Effect of Trehalose on Oxygen Evolution and Electron Transfer in Photosystem 2 Complexes
M. D. Mamedov1,2*, I. O. Petrova1, D. V. Yanykin2, A. A. Zaspa1, and A. Yu. Semenov1,3
1Belozersky Institute of Physical-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: +7 (495) 939-3181; E-mail: mahirmamedov@yandex.ru2Institute of Basic Biological Problems, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: +7 (496) 733-0532; E-mail: ya-d-ozh@rambler.ru
3Semenov Institute of Chemical Physics, Russian Academy of Sciences, ul. Kosygina 4, 117977 Moscow, Russia; fax: +7 (495) 651-2191; E-mail: semenov@genebee.msu.ru
* To whom correspondence should be addressed.
Received July 14, 2014; Revision received September 2, 2014
The pigment–protein complex of photosystem 2 (PS 2) catalyzes the light-driven oxidation of water molecule and the reduction of plastoquinone. In this work, we studied the effect of the disaccharide trehalose, which is unique in its physicochemical properties, on isolated PS 2 complex. It was found that trehalose significantly stimulated the steady-state rate of oxygen evolution. The study of single flash-induced fluorescence decay kinetics demonstrated that trehalose did not affect the rate of QA– oxidation, although it led to an increase in the relative fractions of PS 2 reaction centers capable of QA– oxidation. Trehalose also prevented PS 2 complexes from being inactivated on prolonged storage. We propose that in the presence of trehalose, which affects the extent of hydration, the protein can preferentially exist in a more optimal conformation for effective functioning.
KEY WORDS: photosystem 2, water-oxidizing complex, oxygen evolution, trehalose, chlorophyll fluorescence, plastoquinone oxidationDOI: 10.1134/S0006297915010071
Abbreviations: DCBQ, 2,6-dichloro-p-benzoquinone; DCMU, 3-(3,4-dichlorophenyl)-1,1-dimethylurea; P680, primary electron donor; PS 2, photosystem 2; QA and QB, primary and secondary quinone acceptors; RC, reaction center; WOC, water-oxidizing complex; YZ, redox-active tyrosine 161 of D1 protein.
The major source of energy in the biosphere is sunlight. However, only
oxygenic photosynthetic organisms are able to convert the energy of
photons emitted by the Sun into the energy of chemical bonds of
carbohydrates by splitting water into molecular oxygen and reducing
equivalents (electrons and protons). The pigment–protein complex
of photosystem 2 (PS 2) embedded into the thylakoid membranes of
cyanobacteria and chloroplasts functions as a light-driven
water-plastoquinone oxidoreductase [1, 2]. All the organic and inorganic redox-active
cofactors involved in charge transfer reactions are believed to reside
on the D1 and D2 subunits of the reaction center (RC). A
Mn4CaO5 cluster together with its coordinating
amino acids and four water molecules form a catalytic site where
oxidation of water molecules occurs [3-7]. It should be noted that the water-oxidizing
complex (WOC), which is located on the donor side of the enzyme, is the
most fragile site within PS 2 and is easily susceptible to oxidative
damage.
The use of osmolytes in the stabilization of biomolecules is an old trick of Nature ([8-13] and references therein). In the presence of these small molecules, photosynthetic organisms counteract various stress conditions that they encounter. The osmolytes range from sugars to polyols, amino acids and their derivatives, and so forth. Among these, in recent years trehalose, which is naturally produced by several species of eubacteria, archaea, some fungi, certain invertebrates, and lower plants, has received considerable attention [14, 15].
The specific physical and chemical characteristics of trehalose such as relative inertness of the glycosidic linkage, the existence of both crystalline and amorphous states, thermostability (melting point, 203°C), high glass transition temperature, high stability over a wide pH range (3.5-10.0), and high hydrophilicity (water solubility, 68 g/100 ml) set it apart from other osmolytes such as sucrose or glycerol.
The protective effect of trehalose on the photosynthetic apparatus under abiotic stresses (against dehydration, high salinity, and heat stress) has been demonstrated in transgenic plants ([16, 17] and references therein). Earlier, it was shown that trehalose prevents the release of soluble plastocyanin from thylakoid membranes during freeze–thaw treatment [18]. It was also shown that trehalose protected the chloroplast membranes and PS 2 particles during long and short freezing times [19]. It should be noted that the coupling between electron transfer and protein dynamics has been investigated in detail in photosynthetic bacterial reaction centers incorporated into glassy trehalose matrices [10, 20]. The data suggest that the conformational change stabilizing charge separation between the primary donor (P870+) and the reduced primary quinone acceptor (QA−) is localized around the QA binding pocket.
