Effect of Chaperonin Encoded by Gene 146 on Thermal Aggregation of Lytic Proteins of Bacteriophage EL Pseudomonas aeruginosa
P. I. Semenyuk1*, V. N. Orlov1, and L. P. Kurochkina1,2
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119234 Moscow, Russia; fax: +7 (495) 939-3181; E-mail: psemenyuk@belozersky.msu.ru; orlovvn@belozersky.msu.ru2Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia; fax: +7 (495) 336-6022; E-mail: lpk@ibch.ru
* To whom correspondence should be addressed.
Received June 17, 2014; Revision received September 17, 2014
Investigation of the chaperonin encoded by gene 146 of bacteriophage EL Pseudomonas aeruginosa that we characterized earlier has been continued. To reveal the mechanism of its functioning, new recombinant substrate proteins, fragments of gene product (gp) 183 containing the lysozyme domain were prepared. Their interaction with gp146 was studied. The influence of the phage chaperonin on the thermal aggregation of one of these gp183 fragments and endolysin (gp188) was investigated in both the presence and the absence of ATP by dynamic light scattering. In the absence of ATP, the phage chaperonin forms stable complexes with substrate proteins, thereby protecting them against thermal aggregation. Experimental data obtained for different substrate proteins are analyzed.
KEY WORDS: phage chaperonin, bacteriophage EL, endolysin, protein aggregationDOI: 10.1134/S0006297915020042
Abbreviations: DLS, dynamic light scattering; DSC, differential scanning calorimetry; gp, gene product; ITC, isothermal titration calorimetry.
Chaperonins are proteins that promote the folding of newly synthesized
polypeptide chains into their functionally active form driven by ATP
hydrolysis and prevent aggregation of proteins denatured under heat
shock. Members of this class of chaperones have been found in bacteria
(group I chaperonins, including the well-studied GroEL) as well as in
archaea and eukaryotic cytosol (group II chaperonins) and in
endosymbiotic organelles (group I) [1, 2]. Viruses (for example, bacteriophages λ, T4,
and RB49) were considered until recently to use the host’s GroEL
chaperonin for the folding of some of their proteins. In contrast to
phage λ, which uses the bacterial GroEL/GroES system [3], phages T4 [4] and RB49 [5] encode their own co-chaperonins, GroES orthologs,
working in couple with the host’s GroEL.
However, it turned out that bacterial viruses can also encode GroEL-like proteins [6-9]. Recently, we obtained and characterized for the first time one of the members of this group of proteins, namely gene product (gp) 146 from bacteriophage EL Pseudomonas aeruginosa [10]. The recombinant protein produced by Escherichia coli cells was studied by various physicochemical methods. This protein was found to have architecture similar to that of bacterial GroEL. It consists of two stacked seven-member rings and has an inner cavity. Using serum against gp146, we isolated its complexes with substrate proteins by immunoprecipitation and identified one of them, namely endolysin (gp188), by mass spectrometry. In in vitro experiments, we demonstrated for the first time that gp146 functions as a chaperonin. It protects endolysin against thermal inactivation and suppresses aggregation of thermally unfolded substrate molecules. The phage chaperonin was shown to function in both ATP-dependent and ATP-independent manners [10].
Another protein of phage EL was selected as a potential substrate for further investigation of the phage chaperonin and revealing of the mechanism of its functioning. The lytic bacteriophage EL is known to use a number of enzymes with different substrate specificity for penetration into cells of the Gram-negative bacterium P. aeruginosa and for release of newly synthesized phage particles. One of those enzymes – earlier characterized by us endolysin or gp188 – participates in cell lysis at the end of the phage replication cycle and is required for degradation of the most intractable infection barrier, the peptidoglycan layer. Another protein fulfilling the same function is gp183, which contains a lysozyme domain responsible for peptidoglycan degradation at the beginning of the EL replication cycle [6]. Amino acid sequences of the gp188 and gp183 lytic domains do not share similarity.
In the present work, gp183 fragments containing the lysozyme domain were obtained and characterized. Their interaction with the phage chaperonin was studied. To investigate the mechanism of the phage chaperonin functioning, we comparatively studied the influence of gp146 on thermal aggregation of the gp183 fragment and gp188 (endolysin) in the presence and absence of ATP.
