Role of Loop L5-6 Connecting Transmembrane Segments M5 and M6 in Biogenesis and Functioning of Yeast Pma1 H+-ATPase
V. V. Petrov
Skryabin Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, 142290 Pushchino, Russia; E-mail: vpetrov07@gmail.com
Received May 26, 2014; Revision received July 23, 2014
The L5-6 loop is a short extracytoplasmic stretch (714-DNSLDID) connecting transmembrane segments M5 and M6 and forming along with segments M4 and M8 the core through which cations are transported by H+-, Ca2+-, K+,Na+-, H+,K+-, and other P2-ATPases. To study structure–function relationships within this loop of the yeast plasma membrane Pma1 H+-ATPase, alanine- and cysteine-scanning mutagenesis has been employed. Ala and Cys substitutions for the most conserved residue (Leu717) led to complete block in biogenesis preventing the enzyme from reaching secretory vesicles. The Ala replacement at Asp714 led to five-fold decrease in the mutant expression and loss of its activity, while the Cys substitution blocked biogenesis completely. Replacements of other residues did not lead to loss of enzymatic activity. Additional replacements were made for Asp714 and Asp720 (Asp®Asn/Glu). Of the substitutions made at Asp714, only D714N partially restored the mutant enzyme biogenesis and functioning. However, all mutant enzymes with substituted Asp720 were active. The expressed mutants (34-95% of the wild-type level) showed activity high enough (35-108%) to be analyzed in detail. One of the mutants (I719A) had three-fold reduced coupling ratio between ATP hydrolysis and H+ transport; however, the I719C mutation was rather indistinguishable from the wild-type enzyme. Thus, substitutions at two of the seven positions seriously affected biogenesis and/or functioning of the enzyme. Taken together, these results suggest that the M5-M6 loop residues play an important role in protein stability and function, and they are probably responsible for proper arrangement of transmembrane segments M5 and M6 and other domains of the enzyme. This might also be important for the regulation of the enzyme.
KEY WORDS: yeast, plasma membrane, secretory vesicles, biogenesis, Pma1 H+-ATPase, H+ transport, site-directed mutagenesisDOI: 10.1134/S0006297915010046
Yeast plasma membrane H+-ATPase (Pma1) encoded by the PMA1 gene [1] is a vital enzyme that generates the transmembrane electrochemical proton gradient, thus providing energy for secondary transport systems and maintaining ion homeostasis and intracellular pH. Pma1 ATPase belongs to a widespread and physiologically important group of P2-ATPases, a part of the P-ATPase family. These ions pumps found in pro- and eukaryotic cells use energy from ATP hydrolysis to transport a variety of cations (H+, Na+, K+, Cu+, Ag+, Mg2+, Ca2+, Co2+, Cu2+, Cd2+, Zn2+, Pb2+, Mn2+) across different cellular and intracellular membranes [2, 3].
P2-ATPases have a single large catalytic subunit embedded in the lipid bilayer by four transmembrane hydrophobic segments at the N-terminal and six segments at the C-terminal end of different length and incline. These segments form the enzyme membrane domain (M) that contains residues involved in cation transport. They are connected by five relatively short extracytosolic and more lengthy cytosolic loops, the most important of which are the so-called large (L4-5, between segments M4 and M5) and small (L2-3, between segments M2 and M3) loops; they form domains N (ATP- or nucleotide-binding, which includes the middle of the large loop L4-5), P (phosphorylation, including N- and C-terminal ends of this loop), and A (actuator, including N-terminal part of the enzyme and its small loop L2-3) [4, 5].
These enzymes have a common reaction mechanism in which a conserved Asp residue in the L4-5 loop is phosphorylated by ATP to form an essential covalent intermediate [6]. The stoichiometry of transport varies in members of the family, ranging from 1-2 H+/ATP in the plant and fungal plasma-membrane H+-pumps [7-9] to 2 Ca2+/ATP and 3 Na+/2 K+/ATP in the Ca2+- and Na+,K+-ATPases of animal cells [10]. Determinants of cation specificity and stoichiometry lie in the core of the transmembrane segments M4, M5, M6, and M8 of these enzymes as confirmed by crystal structures of Ca2+- [4, 5] and Na+,K+-ATPase [11, 12] of animal cells and H+-ATPases of fungi [13] and plants [14].
Earlier, site-directed mutagenesis of animal Ca2+- [15-18] and Na+,K+-ATPases [18-21] revealed residues in M4, M5, M6, and M8 segments that are essential for cation transport, and mutagenesis of the yeast plasma membrane H+-ATPase also located several positions in M4 [22], M5 [8, 23], M6 [8, 24, 25], and M8 [8, 9, 26, 27], at which substitutions affect normal functioning and/or biogenesis of the enzyme and also change the coupling between ATP hydrolysis and H+ transport [8, 9, 24-27]. Some replacements of key amino acid residues in these segments of P2 pumps (e.g. in M6 of the yeast endoplasmic reticulum Ca2+,Mn2+-ATPase) can even change specificity of cation transport [28, 29]. Thus, both the membranous and cytosolic parts of these enzymes have been extensively studied [8, 9, 15-32], while less is known about the role of amino acid residues in the extracytosolic part of P2-ATPases facing the cell envelope; the published data mostly refer to study of H+,K+- and Na+,K+-ATPases of animal cells [33-37]. In particular, cysteine-scanning mutagenesis was performed for three extracytosolic loops of Na+,K+-ATPase [33-35]; it was also found that omeprazole-like inhibitors reacted with a Cys residue in the loop between the M5 and M6 segments of the H+,K+-ATPase [37]. As for the fungal H+-ATPase, only the loop between segments M1 and M2 consisting of two amino acid residues and adjacent parts of these segments have been studied [38].
During the reaction cycle, P2-ATPases undergo significant conformational changes; upon ATP and transported cation binding, the enzyme reversibly shifts from conformation E1 with high affinity to substrates (MgATP and transported cation) and low affinity to specific inhibitor orthovanadate (an analog of the reaction product orthophosphate) to conformation E2 with the opposite properties. During the cycle, transmembrane segments M1-M6, which are α-helices, bend, unwind partially, and even exit from the membrane, while the M7-M10 segments are less mobile [5]. It has been shown previously that Ala substitutions of the residues located it the N-terminal half of M6 segment adjacent to the L5-6 loop interfered markedly with the Pma1 ATPase functioning and/or its biogenesis [24, 25]. Since the short extracytosolic L5-6 loop along with M5 and M6 segments form the so-called M5-M6 hairpin [39-41], it is reasonable to expect that the residues in this loop are important for the enzyme structure–function relationships.
The results described here extend the systematic study of the yeast Pma1 H+-ATPase by focusing on the extracytosolic L5-6 loop connecting the C-terminal part of M5 and the N-terminal part of M6 segments of this enzyme, screening for residues that may play a role in ATP hydrolysis and H+ transport or any other aspect of the functioning and/or biogenesis of the enzyme.
MATERIALS AND METHODS
Yeast strain. Saccharomyces cerevisiae strain SY4 (MATa; ura3-52; leu2-3, 112; his4-619; sec6-4; GAL; pma1::YIpGAL-PMA1) [42] was used in this work; it contained both chromosomal (PMA1) and plasmid (pma1) copies of the PMA1 gene, which encodes Pma1 H+-ATPase. In the SY4 strain, the chromosomal copy of the wild-type ATPase gene was under control of the GAL1 promoter (PGAL-PMA1) and the plasmid allele (on the centromeric plasmid YCp2HSE) was under the heat shock-inducible HSE promoter (PHSE-pma1) [42]. The SY4 strain also carried a temperature-sensitive mutation in the SEC6 gene, which blocks the fusion of secretory vesicles with the plasma membrane under heat shock and leads to accumulation of secretory vesicles.
Site-directed mutagenesis. To introduce mutations into a 519 bp BglII-SalI fragment of the pma1 gene that has been previously subcloned into a modified version of the Bluescript plasmid (Stratagene, USA), oligonucleotide-directed mutagenesis kit (Amersham, USA) or polymerase chain reaction were used [8, 9, 43]. After mutagenesis, each fragment was sequenced to verify the presence of the mutation and the absence of unwanted base changes. Then, by the means of endonucleases BglII, HindIII, SacI, and SalI and T4 DNA ligase (New England Biolabs, USA), these fragments were used to replace corresponding fragments of plasmid pPMA1.2, which contains the entire coding sequence of the gene pma1 [42]. To express the Pma1 H+-ATPase in secretory vesicles, a 3.77-kb HindIII-SacI fragment of pPMA1.2 containing the entire coding sequence of the gene was excised by endonucleases HindIII and SacI and with T4 DNA ligase transferred from pPMA1.2 into centromeric plasmid YCp2HSE, where it was placed under the control of two tandemly arranged HSE promoters; this plasmid (YCp2HSE-PMA1) was then used to transform the SY4 cells [42].
