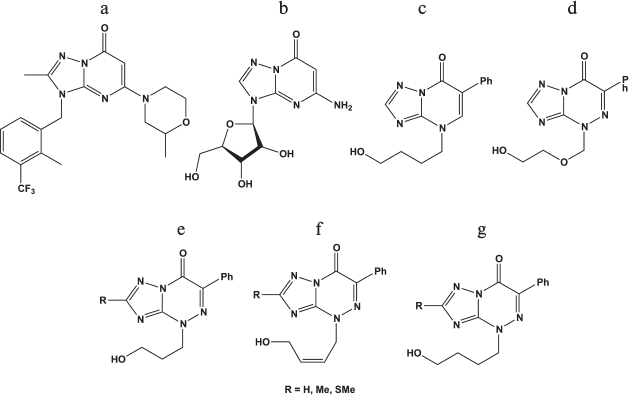
It was shown that acyclic derivatives of triazolopyrimidines display antiherpetic activity in cell culture, while their triphosphates inhibit DNA synthesis catalyzed by the HSV-1 DNA polymerase. The strongest inhibitor was triphosphates of compound (e) with R = SMe [87]. Consequently, one of the targets of these compounds might be the herpes virus DNA polymerase.
Along with the study of herpes replication inhibitors targeting DNA synthesis, intensive search for other viral targets have been performed.
Helicase–primase inhibitors. In the last 10 years, several new classes of compounds manifesting antiherpetic activity due to suppression of the virus helicase–primase complex (UL5, UL52, UL8) were developed. In 2002, the Bayer Company investigated a series of thiazole derivatives as antiherpetic compounds in cellular and animal models with higher efficiency than acyclovir and its derivatives. Leader compound BAY 57-1293 (N-[5-(aminosulfonyl)-4-methyl-1,3-thiazol-2-yl]-N-methyl-2-[4-(2-pyridinyl)phenyl]acetamide) (Fig. 6a) manifested significant antiherpetic properties both in cell lines and in animal models without cross-resistance relative to acyclovir.
Fig. 6. Chemical structures of inhibitors of viral helicase–primase (a-c), adhesion and entry of the virus into the cells (d), and viral ribonucleotide reductase (e).
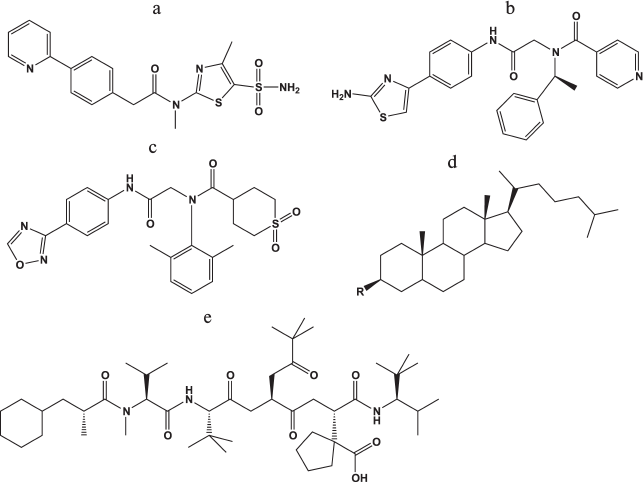
The mechanism of BAY 57-1293 action is the complete blocking of the viral DNA synthesis after the transcription of immediate early genes. Analysis of 10 virus strains resistant to the compound showed that the resistance appears due to mutations in genes coding UL5 and/or UL52 – components of the helicase–primase complex of the virus, which was confirmed by the inhibition of ATPase activity of the complex in vitro. It should be noted that the frequency of appearance of viruses resistant to this compound was lower by an order of magnitude compared with the appearance of resistance to acyclovir.
The only disadvantage of this compound is dose-dependent hyperplasia of the urinary bladder of the rat model after peroral intake of the compound. However, no toxicological effects were observed for dogs under the same conditions. It should be mentioned that primary sulfonamides suppressing dehydratase cause hyperplasia of the epithelium of the urinary bladder in rodents but not in other animals including humans [89]. In addition, BAY 57-1293 considerably more efficiently reduced HSV-2 reactivation in a guinea pig model than valacyclovir during therapy at the early infection stage [90].
In parallel, a series of related compounds that suppresses HSV replication in vitro and in vivo was developed at Boehringer Ingelheim Pharmaceuticals.
The BILS 179 BS compound (Fig. 6b) is approximately 10-fold more effective than ACV in cell culture and suppresses the development of herpetic infection in animals [91].
In contrast with the compounds described above, the oxadiazolylphenyl derivative (ASP2151, Fig. 6c) actively inhibited HSV-1 and HSV-2 as well as VZV [92]. The efficiency of ASP2151 considerably exceeded that of acyclovir, and this compound successfully passed through phase II clinical trials in 2011.
Ribonucleotide reductase inhibitors. Ribonucleotide reductase (RR) is an important enzyme in virus replication in herpetic infection of eyes and for virus reactivation from latent to active state in skin infections. To suppress herpes RR, a class of thiocarbonyl hydrazones was initially proposed. However, it turned out that they inhibit not only the viral enzyme, but also the cellular homolog; therefore, they can be considered only for local application.
Hydroxyurea, which is also a nonselective inhibitor of RR, enhanced the suppression of the HSV replication in Vero cells by ACV and other nucleoside drugs by a factor of 3-4-fold. The effect also manifested for strains resistant to ACV with mutations in both TK and DNA polymerase genes [93].
The BILD 1633 SE compound (Fig. 6e), a peptidomimetic of the C-end of the small subunit of herpes RR, inhibited virus enzyme at the concentration of 3 nM and was active both in wild-type virus and strains resistant to ACV at the concentration of about 0.4 μM with depression of infection for a nude mouse line.