MATERIALS AND METHODS
Cloning and expression of gene 183 fragments. DNA fragments of gene 183 coding the deletion variants gp183-1 and gp183-2 were amplified from EL genomic DNA by PCR using the reverse primer 5′-taa gat ctc gag att caa acg gac ggt ttt atc gg-3′ (the XhoI restriction site is underlined) and forward primers 5′-aaa ttt cat atg gga cgg tcg gat aaa gtt gac-3′ and 5′-aaa ttt cat atg tcg gac acc agt cat ttg gat-3′ (FauNDI restriction sites are underlined), respectively. The 183 gene fragments were cloned into the pET-28b(+) vector (Novagen, USA) between FauNDI and XhoI and expressed in E. coli BL21(DE3). The transformants were grown in 2×TY medium containing 1% glucose and kanamycin (50 µg/ml) at 37°C. When A600 reached 0.6, the cells were harvested by centrifugation at 2500g (Megafuge 2.0 R; Heraeus Instruments, Germany) for 10 min. The pellets were resuspended in fresh medium with antibiotic, induced with 1 mM isopropyl-β-D-thiogalactopyranoside (Helicon, Russia), and incubated at 37°C for 3 h. The cells were harvested by centrifugation at 2500g for 10 min.
Purification of recombinant gp183 fragments. The cell pellets were resuspended in 50 mM Tris-HCl buffer, pH 8.0, and sonicated for 3-5 min (0.5 min pulses with 1-2 min pauses) using a Virsonic 100 disintegrator (Virtis, USA). Cellular debris was removed by centrifugation at 12,000g (Eppendorf, Germany) for 10 min. The recombinant proteins containing a polyhistidine sequence at the N-terminus of the molecule were purified by metal chelate affinity chromatography on His-Select nickel affinity gel (Sigma, USA). The purified proteins were dialyzed against 50 mM Tris-HCl buffer, pH 8.0, and concentrated using an Amicon Ultra-15 centrifugal filter device (MWCO 30000) (Millipore, USA). The concentrations of recombinant proteins were determined spectrophotometrically at 280 nm using theoretical absorption coefficients of 32,320 and 23,840 M–1·cm–1 for gp183-1 and gp183-2, respectively.
Protein activity assay. Enzymatic activity of the gp183 fragments was determined as described earlier [11]. Pseudomonas aeruginosa PAO1 cells with outer membrane permeabilized by chloroform treatment were used as the substrate.
Preparation and analysis of gp146/gp188 complex. Recombinant gp146 and gp188 were prepared as described previously [10]. The mixture of gp146 (1.5 μM) and gp188 (3 μM) was incubated in 50 mM Tris-HCl buffer, pH 7.5, containing 100 mM KCl and 10 mM MgCl2, at 45°C for 1.5 h. After centrifugation (12,000g, 10 min), the samples were mixed with 4× sample buffer and loaded on a gradient (3-6%) polyacrylamide gel. The proteins were separated by native PAGE using 80 mM Mops, pH 7.2, with 1 mM magnesium acetate. Immunoblotting was performed as earlier described [12]. The antigen–antibody complexes were detected with 0.02% 3,3′-diaminobenzidine (Sigma).
Dynamic light scattering (DLS). The thermal aggregation kinetics of the proteins was studied by dynamic light scattering using a ZetaSizer NanoZS instrument (Malvern, UK). Substrate protein concentration was 3 μM in all experiments. The chaperonin concentration was varied to achieve substrate/chaperonin molar ratios of 1 : 0.05, 1 : 0.2, and 1 : 0.5. Total duration of the autocorrelation function was 150 s for every curve point. The experimental curves were smoothed by a fast Fourier filter in MicroCal Origin 7.0 software.
Two different models corresponding to diffusion-limited and reaction-limited aggregation were used to describe the substrate aggregation process. The increase in aggregate hydrodynamic diameter was described in the first case by the equation:
Dh = Dh,0 (1 + K1t)1/df,
where Dh,0 is the diameter of the starting aggregates, K1 is an apparent aggregation constant, df is a fractal dimension that equals to 1.8 for protein systems [13, 14], and in the second case by the equation:
Dh = Dh,0 exp (K2(t – t0)) [15, 16].
Differential scanning calorimetry (DSC). Calorimetric studies were performed using a DASM-4 instrument (Biopribor, Russia) equipped with capillary platinum cells at a scanning rate of 1°C/min. The second heating curve was used as an instrumental baseline because of irreversible denaturation of the proteins. Data processing (including chemical baseline subtraction) was performed using Arina 2 software (Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Russia). All experiments were carried out in 50 mM Tris-HCl buffer, pH 8.0, containing 100 mM KCl and 10 mM MgCl2. The concentration of the individual proteins was 0.5 mg/ml. For investigation of gp146 interaction with substrate, chaperonin concentration was increased to 1 mg/ml.