Isolation of secretory vesicles. To isolate secretory vesicles, the SY4 yeast cells were grown at 23°C in liquid medium containing 2% galactose, 0.67 g/liter YNB (Difco, USA), and 20 mg/liter histidine until mid-exponential phase (A600 ~ 0.7-1.0). Then the cells were washed free from galactose and transferred into the same medium containing 2% glucose. After incubation for 3 h, the yeast was subjected to a heat shock by increasing temperature to 39°C for 2 h. Ten minutes before the end of the heat shock, 10 mM NaN3, which blocks metabolism preventing vesicle fusion to plasma membrane [42], was added to the cell suspension, and the cells were chilled on iced water. Cells were sedimented by centrifugation and spheroplasts were obtained. Spheroplasts were treated with concanavalin A to make plasma membranes heavier as described earlier [42]. After the spheroplasts were obtained, all procedures were performed at 0-4°C. Secretory vesicles containing newly synthesized ATPase were isolated by differential centrifugation and gel filtration [8, 42, 43] or centrifugation in sucrose density gradient [8, 9] and resuspended in 0.8 M sorbitol, 1 mM EDTA, 10 mM triethanolamine-acetic acid, pH 7.2, containing protease inhibitors (2 µg/ml chymostatin and leupeptin, pepstatin, and aprotinin, 1 µg/ml each) as described earlier [8, 43, 44].
Metabolic labeling, immunoprecipitation, and trypsinolysis. To detect the synthesis of mutant ATPases that were unable to reach secretory vesicles, SY4 cells were shifted from galactose to glucose medium as described above and then metabolically labeled with [35S]methionine. Total membranes were isolated and treated with trypsin at trypsin/protein ratio 1 : 20. Then the samples were subjected to SDS-PAGE, immunoprecipitated with anti-Pma1 antibody against closely related Neurospora crassa plasma membrane H+-ATPase [45], and treated as described [46].
Quantitation of expressed ATPase. The amount of ATPase in secretory vesicles was estimated by SDS-PAGE and immunoblotting as described earlier [8, 42, 43]. Blots were treated with affinity-purified polyclonal antibody against Pma1 H+-ATPase and then with 125I-labeled protein A (ICN, USA), and assayed by means of a PhosphorImager equipped with ImageQuant software (Molecular Dynamics, USA). The expression level of mutant Pma1 ATPase in secretory vesicles was calculated relative to a wild-type control run in parallel on the day of preparation [43].
ATPase assay. ATPase activity was measured at 30°C in 0.5 ml of the incubation mix containing 10 mM MgSO4, 5 mM Na2ATP, 50 mM MES-Tris, pH 5.7, 5 mM KN3, and an ATP-regenerating system (5 mM phosphoenolpyruvate and 50 µg/ml pyruvate kinase (40 U/mg protein) (Sigma-Aldrich, USA)) in the absence and presence of 100 µM sodium orthovanadate [9, 42, 43]. To obtain the Ki of the ATPase inhibition by orthovanadate ions, the stock solution of Na3VO4 was boiled in a water bath prior use for 3-5 min to break orthovanadate complexes that are formed during storage. To obtain Km, the true concentration of MgATP was calculated as described [47]. Inorganic phosphate was measured according to [48].
ATP-dependent H+ transport in secretory vesicles was registered at 29°C by fluorescence quenching of the pH-sensitive dye acridine orange on a Hitachi F2000 spectrofluorimeter (excitation, 430 nm; emission, 530 nm) [8, 9, 42, 43]. Secretory vesicles (50-100 µg protein) were suspended in 1.5 ml of 0.6 M sorbitol, 100 mM KCl, 20 mM KNO3, 5 mM Na2ATP, 2 µM acridine orange, 20 mM HEPES-NaOH, pH 6.7; after stabilization of the baseline fluorescence (for 120-150 s), the reaction was started by adding 10 mM MgCl2.
Coupling between H+ transport and ATP hydrolysis. To estimate coupling ratio between H+ transport and ATP hydrolysis, the initial rates of ATP hydrolysis were assayed over a range of MgATP concentrations in parallel with measuring the initial rates of H+ transport under the same concentrations: in 0.1 ml (ATP hydrolysis) or 1.5 ml (H+ pumping) of 0.6 M sorbitol, 20 mM HEPES-KOH, pH 6.7, 100 mM KCl, 20 mM KNO3 containing 0.2-3.0 mM Na2ATP at 29oC [8, 9, 43]. The reaction mixture to assay ATP hydrolysis also contained 5.2-8.0 mM MgCl2. The reaction mixture to assay H+ transport additionally contained 2 µM acridine orange, and the reaction was initiated by addition of 5.2-8.0 mM MgCl2. The reaction of ATP hydrolysis was carried out in the presence and absence of 100 µM Na3VO4 and stopped by addition of 1 ml of 1.25% trichloroacetic acid, and inorganic phosphate was measured as above [48].
Protein assay. Protein was determined by the modified Lowry method [49] with bovine serum albumin used as a standard. Aliquots of the secretory vesicle-resuspending buffer were added to the standard.
RESULTS
Choice of amino acid residues to be studied. The L5-6 loop between transmembrane segments M5 and M6 is one of the five short hydrophilic extracytosolic stretches that connect neighboring transmembrane segments with each other; jointly with M5 and M6, this loop forms the M5-M6 hairpin (Fig. 1) that plays an important role in cation transport [39-41]. According to hydropathy plots [50], the S. cerevisiae Pma1 ATPase L5-6 loop consists of seven amino acid residues, three of which are Asp: 714-DNSLDID. It was shown earlier that the M5 and M6 segments contain amino acid residues whose replacement leads to the impairment of Pma1 ATPase biogenesis and/or loss of activity [8, 24, 25]. Moreover, the N-terminal part of the Pma1 ATPase M6 segment contained an almost uninterrupted stretch of seven amino acid residues 721-LI.FI.IF.D [25] whose replacement impaired biogenesis and/or functioning of the enzyme or even led to loss of activity (Fig. 2). Substitution of one of the residues in this segment (A726S mutation) led to twofold decrease in the coupling of the enzyme between ATP hydrolysis and H+ transport, while substitution of another residue (V723A mutation) led to large changes in kinetics reflecting a shift to conformation E1 [25]. The C-terminal part of M5 contains only two residues whose replacement led to impairment of biogenesis and/or activity of the enzyme [23]. Nevertheless, point replacements of seven residues in this part caused significant (twofold or more) decrease in the coupling ratio of the Pma1 H+ pump ([8], V. V. Petrov, unpublished data). Therefore, it seemed important to study the role of the L5-6 loop connecting the C-terminal part of M5 and the N-terminal part of M6 segments.
Fig. 1. Schematic topology of the S. cerevisiae Pma1 H+-ATPase showing transmembrane segments and extracytosolic loops; the L5-6 loop is marked. Numbers indicate amino acid residues in the cytosolic parts of the enzyme. The total number of residues in S. cerevisiae Pma1 H+-ATPase is 918.
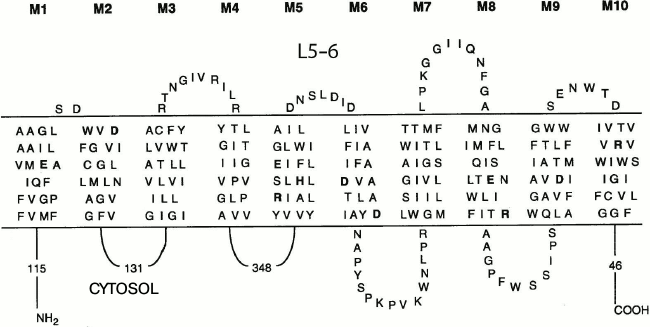
Fig. 2. Amino acid residues in membrane segment M6 of yeast Pma1 ATPase important for the biogenesis and functioning of the enzyme. The N-terminal end of M6 is highlighted in light gray. Ala726, whose substitution for Ser resulted in twofold decrease in coupling ratio between ATP hydrolysis and H+ transport [25], is not shown. Modified version from [25].
