pH Might Play a Role in Regulating the Function of Paired Amphipathic Helices Domains of Human Sin3B by Altering Structure and Thermodynamic Stability
Tauheed Hasan#, Mashook Ali#, Daman Saluja, and Laishram Rajendrakumar Singh*
Dr. B. R. Ambedkar Center for Biomedical Research, University of Delhi, Delhi-110-007, India; fax: +91-11-2766-6248; E-mail: lairksingh@gmail.com; directoracbr@gmail.com; director@acbr.du.ac.in# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received August 19, 2014; Revision received September 29, 2014
Human Sin3B (hSin3B), a transcription regulator, is a scaffold protein that binds to different transcription factors and regulates transcription. It consists of six conserved domains that include four paired amphipathic helices (PAH 1-4), histone deacetylase interaction domain (HID), and highly conserved region (HCR). Interestingly, the PAH domains of hSin3B are significantly homologous to each other, yet each one interacts with a specific set of unique transcription factors. Though various partners interacting with hSin3B PAH domains have been characterized, there is no structural information available on the individual PAH domains of hSin3B. Here we characterize the structure and stability of different PAH domains of hSin3B at both nuclear and physiological pH values by using different optical probes. We found that the native state structure and stability of different PAH domains are different at nuclear pH where hSin3B performs its biological function. We also found that PAH2 and PAH3 behave differently at both nuclear and physiological pH in terms of native state structure and thermodynamic stability, while the structural identity of PAH1 remains unaltered at both pH values. The study indicates that the structural heterogeneity of different PAH domains might be responsible for having a unique set of interacting transcription factors.
KEY WORDS: protein structure, thermodynamic stability, circular dichroism, transcription regulator, scaffold proteinDOI: 10.1134/S0006297915040057
Abbreviations: HCR, highly conserved region; HID, histone deacetylase interaction domain; PAH, paired amphipathic helices domain.
Sin3, a global transcription regulator, helps to regulate many
biological functions including nucleosome remodeling, DNA methylation,
cell proliferation, and apoptosis [1, 2]. Sin3 does not bind to DNA but is a scaffold
protein that helps the transcription of various genes by interacting
with different transcription factors, forming Sin3 complex. The core
complex of Sin3 consists of eight components in humans: SIN3, HDAC1,
HDAC2, RbAp46, RbAp48, SAP30, SAP18, and SDS3 [3,
4]. Sin3 has six distinct conserved domains that
include four repeats of paired amphipathic helices (PAH1-4), histone
deacetylase interaction domain (HID), and the highly conserved region
(HCR) [1]. There is a high degree of similarity
between PAH1 and PAH2 of Sin3 in various organisms, but PAH3 domains
share relatively low levels of sequence identity with the PAH1 and PAH2
domains (25 and 16%, respectively), yet these PAH domains recognize
different sequence motifs, thereby exhibiting high degree of
specificity for their targets [5, 6]. In humans, two isoforms of Sin3 are present
(hSin3A and hSin3B) that are encoded by two separate genes that are
considered to be the result of gene duplication [7]. Human Sin3A and Sin3B proteins are approximately
57% identical throughout the length of their polypeptide chains with
the highest degree of homology localized in the PAH and HID regions [8, 9].
The PAH domains of hSin3B are responsible for interacting with different transcription factors [10-12]. Various interacting partners of PAH1, PAH2, and PAH3 have been identified in humans, and these are responsible for carrying out different biological functions [13]. Some data are also available in the literature about the structural and functional aspects of the PAH domains [2, 14]. In addition, atomic level structural information on Sin3 is available in the literature, but most of the structural studies have been carried out on the PAH domains of mouse or other species than human [6, 14-17]. Therefore, lack of structural information about the different PAH domains of humans impedes systematic analysis with data coming from interaction studies. Here we report investigation of the differences in the structure and stability of different PAH domains of human Sin3B at nuclear pH (6.3-6.8) and physiological pH (7.0) using various spectroscopic tools. We found that there is subtle variation in the structure of the PAH domains of hSin3B at nuclear pH where Sin3 performs its biological functions. We also found that PAH2 and PAH3 behave differently at both the nuclear and physiological pH in terms of native state structure and stability, while the structural identity of PAH1 remains unaltered at both pH values. Our study suggests that the difference in the conformation of native state structure or structural flexibility of the PAH domains might be responsible for interacting with specific binding partners.