In the present work, we studied the effect of trehalose on electron transfer reactions within PS 2 particles from spinach. The data show that this disaccharide significantly stimulates the activity of the water-oxidizing complex of PS 2 and prevents time-dependent inactivation of the protein.
MATERIALS AND METHODS
Oxygen-evolving PS 2 core complexes and PS 2 membrane fragments from spinach were prepared as described in [21] and [22], respectively.
The rate of oxygen evolution under continuous illumination (1000 µmol photon·m–2·s–1) was measured at 25°C using a Clark-type oxygen electrode in medium containing 25 mM MES-NaOH (pH 6.5), 15 mM NaCl, 10 mM CaCl2, 1 mM K3[Fe(CN)6], and 0.1 mM 2,6-dichloro-p-benzoquinone (DCBQ) unless otherwise stated.
Fluorescence transients of PS 2 core particles were measured using an FL3000 fluorometer (Photon Systems Instruments, Czech Republic). The sample was illuminated by continuous light at 625 nm with the intensity at the surface of the cuvette of 2000 µmol photon·m–2·s–1.
To monitor the fluorescence decay after a single saturating flash, actinic and the measuring light pulses were produced by red light-emitting diodes. The duration of the flash was 10 µs. Samples were assayed in medium containing 25 mM MES-NaOH (pH 6.5), 15 mM NaCl, and 10 mM CaCl2 in the absence and in the presence of 1 M trehalose. All samples were dark incubated for 5 min prior to measurement.
The degree of the protein degradation was determined by the pellet content.
Kinetic signals were analyzed using the Origin program package (OriginLab Corporation, USA).
RESULTS AND DISCUSSION
The steady-state oxygen evolving activity of PS 2 complexes was monitored in the presence of trehalose (Fig. 1). The rate of oxygen evolution increased from 825 (in control samples) up to 2060 µmol O2·(mg Chl·h)–1 in the presence of 1 M trehalose in PS 2 core complexes (Fig. 1a). In the case of membrane fragments, the rate of oxygen evolution increased from 740 to 1340 µmol O2·(mg Chl·h)–1, correspondingly (Fig. 1b).
Fig. 1. Effect of trehalose on oxygen evolution in PS 2 core complexes (a) and PS 2 membrane fragments (b) in medium containing 25 mM Mes (pH 6.5), 15 mM NaCl, and 10 mM CaCl2 in the absence (1) or presence of 0.5 M (2) or 1 M (3) trehalose. K3[Fe(CN)6] (1 mM) and DCBQ (0.1 mM) were used as electron acceptors. The concentration of chlorophyll in the samples was 2 µg/ml. The rate of oxygen evolution (µmol O2·(mg Chl·h)–1)) in PS 2 core particles: 1) 825; 2) 1500; 3) 2060; in membrane fragments: 1) 740; 2) 1030; 3) 1340. ↓ and ↑ designate light (λ = 650 nm, 1000 µmol photon·m–2·s–1) on and off, respectively.
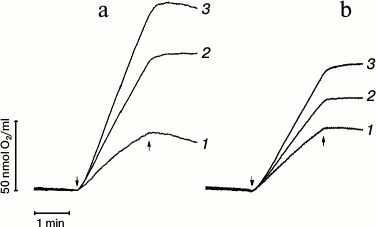
After 2 min of illumination of PS 2 core complexes in the absence of trehalose, the rate of oxygen evolution decreased ~35% in comparison with the initial rate (Fig. 1a, trace 1). In so doing, the decrease in the rate of O2 evolution was ~10% and ~7.5% in the presence of 0.5 M (trace 2) and 1 M trehalose (trace 3), respectively. Similar analysis of the rate of O2 evolution kinetics in case of membrane fragments (Fig. 1b, trace 1) showed that in the absence of trehalose the rate of oxygen evolution in PS 2 cores after 2 min illumination of the samples decreased by ~15% in comparison with the initial rate. After 2 min illumination, but in the presence of 0.5 M trehalose (Fig. 1b, trace 2), the rate of O2 evolution decreased by ~5%, while there was no decrease in the presence of 1 M trehalose (Fig. 1b, trace 3).