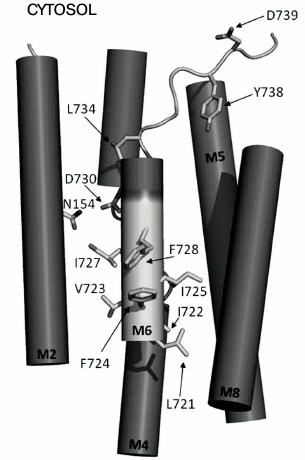
Conservation of studied amino acid residues. Even in closely related Saccharomycetes H+-ATPases, none of the residues that form the L5-6 loop is strictly conserved. However, the homology of the substitutive residues of other H+ pumps in published sequences is very high, being 95-100% for all residues except for Asn715 (53%, Fig. 3). As for identity, it was also high, being 100% for Ser716 and Leu717, 95% for Ile719 (36 Ile/2 Val), 82% for Asp714 (31 Asn/7 Asp), 76% for Asp720 (29 Asp/4 Asn/3 Glu), and 53% for Asp718 (20 Asn/16 Asp/1 Glu); however, it decreased to 29% for Asn715 (11 Asn/5 Gln/4 Glu). The most conserved of these residues is Leu717 considering its presence in other amino acid sequences from ascomycetous Pma ATPases (Fig. 3); the least conserved is Asp715 being represented by eleven Asn, five Glu, four Gln, and even twelve His and six Arg.
Fig. 3. Sequence alignment of amino acid residues from the Saccharomycetes Pma H+-ATPase L5-6 loops. The loop alignments from three ascomycetes – yeast S. pombe and fungi N. crassa and A. flavus – are also shown. The S. cerevisiae Pma1 residues and identical residues in other H+ pumps are highlighted in bold and dark gray; homological residues are highlighted in light gray.

According to these data, the L5-6 residues can be arranged in the following order that takes into account the residue most frequently found in the sequences at position corresponding to the number of a residue in the S. cerevisiae Pma1 ATPase (subscript number indicates the number of the residue in 38 sequences and letter after that indicates the most frequently found residue in this position):
L38L717 ≈ S38S716 > I36I719 > D31N714 > D29D720 > D20N718 > N11N715.
It is reasonable to suggest that conservation of a residue correlates with its role in the structure–function relations in the H+-ATPase.
Constructing and expressing mutants in secretory vesicles. By means of Ala- and Cys-scanning mutagenesis, we constructed point substitutions, one at a time, in the L5-6 loop of the yeast S. cerevisiae Pma1 H+-ATPase. To obtain these substitutions, oligonucleotides carrying the respective substitutions for Ala and Cys codons (Table 1) were synthesized according to the mostly frequently found codons encoding amino acid residues in the yeast plasma membrane H+-ATPase [51]. Then, using site-directed mutagenesis, 519 bp BglII-SalI fragments of the pma1 gene, carrying mutations, were obtained. These fragments were subcloned into the pma1 gene, and then the gene was cloned into centromeric plasmid YCp2HSE under control of the HSE promoter induced by heat shock. This plasmid was then used to transform the S. cerevisiae strain SY4, where the chromosomal wild-type PMA1 gene was under the control of the GAL1 promotor [42]. The yeast was grown at 23°C in medium containing galactose until mid-exponential phase; therefore, the wild-type enzyme was synthesized from the chromosomal PMA1 gene. When the cells were shifted to the glucose-containing medium, the synthesis from the PMA1 gene was interrupted; when the temperature was elevated to 39°C, Pma1 synthesis began from the plasmid pma1 gene (encoding either the wild-type ATPase or a mutant). Strain SY4 also carried temperature-sensitive mutation sec6-4, which blocked the fusion of secretory vesicles to the plasma membrane. Hence, heat shock resulted in accumulation of secretory vesicles containing the newly synthesized enzyme from the plasmid pma1 gene [42]. Preparation of secretory vesicles obtained from the strain carrying plasmid YCp2HSE without gene pma1 (Δpma1; Table 2) was used as control for the presence of plasma membranes. Thus, the use of SY4 cells provided secretory vesicles both carrying the enzyme molecules (regardless of the enzyme activity) and almost without ATPase. In the latter case, metabolic labeling was used to detect the enzyme synthesis.
Secretory vesicles practically free from the plasma membrane contamination (4%, Δpma1; Table 2) were isolated by differential centrifugation and centrifugation in a sucrose density gradient and used to measure the enzyme expression level and activity. Proton transport into secretory vesicles was monitored by fluorescence quenching of the pH-sensitive dye acridine orange as described previously [8, 9, 42, 43]. To obtain coupling ratio between ATP hydrolysis and H+ pumping, parallel measurement of the ATP hydrolysis and H+ transport was performed under the same conditions over a range of MgATP concentrations.
Table 1. Oligonucleotides synthesized to
replace amino acid residues in the domain connecting M5 and M6 segments
of the Pma1 ATPase for Ala, Cys, Glu, and Asn
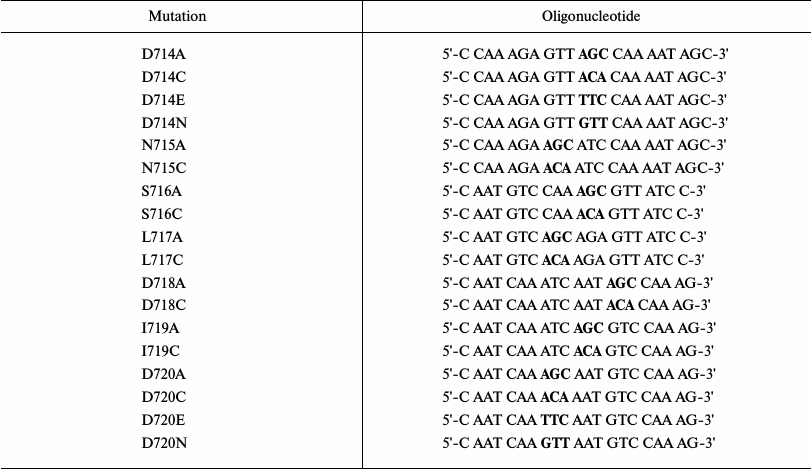
Note: Triplets encoding replacements are shown in bold.
Table 2. Effect of point mutations in the
L5-6 loop on expression of Pma1 ATPase in secretory vesicles, ATP
hydrolysis, and H+ transport1
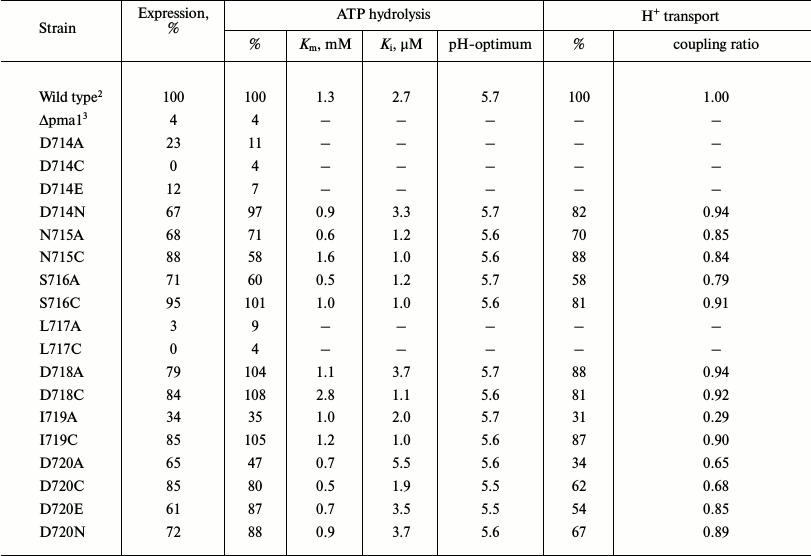
1 Specific expression of 100-kDa ATPase subunit was measured
by quantitative immunoblotting as described under “Materials and
Methods” and is represented as percent of the wild-type control
run in parallel. ATP hydrolysis was assayed at pH 5.7. 100%
corresponded to 4.77 ± 0.68 µmol Pi/min
per mg protein. Km corresponded to mM ATP,
Ki to µM of sodium orthovanadate.
H+ transport was registered by fluorescence quenching of
acridine orange as described under “Materials and Methods”.
Percent of fluorescence quenching (% F) was measured by dividing
difference in fluorescence quenching (ΔF) by its maximum level
(Fmax). Coupling ratio was calculated by the rate of
H+ pumping as a function of the rate of ATP hydrolysis. 100%
corresponded to 743 ± 88% F per mg protein. Data are average of
17-26 experiments for the wild-type enzyme and 2-9 for the mutants.
2 Secretory vesicles were isolated from cells carrying
expression plasmid YCp2HSE with the wild-type pma1 gene
(positive control).
3 Secretory vesicles were isolated from cells carrying
expression plasmid YCp2HSE without pma1 gene (negative
control).
In the case of Asp714 and Asp720, substitutions for Asn and Glu were also made (see below); they were studied as described above.
Mutants carrying alanine substitutions: expression and ATP hydrolysis. Pma1 H+-ATPase is synthesized in endoplasmic reticulum and during maturation moves through Golgi apparatus and secretory vesicles, which deliver enzyme to the plasma membrane. There are at least two points of quality control and if the enzyme has serious defects of folding, it is retained en route to plasma membrane [52-54].