Synergistic action is mentioned for compounds BILD 1633 SE and ACV, since the suppression of viral RR activity led to decrease in the dGTP pool and increase in the ratio of the ACV concentration to dGTP in the cell [94].
Inhibitors of viral attachment and entry into the cell. Heparan sulfate, a herpes virus entry mediator (HVEM), nectines-1 and -2 on the cell surface, and virus glycoproteins gB, gC, gD, gH, and gL are involved in HSV attachment and entry into the cell (see section “Herpes Simplex Virus Type 1: General Description, Life Cycle, and Replication”). It was many times demonstrated that polyanions have antiviral properties in vitro since they resemble heparan sulfate chains and competitively inhibit binding of gB and gC to the cell. For example, a mixture of highly sulfated oligosaccharides of mannose (PI-88) was proposed as an antiherpetic drug; however, it manifested no virucidal properties [95]. Analysis of mutations in resistant viruses, which were cultured in the presence of PI-88, showed that glycoproteins gC, gB, and gD are responsible for the virus sensitivity to the drug [95]. The same authors [96] created a series of compounds based on various oligosaccharides and PI-88 conjugated with hydrophobic aglycone groups, one of which, cholestanyl glycoside of sulfated tetrasaccharide [Manα(1,3)-Manα(1,3)-Manα(1,2)-Man] (Fig. 6d). This compound suppressed not only the virus replication in the cell culture GMK AH-1 (IC50 = 2.1 μg/ml) but also competed with heparan sulfate for binding with viral glycoproteins, which prevented the penetration of the virus into the cell and its further transfer from cell to cell. The compound also inactivated the virus particles, which is apparently the result of destabilization of the viral envelope by the lipophilic cholestanyl group.
A common disadvantage of sulfated oligosaccharides and polysaccharides is the partial destruction of the usually impermeable for pathogens intestine mucus layer [97].
Retrocyclin 2, a short cyclic peptide from the group of Θ-defensins [98], and lactoferrin [99] also protect cells against HSV-1 entry due to binding with virus glycoproteins. In contrast to oligosaccharides, which are suitable for local application only, lactoferrin suppresses the development of skin symptoms of herpetic infection in mice with oral administration [99].
Thus, the number of promising viral targets and classes of compounds with substantial antiherpetic properties considerably increased during the last decade. However, no new effective and low-toxicity clinical drugs against both wild-type viruses and drug-resistant strains have appeared.
GENOTYPIC CHARACTERIZATION OF DRUG-RESISTANT HSV CLINICAL
ISOLATES
As mentioned above, long drug use leads to the emergence of resistant HSV strains, making the disease course uncontrollable. In 95% of cases, ACV resistance is caused by mutations in viral thymidine kinase, which performs the initial phosphorylation step of ACV, followed by two subsequent steps carried out by cellular kinases. The resulting ACVTP acts as a chain terminator of DNA synthesis. Mutations in the DNA polymerase gene, which determine the antiviral drug resistance, are found only in 5%. Resistance can also be induced by simultaneous mutations in both enzymes [100]. The situation is also complicated by the fact that the most clinical isolates appear to be heterogeneous, causing simultaneous coinfection by several variants of ACV-stable variants of the virus.
A large number of mutations are revealed in the thymidine kinase and DNA polymerase genes of HSV clinic isolates and laboratory clones resistant to acyclovir and other drugs [101]; some of them lead to the loss or alteration in enzyme activity or specificity and, consequently, to increased resistance (Fig. 7).
Fig. 7. Arrangement of amino acid substitutions from various clinic isolates and laboratory clones relative to conservative regions and domains: a) of thymidine kinase; b) of HSV-1 DNA polymerase [102, 105].
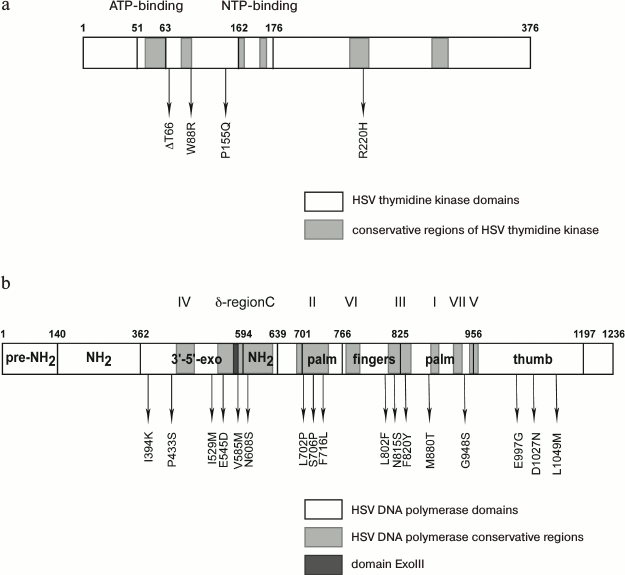
Mutation R220H in thymidine kinase leads to the loss of the virus sensitivity to ACV, PCV, and GCV [102], whose antiviral activity is directly associated with the enzyme function. The authors of paper [103] described similar mutation R220K in the HSV-2 gene of thymidine kinase, which leads (along with other substitutions) to lowering sensitivity to GCV and BVDU by 1-2 orders of magnitude. At the same time, thymidine kinase-independent adenine arabinoside (AraA) suppresses the replication of the mutant virus almost at the same extent as the wild-type HSV-1 [85]. This mutation is also present in the thymidine kinase gene of a laboratory strain resistant to HpACV; however, it does not affect the sensitivity of the virus to HpACV since its mechanism of action is independent of thymidine kinase. Mutation of G59R in the ATP-binding site of the enzyme plays a significant role in lowering its activity (Fig. 7a).
We identified more than 20 mutations in HSV-1 DNA polymerase [102]; some of them are substantial for enzyme functioning (Fig. 7b).