These data indicate that trehalose stabilizes the water-oxidizing complex. It should be noted that further increase in trehalose concentration was not possible due to its limited solubility at room temperature [23].
For further investigations of the effect of trehalose, only PS 2 core complexes were used. These purified complexes transfer electrons from water to the terminal quinone acceptor as well as evolve oxygen. It should be noted that in the presence of 5 µM 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU) the rate of O2 evolution was largely diminished (~75% inhibition) (data not shown).
The fluorescence transients of intact PS 2 core particles, just like in the case of PS 2 membrane fragments [24-28], are characterized by a biphasic (OJ and JP) kinetic pattern. In general, the initial OJ (photochemical) phase reflects the extent of reduction of the primary quinone acceptor QA, whereas the JP phase kinetics and amplitude are a measure of the rate and extent of the reduction of the terminal plastoquinone QB at the PS 2 acceptor side.
Figure 2 shows the fluorescence induction kinetics of dark-adapted PS 2 core particles in the absence (trace 1) and in the presence of 1 M trehalose (trace 2). Addition of trehalose increased the JP-phase amplitude, which is related to an increase in PS 2 RCs capable of reducing the terminal quinone acceptor.
Fig. 2. Fluorescence transients of PS 2 core particles in the absence (1) and presence (2) of 1 M trehalose. Storage of dark-adapted samples during 30 days at 4°C in the absence (3) and in the presence (4) of 1 M trehalose. The sample, in a 2 ml cuvette, was illuminated by continuous light at 625 nm with intensity at the surface of the cuvette 2000 µmol photon·m–2·s–1. The concentration of chlorophyll in the samples was 10 µg/ml. The assay medium as in Fig. 1.
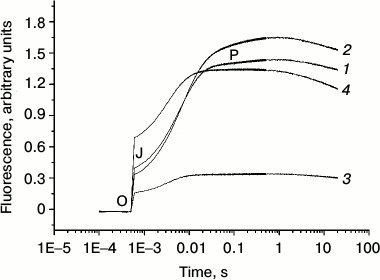
We also investigated the effect of trehalose on the stability of electron transfer within PS 2 complex. Figure 2 demonstrates that 30-day storage of PS 2 core complexes at 4°C resulted in a significant (~2.3-fold) decrease in the OJ and more dramatic (~6-fold) decrease in the JP phases (the JP/OJ ratio diminishes by a factor of ~2.5) (cf. traces 1 and 3). The decrease in the amplitude in the OJ phase is due to lessening of the number of centers capable of QA reduction because of protein inactivation. The more pronounced decrease in the JP phase indicates, at first glance, that the acceptor side of the complex is more sensitive to the storage than the donor side. However, a similar decrease in the OJ phase and the JP/OJ ratio means that the latter parameter directly correlates with the number of centers with functional WOC.
The storage of PS 2 complexes in the presence of trehalose at 4°C for 30 days led to a significant increase in both the OJ and JP phases (4- and 3.7-fold, respectively) compared to the samples in the absence of trehalose stored under the same conditions (cf. traces 3 and 4). It should be noted that under these conditions in the presence of trehalose degradation of proteins was not observed (not shown). However, the JP/OJ ratio remained similar in the absence and in the presence of trehalose (it decreased by less than ~10%). This result indicates that trehalose equally prevents damage to the donor and the acceptor sides of electron transfer in PS 2. Note also that almost 2-fold increase in the OJ phase amplitude after 30-day storage in the presence of trehalose (trace 4) compared to the control (signal after 5 min dark adaptation in the absence of trehalose) (trace 1) means that the fractions of PS 2 RCs capable of QA reduction increases after storage. In fact, the time-dependent degradation of the PS 2 complex results in the extraction of the Mn ions from its binding sites and the corresponding shift of the redox-potential of QA/QA– from low- to high-potential form, which leads to a decrease in the equilibrium constant of electron transfer from QA− to QB [29]. However, storage of samples for 30 days in the presence of trehalose retained oxygen-evolving activity only in ~20% of centers in comparison with control samples. On the other hand, in addition to a stabilizing role of trehalose at the level of the WOC, its mode of action could also include direct protection and/or stabilization of the RC itself. The maintenance of similar JP/OJ ratio suggests that the extent of QA− oxidation by QB depends mainly on the number of centers with functional RC. Thus, we suggest that storage of PS 2 in the presence of trehalose partially prevents the inactivation of the WOC, but it mostly stabilizes the RC structure. Earlier, it was shown that addition of glycinebetaine to RC complex (D1/D2/Cyt b559) protects the capacity for reduction of Cyt b559 from Pheo– and subsequent re-oxidation of Cyt b559 by P680+, i.e. cyclic electron flow around PS 2 against photo- and heat-induced inactivation [30]. We propose a similar mechanism for the effect of trehalose on stabilization of PS 2 core complexes during storage of samples.