According to residue conservation, of the seven Ala mutations studied only the replacement of the most conserved residue (L717A) led to a complete block in ATPase trafficking that prevented the enzyme from reaching the secretory vesicles (Table 2). This behavior is typical for a protein with severe defects in protein folding, causing the abnormal ATPase to be retained in the endoplasmic reticulum [53].
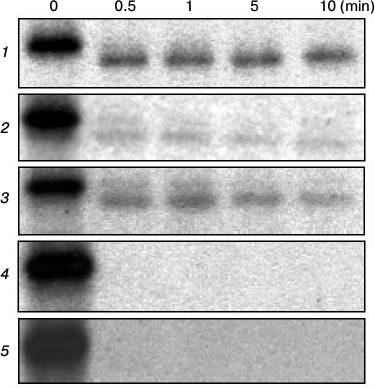
To detect synthesis of mutant ATPases that were unable to reach secretory vesicles, the SY4 cells were transferred from galactose into glucose medium as described above and metabolically labeled with [35S]methionine. Then total membranes were isolated and treated with trypsin at trypsin/protein ratio of 1 : 20, subjected to SDS-PAGE, immunoprecipitated with polyclonal anti-Pma1 antibody, and treated as described [46]. By sensitivity to trypsin, the impairment of protein folding could be detected. Indeed, by contrast to the wild-type enzyme, whose native 100-kDa band remained stable as a 97-kDa band after cleavage of a small fragment even after 10 min of incubation (Fig. 4), the mutant L717A ATPase was completely degraded by 0.5 min pointing to serious defects of folding that led to changes in exposure of trypsin cleavage sites. Both the 100- and 97-kDa bands were completely hydrolyzed.
Fig. 4. Limited trypsinolysis of wild-type and mutant ATPases. Total membranes isolated from cells expressing the wild-type (1), D714A (2), D714E (3), L717A (4), or L717C (5) enzymes were incubated with trypsin at trypsin/protein ratio 1 : 20 for 0, 0.5, 1, 5, or 10 min.
The expression level of the D714A mutant in secretory vesicles was quite low but measurable (23%; Table 2); however, the enzyme possessed very low ATPase activity (11%), which should be considered even lower by taking into account contamination by plasma membrane (4%, Δpma1; Table 2). This made impossible its further study, in particular kinetics and proton transport. Another mutant (I719A) showed significant threefold decrease in both expression and activity (Table 2), nevertheless allowing study of kinetics of ATP hydrolysis (Table 2) and H+ transport (see below and Table 2). The rest of the mutants (N715A, S716A, D718A, and D720A) were rather well expressed (65-79% of the wild-type level; Table 2) and had significant activity (47-104%) allowing further study.
The kinetics of ATP hydrolysis by most of the active mutant enzymes insignificantly differed from that of the wild type; however, in two cases (N715A and S716A) there was more than twofold increase in affinity to MgATP and sensitivity to the specific inhibitor of P-ATPases orthovanadate (Table 2), which could be explained by a shift in the equilibrium between conformations E1 and E2 [52]. Nonetheless, the changes were rather insufficient to explain these results by a shift in conformational state of the enzyme.
Mutants carrying cysteine substitutions: expression and ATP hydrolysis. The next step was to use Cys-scanning mutagenesis to replace all residues in the L5-6 loop with Cys as was done for the Ala mutations (Table 1). Because Cys residues are highly reactive, using these mutants would probe the enzyme topology and conformational changes during the reaction cycle by selective labeling of the introduced Cys residues with sulfhydryl radioactive, fluorescent, or spin-labeled reagents [43, 44, 55, 56]. As seen from Table 2, the result of amino acid substitutions with Cys as compared to Ala was nearly identical only in the case of the D718C mutant. The N715 mutant showed similar but not identical results: its expression was one third higher compared to the Ala substitution, but the ATPase activity was somewhat lower.
In contrast, cells were even more sensitive to the D714C and L717C than to the D714A and L717A replacements: biogenesis of these Cys mutants was completely blocked during the early stages, preventing mutant proteins from reaching secretory vesicles. Metabolic labeling and limited trypsinolysis revealed that their sensitivity to trypsin was the same as that of the enzymes with Ala replacements: D714C ATPase showed similar to D714A fragments (Fig. 4); the L717C mutant enzyme was cleaved by trypsin within 0.5 min in the same way as was seen for the L717A enzyme (Fig. 4).
Finally, S716C, D720C, and especially I719C were better expressed compared to the Ala substitutions (S716C and D720C by 30% and I719C even by 2.5-fold) and had 70% (S716C and D720C) or even threefold (I719C) higher activity than the Ala replacements (Table 2). In the case of S716C, this could probably be explained by the stereochemical similarity of Ser and Cys residues, which was shown by the resemblance of the parameters for expression, activity, and kinetics obtained for the mutant and the wild-type enzyme.
The kinetics of ATP hydrolysis by the Ala and Cys mutant enzymes in the L5-6 loop differed for five positions: Asn715 and Ser716 (Km), Asp718 (Km and Ki), Ile719 and Asp720 (Ki) (Table 2); however, the changes caused by the Cys point mutations were also insufficient to consider pronounced conformational shifts as in the case of Ala substitutions.
Additional substitutions at Asp714 and Asp720. Besides the Pma1 isoform, S. cerevisiae and some other yeast and fungi have less active and expressed at low level Pma2 isoform of ATPase that is 89% identical to the S. cerevisiae Pma1 [57]; Pma1 and Pma2 are 100% homologous to each other in the L5-6 loops of these isoforms. However, the entire loop identity is lower, accounting for 71%: there are Asn residues at positions of Asp714 and Asp720 (Fig. 3). To better understand the role of L5-6 loop and its single amino acid residues, additional substitutions for Asn and Glu (the latter is found in some ascomycetous sequences, e.g. from Aspergillus flavus) were made at Asp714 and Asp720. It should be noted that of the additional replacements at Asp714, only D714N restored biogenesis and activity of the mutant enzyme (Table 2), while the homologous D714E as seriously impaired the ATPase biogenesis as did D714A or D714C. The conserved substitution D714E exhibited the same sensitivity to trypsin as non-conserved D714A (Fig. 4) and D714C (not shown), pointing to serious impairments of the enzyme folding.
Both conserved replacements of Asp720 (D720N and D720E) increased the enzyme activity almost twofold without changing the expression level as compared to D720A. At the same time, non-conserved D720C had expression 20-40% higher that D720N and D720E without changing the enzyme activity. The kinetics of ATP hydrolysis by D714N was similar to that of the wild type, while substitutions at Asp720 were slightly different from each other and the wild type (Table 2). Once again, D720N showed the least change.
ATP-dependent H+ pumping and its coupling to ATP hydrolysis. Taking into account the contribution of the M5 and M6 segments to the transport pathway and their mobility during the reaction cycle, it was of particular interest to determine if any of the mutations in the L5-6 loop connecting these segments affected H+ pumping and its coupling to ATP hydrolysis. The thirteen enzymatically active mutants were further explored for their ability to carry out ATP-dependent H+ transport. To study this, the ATP-dependent quenching of pH-sensitive fluorescent dye acridine orange was used (Table 2 and Fig. 5). In general, the H+ transport level was equal or only slightly lower than the ATP hydrolysis level comparing Ala and Cys mutants, except for the substitutions at the Asp720 residue where H+ transport level was somewhat lower than the ATP hydrolysis level.
Fig. 5. Acridine orange fluorescence quenching at different concentrations of MgATP as an indicator of H+ transport into secretory vesicles by the wild-type enzyme. The numbers indicate ATP concentration in mM; arrow, addition of MgCl2 at concentrations higher than ATP concentration by 5 mM. Percent of fluorescence quenching (% F) was measured by dividing the difference in fluorescence quenching (ΔF) by its maximal level (Fmax).
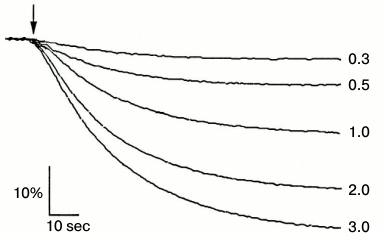
To study H+ transport in more detail and to estimate the enzyme coupling, ATP-dependent quenching was registered at different concentrations of MgATP, while measuring ATP hydrolysis under the same conditions. Since increase in MgATP concentrations changes linearity of fluorescence quenching (Fig. 5) due to interaction between dye, protein, and MgATP [58], the ATP concentrations used did not exceed 2 mM. Then the rate of quenching was plotted as a function of the ATP hydrolysis specific rate, measured in parallel under the same conditions. In every case, there was a roughly linear relationship between the rates of quenching and hydrolysis, as could be expected if the stoichiometry of H+ transport remained constant over the entire range of pump velocities (Fig. 6). The coupling ratio of the wild-type pump was estimated at 1.00, and for most of the mutants this ratio was 0.79-0.94 (Table 2). ATPases with nonconserved substitutions at Asp720 (D720A and D720C) have this coefficient lowered by one third (0.65-0.68; Table 2 and Fig. 6); nonetheless, for the conserved substitutions (D720E and D720N) it was nearly of the wild-type value (Table 2 and Fig. 6).