Isothermal titration calorimetry (ITC). The interaction of chaperonin with a denatured substrate was studied using the isothermal titration microcalorimeter VP-ITC (MicroCal, USA). For this, gp146 solution was titrated with gp183-1 solution in 50 mM Tris-HCl buffer, pH 8.0, containing 100 mM KCl and 10 mM MgCl2, at 37°C. Initial proteins concentrations were 0.5 mg/ml, the injection volume was 20 μl, and the interval between injections was 5 min. Protein solutions were degassed before experiments. The “one set of sites” model was used for fitting of the data using the MicroCal Origin 7.0 software.
ATPase activity assay. The ATPase activity of gp146 was determined by quantitation of released inorganic phosphate (Pi) from ATP by a malachite green colorimetric assay [17]. The gp146 (0.75 μM) in 50 mM Tris-HCl, 100 mM KCl, and 10 mM MgCl2, pH 7.5, was incubated with 1 mM ATP at various temperatures. Aliquots of reaction mixture were removed and mixed with acidic malachite green solution to stop the reaction. After 10-min incubation, the absorbance at 630 nm was read using a Genesys 10uv spectrophotometer (Thermo Electron Corporation, USA). ATPase activity unit was defined as 1 nmol of released Pi per nmol of the chaperonin tetradecamer per minute.
RESULTS AND DISCUSSION
Expression and characterization of gp183 fragments. Since gp183 of phage EL is a relatively large protein (2543 a.a., 280 kDa) and its lytic domain is localized in the C-terminal region, we designed two plasmid constructs for expression of the gp183 fragments deleted from the 5′-terminus in the E. coli cells. The fragments of gp183, gp183-1 (688 a.a., 72.9 kDa) and gp183-2 (581 a.a., 61.4 kDa), obtained by truncation of the N-terminus of the polypeptide chain by 1872 and 1979 amino acids, respectively, were produced by cells in soluble form. The recombinant proteins containing polyhistidine tag at the N-terminus were purified by metal chelate chromatography. Both gp183 fragments were able to lyse the outer membrane-permeabilized P. aeruginosa PAO1 cells, but their enzymatic activity was one order of magnitude less than the activity of gp188 (endolysin) that we characterized previously [10].
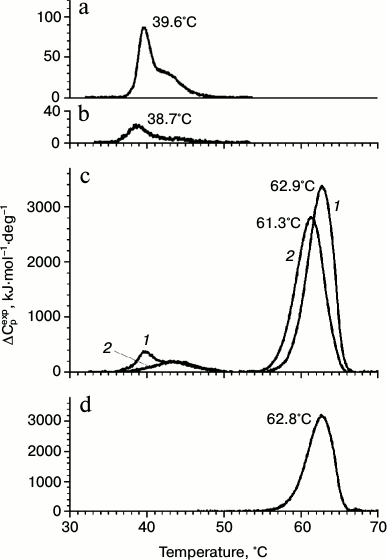
Both gp183 fragments were found by DSC to be more thermolabile than gp188 (Fig. 1). Indeed, the temperature of the denaturation peak maximum for gp183-1 and gp183-2 was 39.6 and 38.7°C, respectively (Fig. 1, a and b), while it was significantly higher for gp188 (54.8°C) [10]. The fragment gp183-1 was chosen for further work because of its higher melting enthalpy, equal to 300.7 kJ/mol versus 99.3 kJ/mol for gp183-2. Moreover, the main component of the denaturation peak for gp183-1 is more cooperative in comparison with that of gp188, which facilitates the use of this protein as a potential substrate for investigation of the phage chaperonin machinery mechanism.
Fig. 1. DSC profiles of gp183-1 (a), gp183-2 (b), the mixture of gp183-1 and gp146 (c) before (curve 1) and after (curve 2) incubation at 39°C for 30 min, as well as free gp146 (d). Protein concentration was 0.5 mg/ml. Curves on panel (c) were calculated per chaperonin tetradecamer.