Redox reactions of quinone acceptors in PS 2 were almost exclusively characterized by measurements of chlorophyll fluorescence decay after single turnover light flashes [31-37]. In dark-adapted samples, the reduction of the primary quinone acceptor QA after a single flash gives rise to an increased fluorescence from PS 2, which decays as QA− is reoxidized [34-37]. In the present work, we measured the reoxidation of QA− to study the kinetics of electron transfer from QA− to the secondary electron quinone acceptor QB in the absence (curve 1) and in the presence (curve 2) of 1 M trehalose (Fig. 3). In PS 2 complex, QA− can be reoxidized by two pathways: due to forward electron transfer to QB and due to recombination with the oxidized components on the donor side.
Fig. 3. Effect of trehalose on the kinetics of QA− reoxidation after a single saturating flash to dark-adapted PS 2 core complexes. The decay of Chl fluorescence in the absence (1) and the presence (2) of 1 M trehalose; 3) conditions as in (1), but in the presence of 5 µM DCMU. Assay medium as in Fig. 1. For other conditions, see “Materials and Methods”.
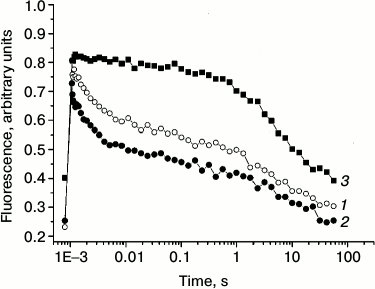
Note that recombination reactions between QA− and the S-states in the WOC or the redox-active tyrosine YZ (in Mn-depleted samples) occur much slower (>50 ms) than the forward electron transfer [5]. Forward electron transfer from QA− to QB was found to be heterogeneous and has to be described with at least two time constants of 0.2-0.8 and 2-3 ms [4, 31-34]. Additionally, this first electron transfer step is influenced by several factors, e.g. pH, dehydration, and temperature [38]. In PS 2 core complexes, the flash-induced fluorescence decay was multi-phasic (table). The kinetics were approximated by three exponential components with the following lifetimes (τ): τ1 ~ 1.6 ms (relative contribution ~52%), τ2 ~ 67 ms (~13%), and τ3 ~ 5.5 s (~35%). The fast phase reflects QA− oxidation by QB, while the two slower kinetic phases can be ascribed to back electron transfer from QA−. The 67-ms phase represents the fraction of PS 2 RCs that lack a functional Mn4CaO5 cluster, in which QA− recombines with oxidized redox-active tyrosine YZ [36, 37]. The slow phase (τ3) is likely due to recombination between QA− and the positively charged species in the WOC in PS 2 centers that are not able to perform the forward electron transfer.
Kinetic parameters for QA– reoxidation after
a single flash in PS 2 core complexes

Note: % in brackets represent relative contribution of the kinetic
component to the total fluorescence decay.
In the presence of trehalose, the flash-induced fluorescence decay was also approximated by three kinetic phases: τ1 ~ 1.4 ms (relative contribution ~64%), τ2 ~ 110 ms (~13%), and τ3 ~ 10 s (~23%) (table). The results obtained in the absence (Fig. 3, curve 1) and in the presence of trehalose (Fig. 3, curve 2) do not reveal significant difference in the lifetimes of these kinetic phases. The main effect of the trehalose is the ~12% increase in the relative contribution of the fast phase (τ1) corresponding to forward electron transfer from QA− at the expense of the slowest phase (τ3) attributable to recombination reaction between QA− and the S2 state of the Mn4CaO5 cluster. The data suggest that trehalose facilitates forward electron transfer from QA−.