Fig. 6. Coupling of H+ transport and ATP hydrolysis by the wild-type (1) and mutant enzymes with replacements at Ile719 (a): I719A (2) and I719C (3), and Asp720 (b): D720A (2), D720N (3), and D720C (4). Initial rates of H+ transport were measured under ATP concentrations of 0.2-2.0 mM; then initial rates of H+ transport were plotted as a function of vanadate-sensitive activity of the Pma1 ATPase (ATP hydrolysis) measured under the same conditions. For details, see “Materials and Methods” and legend to Fig. 5.
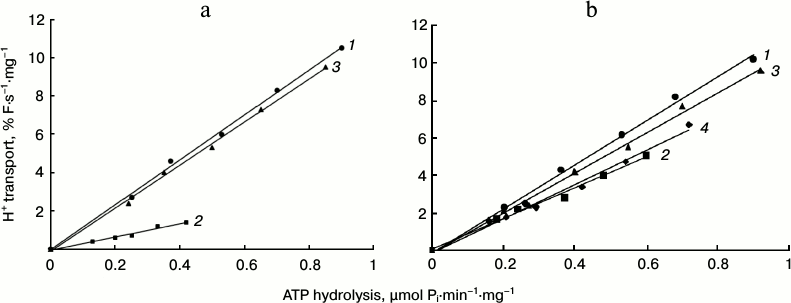
However, the I719A mutant had a ratio significantly lower (0.29) than the wild-type enzyme, which corresponded to three-fold drop in H+ pumping, pointing to a significant but partial uncoupling between ATP hydrolysis and H+ transport (Table 2 and Fig. 6). At the same time, the coupling ratio of the I719C mutant was close to that of the wild type. It should be noted, however, that there is no correlation between ATP hydrolysis and H+ transport level and the coupling ratio. Mutant enzymes with low levels of ATP hydrolysis and H+ transport could be perfectly coupled (L797A mutant in M8 [9]), or vice versa (E803N and E803A in M8 [8, 9]), as well as elevated level of H+ transport (e.g. V723A mutant in M6 segment) is not always evidence for overcoupling of the proton pump [25].
DISCUSSION
Conservation of a residue and structure and functioning of the enzyme. As mentioned above, according to their conservation the L5-6 loop amino acid residues of the yeast S. cerevisiae Pma1 H+-ATPase could be arranged in the following order of homology by aligning them to the published sequences of Saccharomycetes: L717 ≈ S716 → I719 → D714 → D720 → D718 → N715. It may be reasonable to expect a direct correlation between conservation of a residue and the effect of its replacement on the structure, function, and biogenesis of the enzyme. Indeed, substitution of the most conserved residue, Leu717 (38 Leu in 38 amino acid sequences of the Saccharomycetes Pma ATPases; Fig. 3) caused the most dramatic changes in the enzyme expression and activity, resulting in block in biogenesis and loss of activity, while the replacement of the least conserved Asn715 led to the least significant changes (Table 2).
However, conservation of a residue does not always simply predetermine its role in the structure–function organization of the enzyme. Thus, the Ala substitution of Ser716, which is as conserved in Saccharomycetes as Leu717, led to a rather insignificant decrease in expression, ATP-hydrolyzing, and H+-pumping activities by 30-40%, with even lesser effect on the coupling ratio between ATP hydrolysis and H+ pumping; the Cys substitution of this residue was almost indistinguishable from the wild type. Replacement of the next in order of conservation Ile719 (36 Ile/2 Val) caused significant three-fold drop in both expression and ATP-hydrolyzing and H+-pumping activity and also three-fold decrease in coupling ratio, while the Cys substitution did not lead to any significant change. Nevertheless, the replacement of the fourth, middle level of conservation, Asp714 (31 Asn/7 Asp) for Ala, Cys, and even Glu did cause either significant decrease in expression (Asp714→Glu/Ala) with even more pronounced drop in activity or complete block in biogenesis of the mutant enzyme (Asp714→Cys). Changes caused by substitutions of the next residues in the series (Asp720 with similar conservation to Asp714 and, especially, Asp718), were rather small, with the exception of the non-conserved replacement Asp720→Ala, which decreased not only activity but also expression and decreased the enzyme coupling ratio the most markedly of all substitutions at Asp720. Finally, the Ala replacement of the least conserved residue Asn715 lowered expression and activity by a third; for the Cys mutant, the expression was nearly of the wild-type level, while activity decreased by 40%. For both the Ala and Cys mutants, the most important indication of the normal enzyme functioning, the coupling of ATP hydrolysis to H+ transport, was the same and was close to that of the wild type.
It should be also noted that Ala-scanning mutagenesis is considered as a standard approach to replace amino acid residues with less disturbance of the protein structure [59]. It appeared in our case, however, that Cys replacements of three residues, Ser716, Ile719, and Asp718, caused less disturbance of the enzyme structure and function comparing to Ala substitutions (in the case of S716C, evidently, because of the stereochemical similarity of Ser and Cys). In two more cases, for Asn715 and Asp718, the results of the substitution by Ala or Cys were very similar.
Role of charged residues Asp714, Asp718, and Asp720. Besides the results for replacement of Leu717, the most interesting data were found for charged Asp residues in the L5-6 loop – Asp714, Asp718, and Asp720 – that are in the fourth, fifth, and sixth places in the row of conservation. Charged residues are more reactive as compared to neutral, and therefore it was reasonable to expect more pronounced effect of replacing them with neutral Ala or polar Cys. The least conserved of them is Asp718 (20 Asn/16 Asp/1 Glu/1 Ser), which is almost equally represented by Asn and Asp in the majority of the published Saccharomycetes ATPase sequences (Fig. 3). Accordingly, its replacements for Ala or Cys did not cause any essential impairments in the enzyme biogenesis or activity; therefore, its substituting for Asn or Glu as in case of Asp714 and Asp720 and further study were not of interest.
Similarly to Asp718, Asp714 is mostly represented by Asn (31 Asn/7 Asp), but this residue substitution led to completely different results compared to Asp718: all replacements of Asp714 including conservative D714E, except for also conserved D714N, caused complete block in biogenesis or its significant impairment. Results of the replacements of Asp720 (29 Asp/4 Asn/3 Glu/2 His) with homology similar to that of Asp718 were intermediate, although closer to those for the Asp718 replacements. These differences in the consequences of replacements of the same chemically (and close in their order of homology) residues are probably due to their positions in the secondary and tertiary protein structure, in other words, due to their microenvironment and interaction with neighboring residues as well as with lipid and water phases, which may disturb protein structure and stoichiometry of amino acid residues and, respectively, protein folding as proposed [60, 61]. Thus, the first of the aspartate residues, Asp714, is located at the border between hydrophobic and hydrophilic phases; neutral 713-LIAI are next to the residue on the membrane side, and polar 715-NS facing hydrophilic phase are on the cell envelope side (Fig. 1). The next aspartate residue is surrounded by neutral Leu717 and Ile719; polar Ser716 and Asn715 and negatively charged Asp714 and Asp720 are in close proximity to it. Finally, Asp720 is separated from Asp718 by an Ile residue on one side, and on the other side it is located at the border between water and membrane phases, next to membrane-located 721-LIVFI (Figs. 1 and 2), whose substitutions for Ala caused serious impairment of the enzyme functioning [24, 25].
However, substitutions of the Asp718 and Asp720 residues, which should also have changed amino acid residue stoichiometry, led to lesser effects as compared to replacements of Asp714→Ala (Glu/Cys). Such difference might be explained by the idea that in 3D structure Asp714 lies in close proximity to positively charged residues, e.g. Arg320 or Arg323 in the second (L3-4) or Lys781 in the fourth (L7-8) extracytosolic loops (Fig. 1). This should neutralize the charge and stabilize the protein as suggested earlier for Asp739 [25]. Indeed, in a 3D model built for Na+,K+-ATPase, the second and fourth loops seem to be located close to each other [35]. Data obtained for D714E and D714N ruled out a strong ionic bond (salt bridge); however, one might suggest that this could be a weak ionic or a hydrogen bond, since the D714N enzyme properties are almost indistinguishable from the wild type. It should also be taken into consideration that the Pma1 ATPase is an integral transmembrane protein, which interacts with the membrane lipid bilayer; evidently, replacement of even a single residue can cause changes leading to dramatic impairment of its biogenesis and functioning (as in the case of the Asp714 and Leu717 replacements observed in this study). Moreover, consequences of substitution can depend on conditions of expression of mutant ATPases [26, 27] or the chemical nature of a replacement [8, 9, 43, 44].