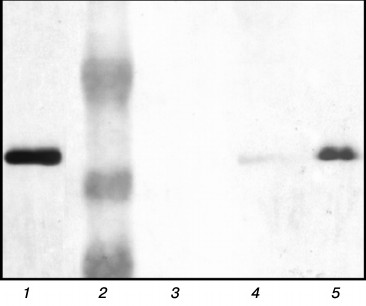
Investigation of chaperonin complexes with substrate proteins. Earlier we investigated the influence of gp146 on endolysin (gp188) aggregation and suggested that gp146 prevents substrate aggregation in the absence of ATP by forming the stable complex with it. In the present work, the formation of this complex was experimentally confirmed. The mixture of gp146 and gp188 incubated at 45°C and also the individual proteins as a control were separated by native PAGE in a gradient gel (data not shown). An additional minor band with low electrophoretic mobility was detected along with the major band of gp146 in the protein pattern after gel staining with Coomassie R-250. At the same time, free positively charged gp188 migrated to the opposite side and was not detected in the gel. The protein bands were cut from a native gel, incubated with SDS-containing sample loading buffer, and analyzed by SDS-PAGE [18]. The separated proteins were detected by staining with Coomassie R-250 (data not shown) as well as transferred onto a nitrocellulose membrane and incubated with serum against recombinant gp188. The gp146 was detected in all bands after Coomassie staining, but its concentration in the minor band of the sample was much lower than in the major band of gp146. Immunoblotting with serum against gp188 revealed that an additional minor band along with gp146 also contains substrate protein (Fig. 2, lane 5). This indicates the existence of the gp146/gp188 complex in the protein mixture subjected to heating. The gp146 major band of the same sample also contained a small amount of gp188 (Fig. 2, lane 4). Unfortunately, gp188 denaturation and correspondingly the binding of its denatured form with the chaperonin at 45°C are slow, which prevented obtaining the gp146/gp188 complex in large quantities. Increasing the temperature also was not helpful because of the high tendency of gp188 to aggregation and thermal inactivation of the chaperonin. Therefore, gp183-1, which denatures with higher cooperativity and at lower temperature than gp188, was selected for further investigation of the interaction of gp146 with substrate protein.
Fig. 2. Analysis of protein bands from native gel by SDS-PAGE in 10% polyacrylamide gel. Protein bands were stained with 3,3′-diaminobenzidine on nitrocellulose membrane after immunoblotting and incubation with serum against gp188. Lanes: 1) control gp188; 2) protein marker (48, 34, and 26 kDa); 3) control gp146; 4) gp146 from sample major band; 5) gp146 from sample minor band.
The influence of chaperonin gp146 on the thermal aggregation of gp183-1 at 38°C was investigated using DLS (Fig. 3). Free gp183-1 aggregation was relatively fast as compared with gp188 (Fig. 3, a and b) and took 9 min to form particles with the diameter of more than 600 nm. However, in the presence of gp146 the aggregation level considerably decreased. The effect of suppression of the aggregation correlated with the chaperonin concentration and became stronger with growth of the gp146/gp183-1 molar ratio (Fig. 3a). This seems to be caused by the formation of a binary complex between the chaperonin and the partially unfolded under heating substrate as in the case of gp188 [10].
Fig. 3. Effect of gp146 on the aggregation of gp183-1 at 38°C (a, c) and gp188 at 45°C (b, d) at various substrate/chaperonin molar ratios in the absence (a, b) and presence (c, d) of ATP. The inset on panel (d) represents the results of the experiment with ATP addition to the mixture of gp188 and gp146; the arrow shows the time when ATP was added. The circles represent experimental data and the solid lines show the best fit by the model. Lines 1-7, 10, and 13-15 correspond to the reaction-limited aggregation regime; lines 8, 9, 11, and 12 correspond to the diffusion-limited aggregation regime.