The substitutions 1394K, P433S, and V585M are localized in the 3′-5′-exonuclease domain of the enzyme, and mutation V585M is located in an ExoIII (572-585) conservative region. The authors [104] showed that mutations in this region cause resistance. Mutation D581A, which is located in the immediate proximity to V585M, leads to almost complete loss of 3′-5′-exonuclease activity of the enzyme, but along with partial retention of polymerase activity [64]. New amino acid substitutions I159M and E545D affecting the enzyme activity were found in the 3′-5′-exonuclease domain of DNA polymerase from an HSV-1 clinical isolate [102]. Mutations in the exonuclease site of the DNA polymerase typically impair the proofreading activity, lower the accuracy of the DNA synthesis, and, consequently, increase the mutation rate.
Mutation N608S is in the conserved δ-region C and leads to resistance of an HSV-1 laboratory strain to ACV and HpACV [105]. A clone with the L702H mutation in the conserved region II of HSV-1 DNA polymerase showed resistance toward acyclovir but remained sensitive or insignificantly resistant towards penciclovir and ganciclovir. The substitution of hydrophobic leucine 702 by proline can promote a conformational change in the β-sheet in the “palm domain” coordinating magnesium ions and triphosphate in the enzyme active site [106]. A similar effect is caused by mutation F761L since it is located near amino acid residue D717 coordinating magnesium ions. When substituting hydrophobic valine 715 by more polar methionine [107] and neighboring hydrophobic phenylalanine 716 by polar lysine [102], sensitivity of the virus to ACV is lost.
According to crystallographic data [53], mutation M880T (Figs. 2B and 7b), which is located rather close to the catalytic triad (D717, D886, and D888) and to the binding site for the phosphate residue of the nucleotide and for magnesium ions, can form steric obstacles for binding phosphate residues and lead to lowering the sensitivity of the virus both to nucleoside analogs and PFA.
Authors of paper [108] showed that the N815S mutant is also resistant to acyclovir and its analogs. According to the crystallographic data [53], side chain N815 is opposed to the base of the nucleotide “entering” the active site of the enzyme. Computer modeling suggested that the side chain of mutant residue S815 has spatial orientation differing from that of the N815 residue of the native strain; the polymerase mutant at this residue does not incorporate ACVMP into the growing polynucleotide chain [109].
Thus, resistance of both clinic isolates and laboratory clones is explained by mutations in both DNA polymerase and thymidine kinase of the herpes virus, and substitutions of the same amino acid residue differently affect the sensitivity of enzymes to various antiherpetic drugs.
CONCLUSION
This review does not pretend to represent all the data on herpes viruses, their interactions with the host cell, and development of antiherpetic drugs overall. From the instant of the creation of the gold standard of antiherpetic therapy, acyclovir, many new effective compounds appeared and another understanding of strategies in the search for drugs has come, one of which is the creation of prodrugs with lowered toxicity, and a multitude of investigations elucidating the details of the interaction of the virus with a cell was implemented [110]. The main purpose of this review was to show that the investigation of the herpes virus and search for inhibitors of its replication still remain a topical problem, which requires further efforts of the chemical, biological, pharmaceutical, and medical communities.
This work was supported by the Russian Foundation for Basic Research (projects 12-04-00581 and 13-04-40307-H) and RAS Presidium (“Molecular and Cell Biology” program).
REFERENCES
1.Bello-Morales, R., Crespillo, A. J., Fraile-Ramos,
A., Tabares, E., Alcina, A., and Lopez-Guerrero, J. A. (2012) Role of
the small GTPase Rab27a during herpes simplex virus infection of
oligodendrocytic cells, BMC Microbiol., 12, 265.
2.Schuppe, H. C., Meinhardt, A., Allam, J. P.,
Bergmann, M., Weidner, W., and Haidl, G. (2008) Chronic orchitis: a
neglected cause of male infertility? Andrologia, 40,
84-91.
3.Cardone, G., Heymann, J. B., Cheng, N., Trus, B.
L., and Steven, A. C. (2012) Procapsid assembly, maturation, nuclear
exit: dynamic steps in the production of infectious herpes virions,
Adv. Exp. Med. Biol., 726, 423-439.
4.Raab-Traub, N. (2012) Novel mechanisms of
EBV-induced oncogenesis, Curr. Opin. Virol., 2,
453-458.
5.Mesri, E. A., Cesarman, E., and Boshoff, C. (2010)
Kaposi’s sarcoma and its associated herpes virus, Nature Rev.
Cancer, 10, 707-719.
6.Webre, J. M., Hill, J. M., Nolan, N. M., Clement,
C., McFerrin, H. E., Bhattacharjee, P. S., Hsia, V., Neumann, D. M.,
Foster, T. P., Lukiw, W. J., and Thompson, H. W. (2012) Rabbit and
mouse models of HSV-1 latency, reactivation, and recurrent eye
diseases, J. Biomed. Biotechnol., 2012, 612316.
7.Grinde, B. (2013) Herpes viruses: latency and
reactivation – viral strategies and host response, J. Oral
Microbiol., 5, 22766; http://dx.doi.org/10.3402/jom.v5i0.22766.
8.Mocarski, E. S., and Roizman, B. (1982) Structure
and role of the herpes simplex virus DNA termini in inversion,
circularization and generation of virion DNA, Cell, 31,
89-97.
9.Grunewald, K., Desai, P., Winkler, D. C., Heymann,
J. B., Belnap, D. M., Baumeister, W., and Steven, A. C. (2003)
Three-dimensional structure of herpes simplex virus from cryoelectron
tomography, Science, 302, 1396-1398.
10.Yudovin-Farber, I., Gurt, I., Hope, R., Domb, A.
J., and Katz, E. (2009) Inhibition of herpes simplex virus by
polyamines, Antiviral Chem. Chemother., 20, 87-98.