Indeed, in the case of mutations with impaired biogenesis, heat shock used in this study could have led to change in interaction of ATPase with lipid bilayer due to modification of the bilayer liquidity (viscosity). Such influence of heat shock was earlier observed in the case of mutations I794A, F796A, Q798A, and I799A in the M8 segment; their biogenesis was blocked due to heat shock [9, 26, 27]. However, when the mutated genes carrying I794A or F796A were integrated into the chromosome and expressed in the absence of heat shock, their biogenesis was restored, although not to the wild-type level, but to that being enough for maintaining growth [9, 26, 27]. Results of this study suggest that the use of heat shock was not critical since mutations D714N and D714E being stereochemically close to each other and the wild type led to absolutely different consequences. In contrast, the Q798A mutant mentioned above was expressed neither in secretory vesicles nor in the plasma membrane, while the ATPase carrying the conserved substitution of this residue (Q798E) was expressed in secretory vesicles at the wild-type level [9].
Now comparing the order of the residue conservation (A) with the effect of the replacement on structure and functioning of the enzyme (B) one can see that the new arrangement is different, being significantly distinguishable for residues Asp714 and, especially, Ser716:
(A) L717 ≈ S716 → I719 → D714 → D720 → D718 → N715
(B) L717 → D714 → I719 → D720 → S716 ≈ N715 → D718.
Three-dimensional organization of the enzyme and mechanisms of proton transport. Thus, at least two of the seven residues in the L5-6 loop appeared to be important for the proper biogenesis, structure, and functioning of the yeast Pma1 H+-ATPase. Leu717 is located in the middle of the L5-6 loop; since the enzyme reaction cycle is accompanied by significant conformational changes [5], substitution of this residue with both smaller Ala or more stereochemically similar Cys might seriously affect the mobility of M5 and M6 (and more likely other) transmembrane segments probably causing protein misfolding and thus retaining the impaired enzyme by the quality control points [53, 54].
Asp714 is also important for structure and, especially, functioning of the enzyme; it probably plays a role similar to Asp739 in M6 (Figs. 1 and 2), which was suggested to neutralize neighboring positive charge, thus stabilizing the protein [25]. However, it can be suggested that Asp714 could be directly involved in H+ transport or other aspects of the enzyme functioning.
It is quite remarkable that S. cerevisiae possesses two isoforms of Pma ATPase, Pma1 and Pma2, which are 89% homologous (in L5-6 loop, 100% homologous and 71% identical). The first is functionally active, i.e. Pma1 functioning changes during cell growth in the presence of glucose or other fermented sugars [58, 62-64], while Pma2 is not very active, dormant, and hardly activated by glucose [57]. Moreover, Asp714 in the Pma2 isoform is substituted with Asn743 (Fig. 3); of all Asp714 replacements, only D714N yielded active enzyme similar to the wild type. Substitutions of the other Asp residue, Asp720, did not lead to such dramatic results; however, D720N and not D720E was characteristically closest to the wild type. As in the case of Asp714, Pma2 has Asn749 instead of Asp720. Therefore, the presence of charged aspartate or polar asparagine residue might influence the ability of the enzyme to respond to changes in growth conditions, causing its activation during fermentation of glucose or transforming into an inactive state under starvation [58, 62, 64]. Changes in the enzyme functioning also occurred when Glu803 in the M8 segment was substituted with Gln; this changed stoichiometry of transport (H+/ATP) and resulted in increasing the amount of transported H+ (H3O+) more than twofold [8, 9, 27].
Of the published amino acid sequences of Pma ATPases, Pma2 isoforms were found so far only in S. cerevisiae, C. glabrata, T. blattae, and A. gossypii (Fig. 3). Amino acid sequences of the L5-6 loops of both isoforms in the last three sequences were 100% identical, only the residue numbers in the sequences were different. There are Asn residues in the place of the S. cerevisiae Asp714 in these sequences, and there are no data about “glucose” activation of the Pma ATPases of these species yet, which supports our suggestion about possible involvement of the L5-6 loop (mostly due to residue Asp714) in enzyme activation during glucose fermentation. Earlier, we suggested similar involvement in the enzyme regulation for the L9-10 loop, which is in close proximity to the regulatory C-terminal end of the Pma1 H+-ATPase [65, 66]. Besides the theoretical implication, this conclusion could be important for many biotechnological processes whose efficiency depends on the plasma membrane H+-ATPase functioning.
Finally, Ile719 replacement by Ala caused significant impairment in the enzyme coupling along with significant decrease in enzyme expression and activity; the milder effect in comparison with L717A is probably explained by the location of this residue, which is situated between Asp718 and Asp720. Substitution of Leu for smaller Ala changes the enzyme three-dimensional structure; this conclusion is supported by the data obtained for substitution of this residue by polar but closer in size Cys.
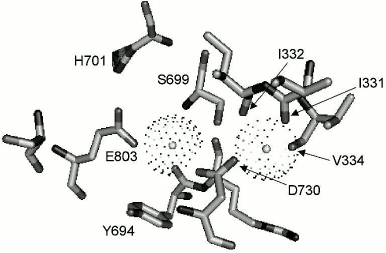
Along with membrane segments M4 and M8, M5 and M6 form cation-binding sites in the sarcoplasmic reticulum Ca2+-ATPase and other P2-ATPases. There are two such sites in SERCA Ca2+-ATPase; they are formed by amino acid residues in the M4, M5, M6, and M8 segments. Two amino acid residues forming Ca2+-binding and transporting sites are found in M5: Asn768, which corresponds to Ser699 in Pma1, and Glu771, which corresponds to Glu703; both are parts of site I. In M6, Thr799 (site I), Asn796 (site II), and Asp800 (both sites) appeared to be such residues [4, 5, 15-17]. In Pma1 ATPase, these residues correspond to Ala729, Ala726, and Asp730. Site-directed mutagenesis earlier revealed that substitutions at residue Glu703 (E703Q, E703L [8]) and Ala726 (A726S [24, 25]) led to partial uncoupling between ATP hydrolysis and H+ transport, while substitutions of Asp730 (D730N and D730V) blocked the enzyme biogenesis, and only the conserved replacement D730E partly restored biogenesis (48% expression as compared to the wild type; however, the enzyme activity was very low) [8]. Substitution S699A did not block biogenesis but did lead to loss of activity [23]. In the sarcoplasmic reticulum Ca2+-ATPase M8 segment, Glu908 (site I) corresponding to Glu803 of the Pma1 ATPase [8, 9] is a residue that is involved in forming the Ca2+-transporting site. In the case of Glu803, none of the substitutions completely blocked trafficking of de novo synthesized mutant ATPases (however, E803S and E803L forms possessed only 8% of the wild-type expression level); nevertheless, these enzymes appeared to be either inactive or had significantly changed coupling between H+ transport and ATP hydrolysis – from almost uncoupled enzymes (E803N, E803A) to more than twofold overcoupling (E803Q) [8, 9, 27]. Finally, in the SERCA Ca2+-ATPase M4 segment, residues Val304, Ala305, Ile307, and Glu309 participate in arrangement of site II; in Pma1 ATPase, these residues correspond to Ile331, Ile332, Val334, and Val336. Thus, not only charged carboxylic and polar carbonyl or amide group of side chains, but also carbonyl groups of the backbone may participate in cation binding and transporting.
The structural model of the Pma1 ATPase H+-transporting sites (Fig. 7) was built by homology modeling using the crystal structure of the plant H+-ATPase [14] and the most trustworthy 3D model of Ca2+-ATPase [5]. The crystal structure of the plant H+-ATPase has a cavity of 380 Å3 [14] with capacity for 12 water molecules, which suggests that it has space for at least two hydrated molecules of hydroxonium [25]. Comparative homology modeling using Ca2+-ATPase 3D structure also shows that the yeast H+ pump has two hydroxonium binding sites [25] and hydroxonium is the ion species that is transported [8, 9, 25]. Despite the opinion that fungal and plant H+-ATPases possess only one H+ (hydroxonium ion H3O+) binding site, the results suggest that these enzymes have two such sites similar to the animal Ca2+-ATPase (Fig. 7), one of which is evidently appeared to be dormant and became active only as a result of mutation [8, 9] or under condition of chronic energy deficiency also caused by mutation [7], and the transported species is the hydrated hydroxonium ion [8, 9, 25, 27] similar to hydrated cations of calcium in Ca2+- or potassium and sodium in Na+,K+-ATPases of animal cells. In Na+,K+-ATPase, the L5-6 loop in involved in regulating the access to the cation-transporting sites [33]. Such involvement could also be suggested for the Pma1 H+-ATPase, since substitutions of the two loop residues (Asp714 and Leu717) cause serious impairment of structure–functional organization of the enzyme, and in the case of Ile719 it leads to partial uncoupling between ATP hydrolysis and proton transport. The presented data also have some practical interest since there is a residue in the L5-6 loop of the H+,K+-ATPase of animal gastric cells that binds omeprazole and similar drugs for ulcer treatment [37], while the Cys substitutions obtained in this work allows continuing the study of the yeast Pma1 ATPase structure–functional relationships and regulation of this enzyme.