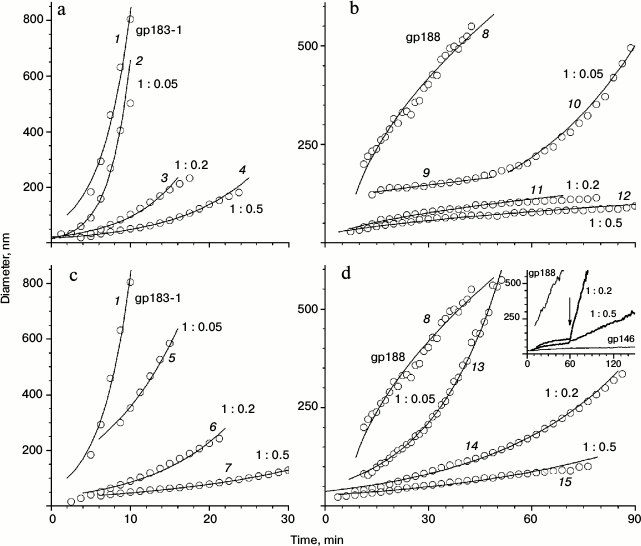
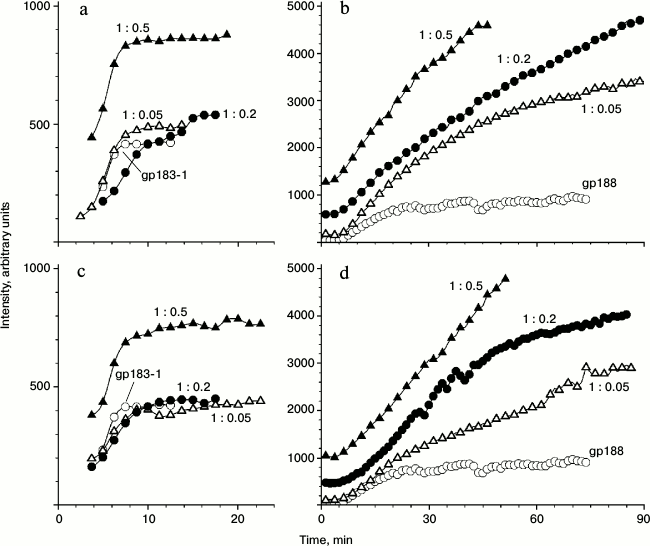
Figure 4 shows the time-dependence of the light scattering intensity for the aggregation of free substrate proteins as well as their mixtures with gp146 at various molar ratios. The scattering intensity curves for gp183-1 reach a plateau relatively fast compared to those for gp188, which seems to be a consequence of different kinetics of denaturation of substrate proteins (Fig. 4, a and b). Observed scattering intensity at high concentrations of gp146 turned out to be higher than that in a case of free substrates. Considering the size of the particles after gp146 addition increased more slowly than that free substrate protein, we suggest that the chaperonin is somehow involved into the aggregation process, otherwise chaperonin addition should alter only kinetics of aggregation but not general behavior. Most likely, the resulting chaperonin complexes with substrate also undergo aggregation. The same picture was observed for the interaction of GroEL with glyceraldehyde-3-phosphate dehydrogenase [19].
Fig. 4. Dependence of light scattering intensity on time for aggregation of gp183-1 at 38°C (a, c) and gp188 at 45°C (b, d) at various substrate/chaperonin molar ratios in the absence (a, b) and presence (c, d) of ATP.
The formation of the chaperonin complex with gp183-1 was demonstrated by DSC (Fig. 1c). Indeed, the melting thermogram of the gp146 and gp183-1 mixture consisted of the sum of the melting thermograms of the individual proteins, while after preliminary incubation of the protein mixture at 39°C for 30 min the denaturation peak of chaperonin shifted to the left by 1.5°C, and the major peak of gp183-1 with a maximum at 39.6°C disappeared. This indicates the denaturation of part of gp183-1 and its binding to gp146 during preliminary incubation of the protein mixture. As shown by gp183-1activity assay, part of the substrate protein was inactivated. The absence of the major peak of the substrate protein on the melting curve indicates that gp183-1 remains in a denatured form in the complex with the chaperonin.
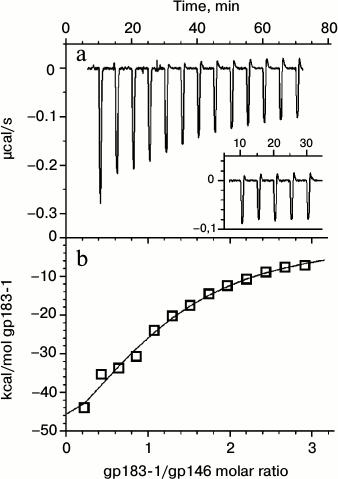
Interaction of the chaperonin with a denatured substrate was also studied by ITC at 37°C (Fig. 5). The choice of the temperature regime for titration was made from the results of the DSC experiments. Heating to 37°C, corresponding to the temperature of the beginning of the gp183-1 melting peak (Fig. 1a), induced substrate denaturation but did not affect the structure of more stable gp146 (Fig. 1d). It was demonstrated that in 50 mM Tris-HCl buffer, pH 8.0, 100 mM KCl, 10 mM MgCl2 at 37°C the denatured gp183-1 actively binds with chaperonin (the association constant is 2.4 ± 0.5 μM–1) at stoichiometric ratio close to 1 : 1. Only one heptameric ring of chaperonin seems to be able to form a stable complex with a substrate, which agrees well with previously obtained data on stoichiometry of the chaperonin interaction with nucleotides, which was close to 1 : 7 [10]. This is probably related to an asymmetric mechanism of chaperonin functioning that is typical, for example, for the GroEL/GroES system [20].