By measuring the flash-induced fluorescence decay kinetics in the presence of DCMU, it is possible to examine the electron transfer on the donor side of PS 2 [36, 37, 39]. DCMU binds to the QB site and inhibits forward electron transfer from QA–. Under these conditions, the recombination kinetics between QA− and the positively charged donor side components (after a single flash, the main component is the S2 state in the WOC) reflects the integrity of the electron transfer chain on the oxidizing side of PS 2 [36, 37]. Under these conditions, the fast phase corresponding to the forward QA− reoxidation reaction disappeared, and the kinetics was approximated by slow components in the time range of seconds (Fig. 3, curve 3). The reason for the lack of an intermediate phase (τ2) in the presence of inhibitor (table) is not clear. The phase with τ3 ~ 2 s (~33%) is likely due to recombination of QA− with partially reduced Mn centers, while the very slow phase (~12 s, ~67%) probably corresponds to PS 2 RCs that were in the S0 state before the flash [40, 41]. It should be noted that, upon addition of trehalose, the time constant and the contribution of the flash-induced fluorescence decay phases in the presence of DCMU (table), which reflect charge recombination between QA− and oxidizing-side components of the PS 2, remained almost the same.
The mechanism of the effect of trehalose on PS 2 complex is not clear. It is known that in governing specific dynamics and functions of proteins, water molecules belonging to the protein hydration shell, to the bulk solvent, or placed inside the protein are critical [10, 20]. Data at extremely low hydration in bacterial RC suggest that a limited number of tightly bound water molecules stabilize the primary light induced charge-separated state on the time scale of 10–2 s [42].
Thus, in this work we have shown that addition of trehalose significantly stimulates the steady-state rate of O2 evolution in both PS 2 membrane fragments and core complexes. By the analysis of the initial rates of oxygen evolution, it was shown that trehalose stabilizes the oxidation of water, and these data are in good agreement with the results obtained by FTIR spectroscopy of isolated PS 2 complex [43].
As in the case of sucrose and glycerol [35], trehalose has no effect on the QA− reoxidation kinetics, although it stimulates this reaction in a greater fraction of PS 2 RCs capable of oxidizing QA−. As noted before, trehalose also prevents the time-dependent degradation and inactivation of PS 2 samples, probably as a result by stabilization of the reaction center. We propose that in the presence of trehalose, which affects the extent of hydration, the protein can preferentially exist in a more optimal conformation for effective functioning.
This work was supported by grants from the Russian Foundation for Basic Research Nos. 14-04-00519, 12-04-00821, and HK-13-04-40299-H. The results presented in Fig. 3 were obtained with support from the Russian Science Foundation (grant 14-14-00789).
REFERENCES
1.Goussias, C., Boussac, A., and Rutherford, A. W.
(2002) Photosystem II and photosynthetic oxidation of water: an
overview, Philos. Trans. R. Soc. London, Ser. B, 357,
1369-1381.
2.Wydrzynski, T. J., and Satoh K. (2005) in
Photosystem II: The Light-Driven Water:Plastoquinone
Oxidoreductase, Springer, New York.
3.Kok, B., Forbush, B., and McGloin, M. (1970)
Cooperation of charges in photosynthetic O2 evolution. I. A
linear four-step mechanism, Photochem. Photobiol., 11,
467-475.
4.Shinkarev, V. P. (2004) in Photosystem II:
Oxygen Evolution and Chlorophyll a Fluorescence Induced by Multiple
Flashes (Papageorgiou, G. C., ed.) Kluwer Academic Publishers,
Dordrecht, pp. 197-229.
5.Renger, G., and Kuhn, P. (2007) Reaction pattern
and mechanism of light induced oxidative water splitting in
photosynthesis, Biochim. Biophys. Acta, 1767,
458-471.
6.Dau, H., and Haumann, M. (2007) Eight steps
preceding O–O bond formation in oxygenic photosynthesis – a
basic reaction cycle of the photosystem II manganese complex,
Biochim. Biophys. Acta, 1767, 472-483.
7.Barber, J. (2008) Photosynthetic generation of
oxygen, Trans. R. Soc. B, 363, 2665-2674.
8.Crowe, J. H., Crowe, L. M., and Jackson, S. A.
(1983) Preservation of structural and functional activity in
lyophilized sarcoplasmic reticulum, Arch. Biochem. Biophys.,
220, 477-484.