11.Radtke, K., Kieneke, D., Wolfstein, A., Michael,
K., Steffen, W., Scholz, T., Karger, A., and Sodeik, B. (2010)
Plus-and-minus-end directed microtubule motors bind simultaneously to
herpes simplex virus capsids using different inner tegument structures,
PLoS Pathog., 6, e1000991.
12.Jovasevic, V., Liang, L., and Roizman, B. (2008)
Proteolytic cleavage of VP1-2 is required for release of herpes simplex
virus 1 DNA into the nucleus, J. Virol., 82,
3311-3319.
13.Ace, C. I., McKee, T. A., Ryan, J. M., Cameron,
J. M., and Preston, C. M. (1989) Construction and characterization of a
herpes simplex virus type 1 mutant unable to trans-induce
immediate-early gene expression, J. Virol., 63,
2260-2269.
14.Barzilai, A., Zivony-Elbom, I., Sarid, R., Noah,
E., and Frenkel, N. (2006) The herpes simplex virus type 1 vhs-UL41
gene secures viral replication by temporarily evading apoptotic
cellular response to infection: Vhs-UL41 activity might require
interactions with elements of cellular mRNA degradation machinery,
J. Virol., 80, 505-513.
15.Gibson, W., and Roizman, B. (1972) Proteins
specified by herpes simplex virus. 8. Characterization and composition
of multiple capsid forms of subtypes 1 and 2, J. Virol.,
10, 1044-1052.
16.Sheaffer, A. K., Newcomb, W. W., Gao, M., Yu, D.,
Weller, S. K., Brown, J. C., and Tenney, D. J. (2001) Herpes simplex
virus DNA cleavage and packaging proteins associate with the procapsid
prior to its maturation, J. Virol., 75, 687-698.
17.Brown, J. C., and Newcomb, W. W. (2011) Herpes
virus capsid assembly: insights from structural analysis, Curr.
Opin. Virol., 1, 142-149.
18.Chowdhury, S., Chouljenko, V. N., Nadheri, M.,
and Kousoulas, K. G. (2013) The amino terminus of herpes simplex virus
type-1 (HSV-1) glycoprotein K (gK) is required for virion entry via the
paired immunoglobulin-like type-2 receptor alpha (PILRalpha), J.
Virol., 87, 3305-3313.
19.Kieff, E. D., Bachenheimer, S. L., and Roizman,
B. (1971) Size, composition, and structure of the deoxyribonucleic acid
of herpes simplex virus subtypes 1 and 2, J. Virol., 8,
125-132.
20.Jenkins, F. J., and Roizman, B. (1986) Herpes
simplex virus 1 recombinants with non-inverting genomes frozen in
different isomeric arrangements are capable of independent replication,
J. Virol., 59, 494-499.
21.Roizman, B., Zhou, G., and Du, T. (2011)
Checkpoints in productive and latent infections with herpes simplex
virus 1: conceptualization of the issues, J. Neurovirol.,
17, 512-517.
22.Jurak, I., Kramer, M. F., Mellor, J. C., van
Lint, A. L., Roth, F. P., Knipe, D. M., and Coen, D. M. (2010) Numerous
conserved and divergent microRNAs expressed by herpes simplex viruses 1
and 2, J. Virol., 84, 4659-4672.
23.Honess, R. W., and Roizman, B. (1974) Regulation
of herpes virus macromolecular synthesis. I. Cascade regulation of the
synthesis of three groups of viral proteins, J. Virol.,
14, 8-19.
24.Chou, J., and Roizman, B. (1986) The terminal a
sequence of the herpes simplex virus genome contains the promoter of a
gene located in the repeat sequences of the L component, J.
Virol., 57, 629-637.
25.Arii, J., Uema, M., Morimoto, T., Sagara, H.,
Akashi, H., Ono, E., Arase, H., and Kawaguchi, Y. (2009) Entry of
herpes simplex virus 1 and other alpha-herpes viruses via the paired
immunoglobulin-like type 2 receptor alpha, J. Virol., 83,
4520-4527.
26.Herold, B. C., Visalli, R. J., Susmarski, N.,
Brandt, C. R., and Spear, P. G. (1994) Glycoprotein C-independent
binding of herpes simplex virus to cells requires cell surface heparan
sulfate and glycoprotein B, J. Gen. Virol., 75,
1211-1222.
27.Gianni, T., Amasio, M., and Campadelli-Fiume, G.
(2009) Herpes simplex virus gD forms distinct complexes with fusion
executors gB and gH/gL in part through the C-terminal profusion domain,
J. Biol. Chem., 284, 17370-17382.
28.Avitabile, E., Forghieri, C., and
Campadelli-Fiume, G. (2009) Cross talk among the glycoproteins involved
in herpes simplex virus entry and fusion: the interaction between gB
and gH/gL does not necessarily require gD, J. Virol., 83,
10752-10760.
29.Baldwin, J., Shukla, D., and Tiwari, V. (2013)
Members of 3-O-sulfotransferases (3-OST) family: a valuable tool from
zebrafish to humans for understanding herpes simplex virus entry,
Open Virol. J., 7, 5-11.
30.Zhou, G., Galvan, V., Campadelli-Fiume, G., and
Roizman, B. (2000) Glycoprotein D or J delivered in trans blocks
apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant
lacking intact genes expressing both glycoproteins, J. Virol.,
74, 11782-11791.
31.Satoh, T., Arii, J., Suenaga, T., Wang, J.,
Kogure, A., Uehori, J., Arase, N., Shiratori, I., Tanaka, S.,
Kawaguchi, Y., Spear, P. G., Lanier, L. L., and Arase, H. (2008)
PILRalpha is a herpes simplex virus-1 entry coreceptor that associates
with glycoprotein B, Cell, 132, 935-944.
32.Sodeik, B., Ebersold, M. W., and Helenius, A.