Fig. 5. Calorimetric titration of chaperonin gp146 with gp183-1 substrate at 37°C. a) Raw data of titration; the inset represents the results of the gp183-1 titration into the buffer; b) dependence of the heat release in every injection on the achieved substrate/chaperonin molar ratio (squares) and fitting by the “one set of sites” model (line).
Effect of chaperonin on substrate protein aggregation in the presence of ATP. Thus, in the absence of ATP, the chaperonin is capable of suppressing aggregation of thermally denatured molecules of both substrates by the formation of stable complexes, but enzymatic activity of the substrate proteins is not preserved.
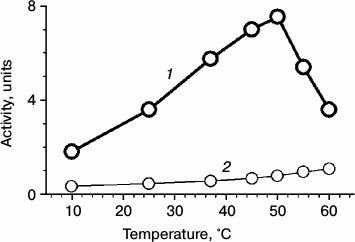
Recently, we found that phage chaperonin is able to function in an ATP-dependent manner [10]. In the present work, we found that the phage chaperonin possesses ATPase activity under physiological conditions. Its value was one order of magnitude less than that of GroEL [21], but comparable with the values of some other group II chaperonins [22]. The ATPase activity of gp146 as well as an activity of other known chaperonins increases with increasing temperature. For gp146, it reaches a maximum value at around 50°C (Fig. 6). On further temperature increase, the activity of ATP hydrolysis sharply decreases because of thermal inactivation of the chaperonin.
Fig. 6. Temperature dependence of ATPase activity of gp146 (1); spontaneous hydrolysis of ATP (2).
A different, namely energy-dependent mechanism of phage chaperonin functioning seems to be activated on ATP addition. In this case, the chaperonin most probably acts by the ATPase cycle resulting in the formation of properly folded and enzymatically active substrate protein molecules. This suggestion is corroborated by results of substrate protein activity assay. Both proteins (gp188 and gp183-1) in the presence of gp146 retain their lytic activity at elevated temperatures only with ATP.
An influence of gp146 on the thermal aggregation of investigated substrates was found to be dramatically different in the presence of ATP (Fig. 3, c and d). Phage chaperonin suppresses gp183-1 aggregation in the presence of ATP more effectively than in its absence, and this feature was observed for various gp146/gp183-1 molar ratios (Fig. 3c). Absolutely another picture was observed for gp188 (Fig. 3d). As shown previously [10], gp188 aggregation in the presence of gp146 in the system containing ATP occurs even faster than that in the absence of ATP, and the pattern for the curves of the growth of size of particles was significantly different (Fig. 3, d and b). Furthermore, addition of ATP to the gp146/gp188 complex obtained by 1-h incubation at 45°C in the absence of ATP resulted in sharp gp188 aggregation (Fig. 3, inset in panel (d)) that did not occur on addition of ATP to the gp146/gp183-1 complex (data not shown). One possible explanation of this difference in gp146 behavior toward investigated substrate proteins is that gp188 and gp183-1 have different affinity to the chaperonin gp146 and are characterized by different kinetics of denaturation under selected conditions. Indeed, at low concentrations of denatured substrate (as in the case of gp188) pronounced anti-aggregation action of chaperonin can be provided even by passive (in the absence of ATP) protection of the substrate against aggregation due to formation of stable long-lived complexes, in which gp146 is not able to refold the denatured gp188 (Fig. 3b). After ATP addition, gp146 seems to act according to an ATPase cycle providing substrate refolding. However, under the influence of temperature, part of a native substrate denatures again, and therefore the system constantly contains a portion of denatured substrate molecules that are able either to be bound to chaperonin or to aggregate with each other. If the substrate affinity to chaperonin is relatively low, aggregation predominates over complex formation, which results in a decrease in apparent efficiency of the anti-aggregation action of the chaperonin. The same is observed in the case of gp188 thermal aggregation (Fig. 3d). On the contrary, with excess of the denatured substrate (as in the case of gp183-1) passive (in the absence of ATP) aggregation suppression by chaperonin is not so effective (Fig. 3a). However, active interaction of chaperonin with a denatured form of substrate enhances its total anti-aggregation activity in the presence of ATP (Fig. 3c). High affinity of the denatured gp183-1 to chaperonin demonstrated the formation of gp146/gp183-1 complex by DSC (Fig. 1c) and ITC (Fig. 5), but it was not achieved for gp188. In turn, the special features of the gp183-1 structure seem to be a reason of its more active (as compared to gp188) interaction with chaperonin. Since the gp183 fragment possesses lytic activity, its C-terminal region containing the lysozyme domain seems to have a compact native structure. On the contrary, the N-terminus structure is most likely partially disordered due to an extended deletion that facilitates the thermal denaturation of this part of the molecule and promotes its binding to the chaperonin.