Fig. 7. Sites of H+ binding and transport. Residues Ile331, Ile332, and Val334 in M4 segment of the Pma1 ATPase correspond to Val304, Ala305, and Ile307 of SERCA Ca2+-ATPase, Ser699 in M5 segment corresponds to Asn768, Asp730 in M6 segment corresponds to Asp800, and Glu803 in M8 segment corresponds to Glu908, which form sites I and II of the sarcoplasmic reticulum SERCA Ca2+-ATPase. Coordinated hydrated H+ (H3O+) ions are shown as circles in spheres. The homological model was built using the crystal structure of Ca2+-ATPase as described in [9, 25]. Modified version from [25].
The author is grateful to Prof. C. W. Slayman (Yale University School of Medicine) who was a scientific adviser for this project, Dr. M. Miranda for constructing some of the mutants, Dr. J. P. Pardo for computer modeling, and K. E. Allen for expert technical assistance.
This work was supported in part by the Russian Foundation for Basic Research, research project No. 12-08-01157-a.
REFERENCES
1.Serrano, R., Kielland-Brandt, M. C., and Fink, G.
R. (1986) Yeast plasma membrane ATPase is essential for growth and has
homology with (Na+,K+)K+- and
Ca2+-ATPases, Nature, 319, 689-693.
2.Lutsenko, S., and Kaplan, J. H. (1995) Organization
of P-type ATPases: significance of structural diversity,
Biochemistry, 34, 15607-15613.
3.Axelsen, K. B., and Palmgren, M. G. (1998)
Evolution of substrate specificities in the P-type ATPase superfamily,
J. Mol. Evol., 46, 84-101.
4.Toyosima, C., Nakasako, M., Nomura, H., and Ogawa,
H. (2000) Crystal structure of the calcium pump of sarcoplasmic
reticulum at 2.6 Å resolution, Nature, 405,
647-655.
5.Toyosima, C., and Nomura, H. (2002) Structural
changes in the calcium pump accompanying the dissociation of calcium,
Nature, 418, 605-611.
6.Goffeau, A., and Slayman, C. W. (1981) The
proton-translocating ATPase of the fungal plasma membrane, Biochim.
Biophys. Acta, 639, 197-223.
7.Warnke, J., and Slayman, C. L. (1980) Metabolic
modulation of stoichiometry in a proton pump, Biochim. Biophys.
Acta, 591, 224-233.
8.Petrov, V. V., Padmanabha, K. P., Nakamoto, R. K.,
Allen, K. E., and Slayman, C. W. (2000) Functional role of charged
residues in the transmembrane segments of the yeast plasma membrane
H+-ATPase, J. Biol. Chem., 275,
15709-15716.
9.Guerra, G., Petrov, V. V., Allen, K. E., Miranda,
M., Pardo, J. P., and Slayman, C. W. (2007) Role of transmembrane
segment M8 in the biogenesis and function of yeast plasma-membrane
H+-ATPase, Biochim. Biophys. Acta,
1768, 2383-2392.
10.Appel, H. J. (2004) How do P-type ATPases
transport ions? Bioelectrochemistry, 63,
149-156.
11.Morth, J. P., Pedersen, B. P., Toustrup-Jensen,
M. S., Sorensen, T. L., Petersen, J., Andersen, J. P., Vilsen, B., and
Nissen, P. (2007) Crystal structure of the sodium-potassium pump,
Nature, 450, 1043-1049.
12.Shinoda, T., Ogawa, H., Cornelius, F., and
Toyosima, C. (2009) Crystal structure of the sodium-potassium pump at
2.4 Å resolution, Nature, 459, 446-450.
13.Auer, M., Scarborough, G. A., and Kuhlbrandt, W.
(1998) Three-dimensional map of the plasma membrane
H+-ATPase in the open conformation, Nature,
392, 840-843.
14.Pedersen, B. P., Buch-Pedersen, M., Morth, J. J.
P., Palmgren, M. G., and Nissen, P. (2007) Crystal structure of the
plasma membrane proton pump, Nature, 450,
1111-1114.
15.Andersen, J. P., and Vilsen, B. (1994) Amino
acids Asn796 and Thr799 of the Ca2+-ATPase of sarcoplasmic
reticulum bind Ca2+ at different sites, J. Biol.
Chem., 269, 15931-15936.
16.Rice, W. J., and MacLennan, D. H. (1996) Scanning
mutagenesis reveals a similar pattern of mutation sensitivity in
transmembrane sequences M4, M5, and M6, but not in M8, of the
Ca2+-ATPase of sarcoplasmic reticulum (SERCA1a), J. Biol.
Chem., 271, 31412-31419.
17.Zhang, Z., Lewis, D., Strock, C., Inesi, G.,
Nakasako, M., Nomura, H., and Toyoshima, C. (2000) Detailed
characterization of the cooperative mechanism of Ca2+
binding and catalytic activation in the Ca2+ transport
(SERCA) ATPase, Biochemistry, 39, 8758-8767.
18.Vilsen, B., and Andersen, J. P. (1998) Mutation
to the glutamate in the fourth membrane segment of
Na+,K+-ATPase and Ca2+-ATPase affects
cation binding from both sides of the membrane and destabilizes the
occluded enzyme forms, Biochemistry, 37,
10961-10971.
19.Jewell-Motz, E. A., and Lingrel, J. B. (1993)
Site-directed mutagenesis of the Na,K-ATPase: consequences of
substitutions of negatively-charged amino acids localized in the
transmembrane domains, Biochemistry, 32,
13523-13530.
20.Kuntzweiler, T. A., Arguello, J. M., and Lingrel,
J. B. (1996) Asp804 and Asp808 in the transmembrane domain of the
Na,K-ATPase α subunit are cation coordinating residues, J.
Biol. Chem., 271, 29682-29687.
21.Nielsen, J. M., Pedersen, P. A., Karlish, S. J.
D., and Jorgensen, P. L. (1998) Importance of intramembrane carboxylic
acids for occlusion of K+ ions at equilibrium in renal
Na,K-ATPase, Biochemistry, 37, 1961-1968.
22.Ambesi, A., Pan, R. L., and Slayman, C. W. (1996)
Alanine-scanning mutagenesis along membrane segment 4 of the yeast
plasma membrane H+-ATPase. Effects on structure and
function, J. Biol. Chem., 271, 22999-23005.
23.Dutra, M. B., Ambesi, A., and Slayman, C. W.
(1998) Structure-function relationships in membrane segment 5 of the
yeast Pma1 H+-ATPase, J. Biol. Chem., 273,
17411-17417.
24.Miranda-Arango, M., Pardo, J. P., and Petrov, V.
V. (2009) Role of transmembrane segment M6 in the biogenesis and
function of the yeast Pma1 H+-ATPase, J. Biomol. Struct.
Dyn., 26, 866-868.
25.Miranda, M., Pardo, J. P., and Petrov, V. V.
(2011) Structure-function relationships in membrane segment 6 of the
yeast plasma membrane Pma1 H+-ATPase, Biochim. Biophys.
Acta, 1808, 1781-1789.
26.Petrov, V. V. (2009) Heat shock affects
functioning of the yeast Pma1 H+-ATPase, J. Biomol.
Struct. Dyn., 26, 857-858.
27.Petrov, V. V. (2010) Point mutations in Pma1
H+-ATPase of Saccharomyces cerevisiae: influence on
its expression and activity, Biochemistry (Moscow),
75, 1055-1063.
28.Wei, Y., Chen, J., Rosas, G., Tompkins, D. A.,
Holt, P. A., and Rao, R. (2000) Phenotypic screening of mutations in
Pmr1, the yeast secretory pathway
Ca2+/Mn2+-ATPase, reveals residues critical for
ion selectivity and transport, J. Biol. Chem., 275,
23927-23932.
29.Mandal, D., Woolf, T. B., and Rao, R. (2000)
Manganese selectivity of pmr1, the yeast secretory pathway ion pump, is
defined by residue Gln783 in transmembrane segment 6. Residue Asp778 is
essential for cation transport, J. Biol. Chem., 275,
23933-23938.
30.MacLennan, D. H., Rice, W. J., and Green, N. M.
(1997) The mechanism of Ca2+ transport by sarco(endo)plasmic
reticulum Ca2+-ATPases, J. Biol. Chem., 272,
28815-28818.