(1997) Microtubule-mediated transport of incoming herpes simplex virus
1 capsids to the nucleus, J. Cell Biol., 136,
1007-1021.
33.Ojala, P. M., Sodeik, B., Ebersold, M. W., Kutay,
U., and Helenius, A. (2000) Herpes simplex virus type 1 entry into host
cells: reconstitution of capsid binding and uncoating at the nuclear
pore complex in vitro, Mol. Cell. Biol., 20,
4922-4931.
34.Abaitua, F., and O’Hare, P. (2008)
Identification of a highly conserved, functional nuclear localization
signal within the N-terminal region of herpes simplex virus type 1
VP1-2 tegument protein, J. Virol., 82, 5234-5244.
35.Copeland, A. M., Newcomb, W. W., and Brown, J. C.
(2009) Herpes simplex virus replication: roles of viral proteins and
nucleoporins in capsid-nucleus attachment, J. Virol., 83,
1660-1668.
36.Calle, A., Ugrinova, I., Epstein, A. L., Bouvet,
P., Diaz, J. J., and Greco, A. (2008) Nucleolin is required for an
efficient herpes simplex virus type 1 infection, J. Virol.,
82, 4762-4773.
37.Everett, R. D., Freemont, P., Saitoh, H., Dasso,
M., Orr, A., Kathoria, M., and Parkinson, J. (1998) The disruption of
ND10 during herpes simplex virus infection correlates with the Vmw110-
and proteasome-dependent loss of several PML isoforms, J.
Virol., 72, 6581-6591.
38.Simpson-Holley, M., Colgrove, R. C., Nalepa, G.,
Harper, J. W., and Knipe, D. M. (2005) Identification and functional
evaluation of cellular and viral factors involved in the alteration of
nuclear architecture during herpes simplex virus 1 infection, J.
Virol., 79, 12840-12851.
39.Jenkins, H. L., and Spencer, C. A. (2001) RNA
polymerase II holoenzyme modifications accompany transcription
reprogramming in herpes simplex virus type 1-infected cells, J.
Virol., 75, 9872-9884.
40.Hardy, W. R., and Sandri-Goldin, R. M. (1994)
Herpes simplex virus inhibits host cell splicing, and regulatory
protein ICP27 is required for this effect, J. Virol., 68,
7790-7799.
41.Matis, J., and Kudelova, M. (2001) Early shutoff
of host protein synthesis in cells infected with herpes simplex
viruses, Acta Virol., 45, 269-277.
42.Neumann, L., Kraas, W., Uebel, S., Jung, G., and
Tampe, R. (1997) The active domain of the herpes simplex virus protein
ICP47: a potent inhibitor of the transporter associated with antigen
processing, J. Mol. Biol., 272, 484-492.
43.Mackem, S., and Roizman, B. (1982) Structural
features of the herpes simplex virus alpha gene 4, 0, and 27
promoter-regulatory sequences which confer alpha regulation on chimeric
thymidine kinase genes, J. Virol., 44, 939-949.
44.Herrera, F. J., and Triezenberg, S. J. (2004)
VP16-dependent association of chromatin-modifying coactivators and
underrepresentation of histones at immediate-early gene promoters
during herpes simplex virus infection, J. Virol., 78,
9689-9696.
45.Orzalli, M. H., DeLuca, N. A., and Knipe, D. M.
(2012) Nuclear IFI16 induction of IRF-3 signaling during herpesviral
infection and degradation of IFI16 by the viral ICP0 protein, Proc.
Natl. Acad. Sci. USA, 109, E3008-3017.
46.Boutell, C., and Everett, R. D. (2012) Regulation
of alpha-herpes virus infections by the ICP0 family of proteins, J.
Gen. Virol., 94, 465-481.
47.Takaoka, A., Wang, Z., Choi, M. K., Yanai, H.,
Negishi, H., Ban, T., Lu, Y., Miyagishi, M., Kodama, T., Honda, K.,
Ohba, Y., and Taniguchi, T. (2007) DAI (DLM-1/ZBP1) is a cytosolic DNA
sensor and an activator of innate immune response, Nature,
448, 501-505.
48.Pham, T. H., Kwon, K. M., Kim, Y. E., Kim, K. K.,
and Ahn, J. H. (2013) DNA sensing-independent inhibition of herpes
simplex virus type-1 replication by DAI/ZBP1, J. Virol.,
87, 3076-3086.
49.Guo, L., Wu, W. J., Liu, L. D., Wang, L. C.,
Zhang, Y., Wu, L. Q., Guan, Y., and Li, Q. H. (2012) Herpes simplex
virus 1 ICP22 inhibits the transcription of viral gene promoters by
binding to and blocking the recruitment of P-TEFb, PloS one,
7, e45749.
50.Yager, D. R., and Marcy, A. I. (1990) Translation
regulation of herpes simplex virus DNA polymerase, J. Virol.,
64, 2217-2225.
51.Skaliter, R., and Lehman, I. R. (1994) Rolling
circle DNA replication in vitro by a complex of herpes simplex
virus type 1-encoded enzymes, Proc. Natl. Acad. Sci. USA,
91, 10665-10669.
52.Zuccola, H. J., Filman, D. J., Coen, D. M., and
Hogle, J. M. (2000) The crystal structure of an unusual processivity
factor, herpes simplex virus UL42, bound to the C terminus of its
cognate polymerase, Mol. Cell, 5, 267-278.
53.Liu, S., Knafels, J. D., Chang, J. S., Waszak, G.
A., Baldwin, E. T., Deibel, M. R., Jr., Thomsen, D. R., Homa, F. L.,
Wells, P. A., Tory, M. C., Poorman, R. A., Gao, H., Qiu, X., and
Seddon, A. P. (2006) Crystal structure of the herpes simplex virus 1
DNA polymerase, J. Biol. Chem., 281, 18193-18200.