9.Sun, W. Q., and Davidson, P. (1998) Protein
inactivation in amorphous sucrose and trehalose matrices: effects of
phase separation and crystallization, Biochim. Biophys. Acta,
1425, 235-244.
10.Francia, F., Dezi, M., Mallardi, A., Palazzo, G.,
Cordone, L., and Venturoli, G. (2008) Protein–matrix
coupling/uncoupling in “dry” systems of photosynthetic
reaction center embedded in trehalose/sucrose: the origin of trehalose
peculiarity, J. Am. Chem. Soc., 130, 10240-10246.
11.Jain, N. K., and Roy, I. (2009) Effect of
trehalose on protein structure, Protein Sci., 18,
24-36.
12.Allakhverdiev, S. I., Los, D. A., Mohanty, P.,
Nishiyama, Y., and Murata, N. (2007) Glycinebetaine alleviates the
inhibitory effect of moderate heat stress on the repair of photosystem
II during photoinhibition, Biochim. Biophys. Acta, 1767,
1363-1371.
13.Chen, T. H., and Murata, N. (2011) Glycinebetaine
protects plants against abiotic stress: mechanisms and biotechnological
applications, Plant Cell Environ., 34, 1-20.
14.Fernandez, O., Bethencourt, L., Quero, A.,
Sangwan, R. S., and Clement, C. (2010) Trehalose and plant stress
responses: friend or foe? Trend Plant Sci., 15,
409-417.
15.Ohtake, S., and Wang, Y. J. (2011) Trehalose:
current use and future applications, J. Pharm. Sci., 100,
2020-2053.
16.Jun, S.-S., Choi, H. J., Lee, H. Y., and Hong,
Y-N. (2008) Altered physiology in trehalose-producing transgenic
tobacco plants: enhanced tolerance to drought and salinity stresses,
J. Plant Biol., 51, 327-336.
17.Garg, A. K., Kim, J.-K., Owens, T. G., Ranwala,
A. P., Choi, Y. D., Kochian, L. V., and Ray, J. Wu. (2002) Trehalose
accumulation in rice plants confers high tolerance levels to different
abiotic stresses, Proc. Natl. Acad. Sci. USA, 99,
15898-15903.
18.Hincha, D. K., Hofner, R., Schwab, K. B., Heber,
U., and Schmitt, J. M. (1987) Membrane rupture is the common cause of
damage to chloroplast membranes in leaves injured by freezing or
excessive wilting, Plant Physiol., 83, 251-253.
19.Apostolova, E., Bushova, M., and Tenchov, B.
(2005) in Research Photosynthesis (Murata, N., ed.) Vol. IV,
Kluwer Academic Publishers, pp. 165-168.
20.Francia, E., Malferrari, M., Sacquin-Mora, S.,
and Venturoli, G. (2009) Charge recombination kinetics and protein
dynamics in wild type and carotenoid-less bacterial reaction centers:
studies in trehalose glasses, J. Phys. Chem., 113,
10389-10398.
21.Haag, E., Irrgang, K. D., Boekema, E. J., and
Renger, G. (1990) Functional and structural analysis of photosystem II
core complexes from spinach with high oxygen evolution capacity,
Eur. J. Biochem., 189, 47-53.
22.Ford, R. C., and Evans, M. C. W. (1983) Isolation
of a photosystem 2 preparation from higher plants with highly enriched
oxygen evolution activity, FEBS Lett., 160, 159-164.
23.Lerbret, A., Bordat, P., Affouard, F., Descamps,
M., and Migliardo, F. (2005) How homogeneous are the trehalose,
maltose, and sucrose water solutions? An insight from molecular
dynamics simulations, J. Phys. Chem. B, 109,
11046-11057.
24.Govindjee (1995) Sixty-three years since Kautsky:
chlorophyll a fluorescence, Aust. J. Plant Physiol., 22,
131-160.
25.Pospisil, P., and Dau, H. (2000) Chlorophyll
fluorescence transients of photosystem II membrane particles as a tool
for studying photosynthetic oxygen evolution, Photosynth. Res.,
65, 41-52.
26.Bukhov, N. G., Egorova, E. A., Govindacharya, S.,
and Carpentier, R. (2004) Changes in polyphasic chlorophyll a
fluorescence induction curve upon inhibition of donor or acceptor side
of photosystem II in isolated thylakoids, Biochim. Biophys.
Acta, 1657, 121-130.