31.Morsomme, P., Slayman, C. W., and Goffeau, A.
(2000) Mutagenic study of the structure, function and biogenesis of the
yeast plasma membrane H+-ATPase, Biochim. Biophys.
Acta, 1469, 133-157.
32.Jorgensen, P. L., Hakansson, K. O., and Karlish,
S. J. (2003) Structure and mechanism of Na,K-ATPase: functional sites
and their interactions, Annu. Rev. Physiol., 65,
817-849.
33.Capendeguy, O., and Horisberger, J.-D. (2005) The
role of the third extracellular loop of the
Na+,K+-ATPase α-subunit in a luminal gating
mechanism, J. Physiol., 565, 207-218.
34.Capendeguy, O., Chodanovski, P., Michielin, O.,
and Horisberger, J.-D. (2006) Access of extracellular cations to their
binding sites in Na,K-ATPase: role of the second extracellular loop of
the α-subunit, J. Gen. Physiol., 127,
341-352.
35.Capendeguy, O., Iwaszkiewicz, J., Michielin, O.,
and Horisberger, J.-D. (2008) The fourth extracellular loop of the
α-subunit of Na,K-ATPase: functional evidence for close proximity
with the second extracellular loop, J. Biol. Chem.,
283, 27850-27858.
36.Munson, K. B., Gutierrez, C., Balaji, V. N.,
Ramnarayan, K., and Sachs, G. (1991) Identification of an
extracytoplasmic region of H+,K+-ATPase labeled
by a K+-competitive photoaffinity inhibitor, J. Biol.
Chem., 266, 18976-18988.
37.Besancon, M., Simon, A., Sachs, G., and Shin, J.
M. (1997) Sites of reaction of the gastric H,K-ATPase with
extracytoplasmic thiol reagents, J. Biol. Chem., 272,
22438-22446.
38.Seto-Young, D., Na, S., Monk, B. C., Haber, J.
E., and Perlin, D. S. (1994) Mutational analysis of the first
extracellular loop region of the H+-ATPase from
Saccharomyces cerevisiae, J. Biol. Chem., 269,
23988-23995.
39.Lutsenko, S., Anderko, R., and Kaplan, J. H.
(1995) Membrane disposition of the M5-M6 hairpin of
Na+,K+-ATPase alpha subunit is ligand dependent,
Proc. Natl. Acad. Sci. USA, 92, 7936-7940.
40.Gatto, C., Lutsenko, S., Shin, J. M., Sachs, G.,
and Kaplan, J. H. (1999) Stabilization of the H,K-ATPase M5M6 membrane
hairpin by K+ ions. Mechanistic significance for
P2-type ATPases, J. Biol. Chem., 274,
13737-13740.
41.Mikhailova, L., Mandal, A. K., and Arguello, J.
M. (2002) Catalytic phosphorylation of Na,K-ATPase drives the outward
movement of its cation-binding H5-H6 hairpin, Biochemistry,
41, 8195-8202.
42.Nakamoto, R. K., Rao, R., and Slayman, C. W.
(1991) Expression of the yeast plasma membrane H+-ATPase in
secretory vesicles. A new strategy for directed mutagenesis, J.
Biol. Chem., 266, 7940-7949.
43.Petrov, V. V., and Slayman, C. W. (1995)
Site-directed mutagenesis of the yeast PMA1 H+-ATPase.
Structural and functional role of cysteine residues, J. Biol.
Chem., 270, 28535-28540.
44.Petrov, V. V. (2009). Functioning of
Saccharomyces cerevisiae Pma1 H+-ATPase carrying the
minimal number of cysteine residues, Biochemistry (Moscow),
74, 1155-1163.
45.Hager, K. M., Mandala, S. M., Davenport, J. W.,
Speicher, D. W., Benz, E. J., Jr., and Slayman, C. W. (1986) Amino acid
sequence of the plasma membrane ATPase of Neurospora crassa:
deduction from genomic and cDNA sequences, Proc. Natl. Acad. Sci.
USA, 83, 7693-7697.
46.Nakamoto, R. K., Verjovski-Almeida, S., Allen, K.
E., Ambesi, A., Rao, R., and Slayman, C. W. (1998) Substitutions of
aspartate 378 in the phosphorylation domain of the yeast PMA1
H+-ATPase disrupt protein folding and biogenesis, J.
Biol. Chem., 273, 7338-7344.
47.Fabiato, A., and Fabiato, F. (1979) Calculator
programs for computing the composition of the solutions containing
multiple metals and ligands used for experiments in skinned muscle
cells, J. Physiol. (Paris), 75, 463-505.
48.Fiske, C. H., and Subbarow, Y. (1925) The
colorimetric determination of phosphorus, J. Biol. Chem.,
66, 375-400.
49.Bensadoun, A., and Weinstein, D. (1976) Assay of
proteins in the presence of interfering materials, Anal.
Biochem., 70, 241-250.
50.Kyte, J., and Doolittle, R. F. (1982) A simple
method for displaying the hydropathic character of a protein, J.
Mol. Biol., 157, 105-132.
51.Serrano, R. (1988) Structure and function of
proton translocating ATPase in plasma membranes of plants and fungi,
Biochim. Biophys. Acta, 947, 1-28.
52.Ambesi, A., Miranda, M., Petrov, V. V., and
Slayman, C. W. (2000) Biogenesis and function of the yeast
plasma-membrane H+-ATPase, J. Exp. Biol., 203,
156-160.
53.Ferreira, T., Mason, A. B., Pypaert, M., Allen,
K. E., and Slayman, C. W. (2002) Quality control in the yeast secretory
pathway: a misfolded PMA1 H+-ATPase reveals two checkpoints,
J. Biol. Chem., 277, 21027-21040.
54.Mason, A. B., Allen, K. E., and Slayman, C. W.
(2014) C-terminal truncations of the Saccharomyces
cerevisiae PMA1 H+-ATPase have major impacts on protein
trafficking, quality control, and function, Eukaryot. Cell,
13, 43-52.
55.Petrov, V. V., Pardo, J. P., and Slayman, C. W.
(1997) Reactive cysteines of the yeast plasma membrane
H+-ATPase (PMA1): mapping the sites of inactivation by
N-ethylmaleimide, J. Biol. Chem., 277, 1688-1693.
56.Petrov, V. V. (2012) Cysteine residues of the
yeast Pma1 H+-ATPase: structural and functional role, in
Cysteine: Biosynthesis, Chemical Structure and Toxicity
(Chorkina, F. V., and Karataev, A. I., eds.) Nova Science Publishers,
Hauppauge, N.Y., pp. 31-60.
57.Supply, P., Wach, A., and Goffeau, A. (1993)
Enzymatic properties of the PMA2 plasma membrane-bound
H+-ATPase of Saccharomyces cerevisiae, J. Biol.
Chem., 268, 19753-19759.
58.Petrov, V. V. (1987) Investigation of Ion and
Metabolite Transport into Yeast Saccharomyces carlsbergensis Plasma
Membrane Vesicles: Ph. D. Thesis [in Russian], Pushchino.
59.Morrison, K. L., and Weiss, G. A. (2001)
Combinatorial alanine-scanning, Curr. Opin. Chem. Biol.,
5, 302-307.
60.Mittal, A., Jayaram, B., Shenoy, S., and Bawa, T.
S. (2010) A stoichiometry driven universal spatial organization of
backbones of folded proteins: are there Chargaff’s rules for
protein folding? J. Biomol. Struct. Dyn., 28,
133-142.
61.Mittal, A., and Jayaram, B. (2011) Backbones of
folded proteins reveal novel invariant amino acid neighborhoods, J.
Biomol. Struct. Dyn., 28, 443-454.
62.Serrano, R. (1983) In vivo glucose
activation of the yeast plasma membrane ATPase, FEBS Lett.,
156, 11-14.
63.Sychrova, H., and Kotyk, A. (1985) Condition of
activation of the yeast plasma membrane ATPase, FEBS Lett.,
183, 21-24.
64.Okorokov, L. A., and Petrov, V. V. (1986)
Isolation of plasma membrane vesicles from the yeast Saccharomyces
carlsbergensis suitable for solute transport studies, Biol.
Membr. (Moscow), 3, 549-556.
65.Tomashevsky, A. A., and Petrov, V. V. (2011)
Point mutation in M9-M10 loop of the yeast Pma1 H+-ATPase
affects both ATPase functioning and polyphosphate (PolyP) distribution,
J. Biomol. Struct. Dyn., 28, 1025-1026.
66.Tomashevski, A. A., and Petrov, V. V. (2013)
Point mutations in the yeast Pma1 H+-ATPase affect
polyphosphate (PolyP) distribution, J. Biomol. Struct. Dyn.,
31, 123-124.