54.Weller, S. K., and Coen, D. M. (2012) Herpes
simplex viruses: mechanisms of DNA replication, Cold Spring Harbor
Perspect. Biol., 4, a013011.
55.Burch, A. D., and Weller, S. K. (2005) Herpes
simplex virus type 1 DNA polymerase requires the mammalian chaperone
hsp90 for proper localization to the nucleus, J. Virol.,
79, 10740-10749.
56.Sandbaumhuter, M., Dohner, K., Schipke, J., Binz,
A., Pohlmann, A., Sodeik, B., and Bauerfeind, R. (2013) Cytosolic
herpes simplex virus capsids not only require binding inner tegument
protein pUL36 but also pUL37 for active transport prior to secondary
envelopment, Cell. Microbiol., 15, 248-269.
57.Pasdeloup, D., McElwee, M., Beilstein, F.,
Labetoulle, M., and Rixon, F. J. (2012) Herpes virus tegument protein
pUL37 interacts with dystonin/BPAG1 to promote capsid transport on
microtubules during egress, J. Virol., 87, 2857-2867.
58.Ibiricu, I., Maurer, U. E., and Grunewald, K.
(2013) Characterization of herpes simplex virus type 1 L-particle
assembly and egress in hippocampal neurons by electron cryotomography,
Cell. Microbiol., 15, 285-291.
59.Stegen, C., Yakova, Y., Henaff, D., Nadjar, J.,
Duron, J., and Lippe, R. (2013) Analysis of virion-incorporated host
proteins required for herpes simplex virus type 1 infection through a
RNA interference screen, PloS one, 8, e53276.
60.Deshmane, S. L., and Fraser, N. W. (1989) During
latency, herpes simplex virus type 1 DNA is associated with nucleosomes
in a chromatin structure, J. Virol., 63, 943-947.
61.Digard, P., Bebrin, W. R., Weisshart, K., and
Coen, D. M. (1993) The extreme C terminus of herpes simplex virus DNA
polymerase is crucial for functional interaction with processivity
factor UL42 and for viral replication, J. Virol., 67,
398-406.
62.Stow, N. D. (1993) Sequences at the C-terminus of
the herpes simplex virus type 1 UL30 protein are dispensable for DNA
polymerase activity but not for viral origin-dependent DNA replication,
Nucleic Acids Res., 21, 87-92.
63.Loregian, A., Piaia, E., Cancellotti, E., Papini,
E., Marsden, H. S., and Palu, G. (2000) The catalytic subunit of herpes
simplex virus type 1 DNA polymerase contains a nuclear localization
signal in the UL42-binding region, Virology, 273,
139-148.
64.Kuhn, F. J., and Knopf, C. W. (1996) Herpes
simplex virus type 1 DNA polymerase. Mutational analysis of the
3′-5′-exonuclease domain, J. Biol. Chem.,
271, 29245-29254.
65.Crute, J. J., and Lehman, I. R. (1989) Herpes
simplex-1 DNA polymerase. Identification of an intrinsic
5′-3′ exonuclease with ribonuclease H activity, J. Biol.
Chem., 264, 19266-19270.
66.Bogani, F., and Boehmer, P. E. (2008) The
replicative DNA polymerase of herpes simplex virus 1 exhibits
apurinic/apyrimidinic and 5′-deoxyribose phosphate lyase
activities, Proc. Natl. Acad. Sci. USA, 105,
11709-11714.
67.Bogani, F., Corredeira, I., Fernandez, V.,
Sattler, U., Rutvisuttinunt, W., Defais, M., and Boehmer, P. E. (2010)
Association between the herpes simplex virus-1 DNA polymerase and
uracil DNA glycosylase, J. Biol. Chem., 285,
27664-27672.
68.Terrell, S. L., and Coen, D. M. (2012) The
pre-NH(2)-terminal domain of the herpes simplex virus 1 DNA polymerase
catalytic subunit is required for efficient viral replication, J.
Virol., 86, 11057-11065.
69.Coen, D. M., and Schaffer, P. A. (2003)
Anti-herpes virus drugs: a promising spectrum of new drugs and drug
targets, Nature Rev. Drug Discov., 2, 278-288.
70.De Clercq, E., and Field, H. J. (2006) Antiviral
prodrugs – the development of successful prodrug strategies for
antiviral chemotherapy, Brit. J. Pharmacol., 147,
1-11.
71.Prusoff, W. H. (1959) Synthesis and biological
activities of iododeoxyuridine, an analog of thymidine, Biochim.
Biophys. Acta, 32, 295-296.
72.Elion, G. B., Furman, P. A., Fyfe, J. A., de
Miranda, P., Beauchamp, L., and Schaeffer, H. J. (1977) Selectivity of
action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine,
Proc. Natl. Acad. Sci. USA, 74, 5716-5720.
73.Schaeffer, H. J., Beauchamp, L., de Miranda, P.,
Elion, G. B., Bauer, D. J., and Collins, P. (1978)
9-(2-Hydroxyethoxymethyl)guanine activity against viruses of the herpes
group, Nature, 272, 583-585.
74.Elion, G. B. (1993) Acyclovir: discovery,
mechanism of action, and selectivity, J. Med. Virol., Suppl. 1,
2-6.
75.Martin, J. C., Dvorak, C. A., Smee, D. F.,
Matthews, T. R., and Verheyden, J. P. (1983)
9-[(1,3-Dihydroxy-2-propoxy)methyl]guanine: a new potent and selective
antiherpes agent, J. Med. Chem., 26, 759-761.
76.Thust, R., Tomicic, M., Klocking, R.,
Voutilainen, N., Wutzler, P., and Kaina, B. (2000) Comparison of the
genotoxic and apoptosis-inducing properties of ganciclovir and
penciclovir in Chinese hamster ovary cells transfected with the
thymidine kinase gene of herpes simplex virus-1: implications for gene
therapeutic approaches, Cancer Gene Ther., 7,
107-117.