The suggestions described above are confirmed by the results of mathematical analysis of the aggregation kinetics. In contrast to gp183-1, the mechanism of gp188 aggregation in the presence of gp146 changes on addition of ATP (Fig. 3). Thus, the hydrodynamic diameter of the particle trend curves of the free gp188 aggregation can be described by the equation:
Dh = Dh,0 (1 + K1t)1/df,
corresponding to the diffusion-limited aggregation regime when every collision of aggregating particles causes its adhesion [13, 14]. The presence of chaperonin with ATP changes the aggregation regime to the reaction-limited one, when denatured substrate molecules aggregate not after every collision with other molecules. This regime is described by the equation:
Dh = Dh,0 exp (K2(t – t0)) [15, 16].
The same change in the mechanism was shown for glyceraldehyde-3-phosphate dehydrogenase aggregation in the presence of the chaperones alpha-crystallin and GroEL [19]. The gp146 suppresses gp183-1 aggregation in the absence of ATP in a similar manner, though less effectively, while the gp188 aggregation regime becomes diffusion-limited. This seems to occur because of relatively slow (under these conditions) accumulation of denatured gp188, which interacts with the chaperonin much less efficiently than gp183-1. As a result, the denatured gp188 molecules more readily interact with already formed aggregates than with free chaperonin. This suggestion is corroborated by the fact that aggregation rate decreases rather slowly when the chaperonin concentration increases, and even at the gp188/gp146 molar ratio of 1 : 1 aggregation of the substrate is still noticeable (data not shown).
This work was financially supported by the Russian Foundation for Basic Research, grant No. 14-04-00832.
REFERENCES
1.Horwich, A. L. (2013) Chaperonin-mediated protein
folding, J. Biol. Chem., 288, 23622-23632.
2.Iizuka, R., Yoshida, T., Ishii, N., Zako, T.,
Takahashi, K., Maki, K., Inobe, T., Kuwajima, K., and Yohda, M. (2005)
Characterization of archaeal group II chaperonin-ADP-metal fluoride
complexes: implications that group II chaperonins operate as a
“two-stroke engine”, J. Biol. Chem., 280,
40375-40383.
3.Georgopoulos, C. P., Hendrix, R. W., Casjens, S.
R., and Kaiser, A. D. (1973) Host participation in bacteriophage lambda
head assembly, J. Mol. Biol., 76, 45-60.
4.Van der Vies, S. M., Gatenby, A. A., and
Georgopoulos, C. (1994) Bacteriophage T4 encodes a co-chaperonin that
can substitute for Escherichia coli GroES in protein folding,
Nature, 368, 654-656.
5.Ang, D., Richardson, A., Mayer, M. P., Keppel, F.,
Krisch, H., and Georgopoulos, C. (2001) Pseudo-T-even bacteriophage
RB49 encodes CocO, a cochaperonin for GroEL, which can substitute for
Escherichia coli’s GroES and bacteriophage T4’s
gp31, J. Biol. Chem., 276, 8720-8726.
6.Hertveldt, K., Lavigne, R., Pleteneva, E., Sernova,
N., Kurochkina, L., Korchevskii, R., Robben, J., Mesyanzhinov, V.,
Krylov, V. N., and Volckaert, G. (2005) Genome comparison of
Pseudomonas aeruginosa large phages, J. Mol. Biol.,
354, 536-545.
7.Kiljunen, S., Hakala, K., Pinta, E., Huttunen, S.,
Pluta, P., Gador, A., Lönnberg, H., and Skurnik, M. (2005)
Yersiniophage phiR1-37 is a tailed bacteriophage having a 270 kb DNA
genome with thymidine replaced by deoxyuridine, Microbiology,
151, 4093-4102.