27.Zhu, X-G., Govindjee, Baker, N. R., deSturler,
E., Ort, D. R., and Long, S. P. (2005) Chlorophyll a fluorescence
induction kinetics in leaves predicted from a model describing each
discrete step of excitation energy and electron transfer associated
with photosystem II, Planta, 223, 114-133.
28.Petrova, I. O., Kurashov, V. N., Semenov, A. Yu.,
and Mamedov, M. D. (2011) Manganese-depleted/reconstituted photosystem
II core complexes in solution and liposomes, J. Photochem.
Photobiol. B: Biol., 104, 372-376.
29.Krieger, A., Rutherford, A. W., and Johnson, G.
N. (1995) On the determination of redox midpoint potential of the
primary quinone electron acceptor, QA, in photosystem II,
Biochim. Biophys. Acta, 1229, 193-201.
30.Allakhverdiev, S. I., Hayashi, H., Nishiyama, Y.,
Ivanov, A. G., Aliev, J. A., Klimov, V. V., Murata, N., and Carpentier,
R. (2003) Glycinebetaine protects the D1/D2/Cyt b559 complex of
photosystem II against photo-induced and heat-induced inactivation,
J. Plant Physiol., 160, 41-49.
31.Bowes, J. M., and Crofts, A. R. (1980) Binary
oscillations in the rate of reoxidation of the primary acceptor of
photosystem II, Biochim. Biophys. Acta, 590, 373-384.
32.Eaton-Rye, J. J., and Govindjee (1988)
Electron-transfer through the quinone acceptor complex of photosystem
II after one or two actinic flashes in bicarbonate-depleted spinach
thylakoid membranes, Biochim. Biophys. Acta, 935,
248-257.
33.De Wijn, R., and van Gorkom, H. J. (2001)
Kinetics of electron transfer from Q(a) to Q(b) in photosystem II,
Biochemistry, 40, 11912-11922.
34.Rova, M., Mamedov, F., Magnuson, A., Fredriksson,
P.-O., and Styring, S. (1998) Coupled activation of the donor and the
acceptor side of photosystem II during photoactivation of the oxygen
evolving cluster, Biochemistry, 37, 11039-11045.
35.Halverson, K. M., and Barry, B. A. (2003) Sucrose
and glycerol effects on photosystem II, Biophys. J., 85,
1317-1325.
36.Sigfridsson, K. G. V., Bernat, G., Mamedov, F.,
and Styring, S. (2004) Molecular interference of Cd(2+) with
photosystem II, Biochim. Biophys. Acta, 1659, 19-31.
37.Roose, J. L., Frankel, L. K., and Bricker, T. M.
(2010) Documentation of significant electron transport defects on the
reducing side of photosystem II upon removal of the PsbP and PsbQ
extrinsic proteins, Biochemistry, 49, 36-41.
38.Muh, F., Glockner, C., Hellmich, J., and Zouni,
A. (2012) Light-induced quinone reduction in photosystem II,
Biochim. Biophys. Acta, 1817, 44-65.
39.Renger, G. (1999) in Concepts in Photobiology:
Photosynthesis and Photomorphogenesis, Molecular Mechanism of Water
Oxidation (Singhal, G. S., Renger, G., Govindjee, Irrgang, K.-D.,
and Sopory, S. K., eds.) Kluwer Academic Publishers, Dordrecht, Narosa
Publishing Co., Delhi, pp. 292-329.
40.Rappaport, F., Guergova-Kuras, M., Nixon, P. J.,
Diner, B. A., and Lavergne, J. (2002) Kinetics and pathways of charge
recombination in photosystem II, Biochemistry, 41,
8518-8527.
41.Mamedov, F., Rintamaki, E., Aro, E.-M.,
Andersons, B., and Styring, S. (2002) Influence of protein
phosphorylation on the electron-transport properties of photosystem II,
Photosynth. Res., 74, 61-72.
42.Malferrari, M., Francia, F., and Venturoli, G.
(2011) Coupling between electron transfer and protein-solvent dynamics:
FTIR and laser-flash spectroscopy studies in photosynthetic reaction
center films at different hydration levels, J. Phys. Chem.,
115, 14732-14750.
43.Polander, P. C., and Barry, B. A. (2012) A
hydrogen-bonding network plays a catalytic role in photosynthetic
oxygen evolution, Proc. Natl. Acad. Sci. USA, 109,
6112-6117.