77.Boyd, M. R., Bacon, T. H., Sutton, D., and Cole,
M. (1987) Anti-herpes virus activity of
9-(4-hydroxy-3-hydroxy-methylbut-1-yl)guanine (BRL 39123) in cell
culture, Antimicrob. Agents Chemother., 31,
1238-1242.
78.Thackray, A. M., and Field, H. J. (1998)
Famciclovir and valaciclovir differ in the prevention of herpes simplex
virus type 1 latency in mice: a quantitative study, Antimicrob.
Agents Chemother., 42, 1555-1562.
79.Oberg, B. (1989) Antiviral effects of
phosphonoformate (PFA, foscarnet sodium), Pharmacol. Therap.,
40, 213-285.
80.Helgstrand, E., Eriksson, B., Johansson, N. G.,
Lannero, B., Larsson, A., Misiorny, A., Noren, J. O., Sjoberg, B.,
Stenberg, K., Stening, G., Stridh, S., and Oberg, B. (1978) Trisodium
phosphonoformate, a new antiviral compound, Science, 201,
819-821.
81.De Clercq, E., Andrei, G., Snoeck, R., De Bolle,
L., Naesens, L., Degreve, B., Balzarini, J., Zhang, Y., Schols, D.,
Leyssen, P., Ying, C., and Neyts, J. (2001) Acyclic/carbocyclic
guanosine analogues as anti-herpes virus agents, Nucleosides
Nucleotides Nucl. Acids, 20, 271-285.
82.Ivanov, A. V., Andronova, B. L., Galegov, G. A.,
and Jasko, M. V. (2005) Synthesis and antiherpetic activity of (Z)- and
(E)-isomers of 9-(3-phosphonomethoxyprop-1-enyl)adenine, Bioorg.
Khim., 31, 65-72.
83.Korovina, A. N., Jasko, M. V., Ivanov, A. V.,
Khandazhynskaya, A. L., Kramarov, E. V., Kornilaeva, G. V., and
Kukhanova, M. K. (2008) Novel herpes simplex virus and human
immunodeficiency virus inhibitors based on phosphonate nucleoside
analogs, Moscow Univ. Chem. Bull., 63, 85-88.
84.Karpenko, I. L., Jasko, M. V., Andronova, V. L.,
Ivanov, A. V., Kukhanova, M. K., Galegov, G. A., and Skoblov, Y. S.
(2003) Synthesis and antiherpetic activity of acyclovir phosphonates,
Nucleosides Nucleotides Nucl. Acids, 22, 319-328.
85.Skoblov, Y. S., Karpenko, I. L., Jasko, M. V.,
Kukhanova, M. K., Andronova, V. L., Galegov, G. A., Sidorov, G. V., and
Myasoedov, N. F. (2007) Cell metabolism of acyclovir phosphonate
derivatives and anti-herpes virus activity of their combinations with
alpha2-interferon, Chem. Biol. Drug Design, 69,
429-434.
86.Sanchez, R. M., Erhard, K., Hardwicke, M. A.,
Lin, H., McSurdy-Freed, J., Plant, R., Raha, K., Rominger, C. M.,
Schaber, M. D., Spengler, M. D., Moore, M. L., Yu, H., Luengo, J. I.,
Tedesco, R., and Rivero, R. A. (2012) Synthesis and structure-activity
relationships of 1,2,4-triazolo[1,5-a]pyrimidin-7(3H)-ones as novel
series of potent beta isoform selective phosphatidylinositol 3-kinase
inhibitors, Bioorg. Med. Chem. Lett., 22, 3198-3202.
87.Deev, S. L., Yasko, M. V., Karpenko, I. L.,
Korovina, A. N., Khandazhinskaya, A. L., Andronova, V. L., Galegov, G.
A., Shestakova, T. S., Ulomskii, E. N., Rusinov, V. L., Chupakhin, O.
N., and Kukhanova, M. K. (2010) 1,2,4-Triazoloazine derivatives as a
new type of herpes simplex virus inhibitors, Bioorg. Chem.,
38, 265-270.
88.Revankar, G. R., and Robins, R. K. (1975)
Synthesis and biological activity of some nucleosides resembling
guanosine: imidazo(1,2-alpha)pyrimidine nucleosides, Ann. N. Y.
Acad. Sci., 255, 166-176.
89.Kleymann, G., Fischer, R., Betz, U. A., Hendrix,
M., Bender, W., Schneider, U., Handke, G., Eckenberg, P., Hewlett, G.,
Pevzner, V., Baumeister, J., Weber, O., Henninger, K., Keldenich, J.,
Jensen, A., Kolb, J., Bach, U., Popp, A., Maben, J., Frappa, I.,
Haebich, D., Lockhoff, O., and Rubsamen-Waigmann, H. (2002) New
helicase-primase inhibitors as drug candidates for the treatment of
herpes simplex disease, Nature Med., 8, 392-398.
90.Baumeister, J., Fischer, R., Eckenberg, P.,
Henninger, K., Ruebsamen-Waigmann, H., and Kleymann, G. (2007) Superior
efficacy of helicase-primase inhibitor BAY 57-1293 for herpes infection
and latency in the guinea pig model of human genital herpes disease,
Antiviral Chem. Chemother., 18, 35-48.
91.Crute, J. J., Grygon, C. A., Hargrave, K. D.,
Simoneau, B., Faucher, A. M., Bolger, G., Kibler, P., Liuzzi, M., and
Cordingley, M. G. (2002) Herpes simplex virus helicase-primase
inhibitors are active in animal models of human disease, Nature
Med., 8, 386-391.