8.Cornelissen, A., Hardies, S. C., Shaburova, O. V.,
Krylov, V. N., Mattheus, W., Kropinski, A. M., and Lavigne, R. (2012)
Complete genome sequence of the giant virus OBP and comparative genome
analysis of the diverse φKZ-related phages, J. Virol.,
86, 1844-1852.
9.Jang, H. B., Fagutao, F. F., Nho, S. W., Park, S.
B., Cha, I. S., Yu, J. E., Lee, J. S., Im, S. P., Aoki, T., and Jung,
T. S. (2013) Phylogenomic network and comparative genomics reveal a
diverged member of the φKZ-related group, marine vibrio phage
φJM-2012, J. Virol., 87, 12866-12878.
10.Kurochkina, L. P., Semenyuk, P. I., Orlov, V. N.,
Robben, J., Sykilinda, N. N., and Mesyanzhinov, V. V. (2012) Expression
and functional characterization of the first bacteriophage-encoded
chaperonin, J. Virol., 86, 10103-10111.
11.Briers, Y., Lavigne, R., Volckaert, G., and
Hertveldt, K. (2007) A standardized approach for accurate
quantification of murein hydrolase activity in high-throughput assays,
J. Biochem. Biophys. Methods, 70, 531-533.
12.Zhemaeva, L. V., Sykilinda, N. N., Navruzbekov,
G. A., Kurochkina, L. P., and Mesyanzhinov, V. V. (2000) Structure and
folding of bacteriophage T4 gene product 9 triggering infection. II.
Study of conformational changes of gene product 9 mutants using
monoclonal antibodies, Biochemistry (Moscow), 65,
1068-1074.
13.Lin, M. Y., Lindsay, H. M., Weitz, D. A., Ball,
R. C., Klein, R., and Meakin, P. (1989) Universality of fractal
aggregates as probed by light-scattering, Proc. Roy. Soc. L. Ser.
Math. Phys. Eng. Sci., 423, 71-87.
14.Khanova, H. A., Markossian, K. A., Kurganov, B.
I., Samoilov, A. M., Kleimenov, S. Y., Levitsky, D. I., Yudin, I. K.,
Timofeeva, A. C., Muranov, K. O., and Ostrovsky, M. A. (2005) Mechanism
of chaperone-like activity. Suppression of thermal aggregation of
βL-crystallin by α-crystallin, Biochemistry,
44, 15480-15487.
15.Weitz, D. A., Huang, J. S., Lin, M. Y., and Sung,
J. (1985) Limits of the fractal dimension for irreversible kinetic
aggregation of gold colloids, Phys. Rev. Lett., 54,
1416-1419.
16.Khanova, H. A., Markossian, K. A., Kleimenov, S.
Y., Levitsky, D. I., Chebotareva, N. A., Golub, N. V., Asryants, R. A.,
Muronetz, V. I., Saso, L., Yudin, I. K., Muranov, K. O., Ostrovsky, M.
A., and Kurganov, B. I. (2007) Effect of α-crystallin on thermal
denaturation and aggregation of rabbit muscle
glyceraldehyde-3-phosphate dehydrogenase, Biophys. Chem.,
125, 521-531.
17.Baykov, A. A., Evtushenko, O. A., and Avaeva, S.
M. (1988) A malachite green procedure for orthophosphate determination
and its use in alkaline phosphatase-based enzyme immunoassay, Anal.
Biochem., 171, 266-270.
18.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
19.Markossian, K. A., Golub, N. V., Chebotareva, N.
A., Asryants, R. A., Naletova, I. N., Muronetz, V. I., Muranov, K. O.,
and Kurganov, B. I. (2010) Comparative analysis of the effects of
α-crystallin and GroEL on the kinetics of thermal aggregation of
rabbit muscle glyceraldehyde-3-phosphate dehydrogenase, Protein
J., 29, 11-25.
20.Hartl, F. U., and Hayer-Hartl, M. (2002)
Molecular chaperones in the cytosol: from nascent chain to folded
protein, Science, 295, 1852-1858.
21.Li, Y., Zheng, Z., Ramsey, A., and Chen, L.
(2010) Analysis of peptides and proteins in their binding to GroEL,
J. Pept. Sci., 16, 693-700.
22.Gutsche, I., Essen, L. O., and Baumeister, W.
(1999) Group II chaperonins: new TRiC(k)s and turns of a protein
folding machine, J. Mol. Biol., 293, 295-312.