92.Katsumata, K., Chono, K., Sudo, K., Shimizu, Y.,
Kontani, T., and Suzuki, H. (2011) Effect of ASP2151, a herpes virus
helicase-primase inhibitor, in a guinea pig model of genital herpes,
Molecules, 16, 7210-7223.
93.Sergerie, Y., and Boivin, G. (2008) Hydroxyurea
enhances the activity of acyclovir and cidofovir against herpes simplex
virus type 1 resistant strains harboring mutations in the thymidine
kinase and/or the DNA polymerase genes, Antiviral Res.,
77, 77-80.
94.Duan, J., Liuzzi, M., Paris, W., Lambert, M.,
Lawetz, C., Moss, N., Jaramillo, J., Gauthier, J., Deziel, R., and
Cordingley, M. G. (1998) Antiviral activity of a selective
ribonucleotide reductase inhibitor against acyclovir-resistant herpes
simplex virus type 1 in vivo, Antimicrob. Agents
Chemother., 42, 1629-1635.
95.Ekblad, M., Adamiak, B., Bergefall, K., Nenonen,
H., Roth, A., Bergstrom, T., Ferro, V., and Trybala, E. (2007)
Molecular basis for resistance of herpes simplex virus type 1 mutants
to the sulfated oligosaccharide inhibitor PI-88, Virology,
367, 244-252.
96.Ekblad, M., Adamiak, B., Bergstrom, T.,
Johnstone, K. D., Karoli, T., Liu, L., Ferro, V., and Trybala, E.
(2010) A highly lipophilic sulfated tetrasaccharide glycoside related
to muparfostat (PI-88) exhibits virucidal activity against herpes
simplex virus, Antiviral Res., 86, 196-203.
97.Johansson, M. E., Gustafsson, J. K., Sjoberg, K.
E., Petersson, J., Holm, L., Sjovall, H., and Hansson, G. C. (2010)
Bacteria penetrate the inner mucus layer before inflammation in the
dextran sulfate colitis model, PloS one, 5, e12238.
98.Yasin, B., Wang, W., Pang, M., Cheshenko, N.,
Hong, T., Waring, A. J., Herold, B. C., Wagar, E. A., and Lehrer, R. I.
(2004) Theta defensins protect cells from infection by herpes simplex
virus by inhibiting viral adhesion and entry, J. Virol.,
78, 5147-5156.
99.Berlutti, F., Pantanella, F., Natalizi, T.,
Frioni, A., Paesano, R., Polimeni, A., and Valenti, P. (2011) Antiviral
properties of lactoferrin – a natural immunity molecule,
Molecules, 16, 6992-7018.
100.Coen, D. M. (1991) The implications of
resistance to antiviral agents for herpes virus drug targets and drug
therapy, Antiviral Res., 15, 287-300.
101.Andrei, G., Georgala, A., Topalis, D., Fiten,
P., Aoun, M., Opdenakker, G., and Snoeck, R. (2013) Heterogeneity and
evolution of thymidine kinase and DNA polymerase mutants of herpes
simplex virus type 1: implications for antiviral therapy, J. Infect.
Dis., 207, 1295-1305.
102.Korovina, A. N., Gus’kova, A. A.,
Skoblov, M. Iu., Andronova, V. L., Galegov, G. A., Kochetkov, S. N.,
Kukhanova, M. K., and Skoblov, Iu. S. (2010) Analysis of mutations in
DNA polymerase and thymidine kinase genes of herpes simplex virus
clinical isolates resistant to antiherpetic drugs, Mol. Biol.
(Moscow), 44, 488-496.
103.Suzutani, T., Saijo, M., Nagamine, M.,
Ogasawara, M., and Azuma, M. (2000) Rapid phenotypic characterization
method for herpes simplex virus and Varicella–Zoster virus
thymidine kinases to screen for acyclovir-resistant viral infection,
J. Clin. Microbiol., 38, 1839-1844.
104.Hwang, Y. T., Smith, J. F., Gao, L., and Hwang,
C. B. (1998) Mutations in the Exo III motif of the herpes simplex virus
DNA polymerase gene can confer altered drug sensitivities,
Virology, 246, 298-305.
105.Gus’kova, A. A., Skoblov, M. Y.,
Korovina, A. N., Yasko, M. V., Karpenko, I. L., Kukhanova, M. K.,
Andronova, V. L., Galegov, G. A., and Skoblov, Y. S. (2009)
Antiherpetic properties of acyclovir 5′-hydrogenphosphonate and
the mutation analysis of herpes virus resistant strains, Chem. Biol.
Drug Design, 74, 382-389.
106.Suzutani, T., Ishioka, K., De Clercq, E.,
Ishibashi, K., Kaneko, H., Kira, T., Hashimoto, K., Ogasawara, M.,
Ohtani, K., Wakamiya, N., and Saijo, M. (2003) Differential mutation
patterns in thymidine kinase and DNA polymerase genes of herpes simplex
virus type 1 clones passaged in the presence of acyclovir or
penciclovir, Antimicrob. Agents Chemother., 47,
1707-1713.
107.Bestman-Smith, J., and Boivin, G. (2003) Drug
resistance patterns of recombinant herpes simplex virus DNA polymerase
mutants generated with a set of overlapping cosmids and plasmids, J.
Virol., 77, 7820-7829.
108.Matthews, J. T., Carroll, R. D., Stevens, J.
T., and Haffey, M. L. (1989) In vitro mutagenesis of the herpes
simplex virus type 1 DNA polymerase gene results in altered drug
sensitivity of the enzyme, J. Virol., 63, 4913-4918.
109.Matthews, J. T., Terry, B. J., and Field, A. K.
(1993) The structure and function of the HSV DNA replication proteins:
defining novel antiviral targets, Antivir. Res., 20,
89-114.
110.Vere Hodge, R. A., and Field, H. J. (2013)
Antiviral agents for herpes simplex virus, Adv. Pharmacol.,
67, 1-38.