MATERIALS AND METHODS
Cloning, expression, and purification of proteins. The human fetal brain cDNA library was amplified according to the instruction of the supplier (Clontech Laboratory Inc., USA) and used for PCR amplification of various PAH domains of hSin3B using primers for PAH1 (5′- AGCTGCGGATCCACGTAGAAGACG-3′ and 5′-TCTAGTC TCGAGCCGAGGGGAAGAAAAG-3′), PAH2 (5′-CATAGGGGATCCTGGAGTCCGATTC-3′ and 5′-GTGCATCTCGAGCGGCCCGTTTCCTGT-3′), and PAH3 (5′-CAGTGGG GATCCACGGGACTCTGCAG-3′ and 5′-ATGGAACTCGAGAAGGACAGCTCTTTTACC-3′) with a BamHI restriction site in the forward primer and XhoI in reverse primer. The PCR amplified product was ligated to TA vector (Promega, USA) and then subcloned into pGEX-5x3 expression vectors using the restriction sites. The ligation product was transformed into E. coli DH5α competent cells and grown overnight on a Luria–Bertani (LB) agar plate containing ampicillin (100 µg/ml). A recombinant clone for each PAH domain was subjected to DNA sequencing to confirm the reading frame and sequence. For purification of the PAH domain, the plasmids were transformed into E. coli BL21(DE3) competent cells, and a single colony was grown overnight in LB medium containing 100 µg/ml ampicillin at 37°C with shaking at 250-300 rpm [18]. Fresh culture (1 liter) was inoculated with 10 ml of the overnight culture, and it was vigorously agitated for 3-4 h at 250 rpm until the OD600 reached 0.5-0.6. Overexpression of PAH domains was induced with 0.5 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) at 37°C with vigorous agitation for 4 h before harvesting the cells by centrifugation at 8000g for 20 min at 4°C. The cell pellet was frozen and stored at –80°C and used within one week.
Cells were lysed in buffer containing 50 mM Tris (pH 8.0) (MP Biomedicals, USA), 250 mM NaCl (G-Biosciences, USA), 0.1% NP-40 (Sigma, USA), and 1% bacterial protease inhibitor cocktail (Sigma). The cells were lysed by giving six cycles of sonication at 60% efficiency. The bacterial lysate was centrifuged at 8000g for 20 min at 4°C to remove the insoluble fraction. Supernatant (4 ml) was mixed with 2 ml bed volume of glutathione-Sepharose (GE Healthcare, USA) and incubated for 30 min at 4°C with gentle shaking. The glutathione-Sepharose was washed five times with 10 ml of phosphate buffered saline (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4). Approximately 4 mg of the fusion protein was incubated with 40 µl of Factor Xa enzyme (1 µg/µl) (GE Healthcare) at 22°C for 16 h in 4 ml of cleavage buffer (50 mM Tris, pH 8.0, 150 mM NaCl, and 1 mM CaCl2) to cleave the GST tag from the fusion protein. The suspension was centrifuged at 500g for 5 min to pellet the resin. The supernatant contains the purified protein and Factor Xa. Factor Xa was removed from the protein using a HiTrap Benzamidine FF (high sub) column (GE Healthcare). This column captures the Factor Xa, thus enabling the collection of pure protease-free protein in the eluate. Protein solutions were dialyzed extensively against 0.1 M KCl at pH 7.0 in the cold (4°C) to remove the salts and then lyophilized for later experiments.
Thermal denaturation studies. Thermal denaturation of the protein was studied using a Jasco J-810 spectropolarimeter equipped with a Peltier-type temperature controller at heating rate of 1°C/min. This scan rate was found to provide adequate time for equilibration. Each sample was heated from 20 to 85°C. The change in absorbance with increasing temperature was followed at 222 nm. About 500 data points of each transition curve were collected. Measurements were repeated at least three times. After denaturation, the protein sample was immediately cooled to measure reversibility of the reaction. Each heat-induced transition curve was analyzed for Tm (midpoint of denaturation) and ΔHm (enthalpy change at Tm) using a nonlinear least squares method according to the relation:
where y(T) is the optical property at temperature T (K); yN(T) and yD(T) are the optical properties of the native and denatured protein molecules at T (K), respectively; and R is the gas constant. In the analysis of the transition curve, it was assumed that a parabolic function describes the dependence of the optical properties of the native and denatured protein molecules (i.e. yN(T) = aN + bNT + cNT2 and yD(T) = aD + bDT + cDT2, where aN, bN, cN, aD, bD, and cD are temperature-independent coefficients). A plot of ΔHm versus Tm at each pH gave the value of ΔCp, the change in heat capacity at constant pressure. The value of Gibbs free energy change at any temperature T, ΔGD(T), was estimated using the Gibbs–Helmholtz equation (Eq. (2)) with values of ΔHm, Tm, and ΔCp:
Circular dichroism (CD) measurements. CD measurements were made using a Jasco J-810 spectropolarimeter equipped with a Peltier-type temperature controller with six accumulations. Protein concentration used for the far UV CD measurements was 0.6 mg/ml. A cell of 0.1-cm pathlength was used for the measurements. The protein samples were preincubated overnight at the desired solvent conditions. The necessary blank was subtracted from each measurement. All readings were made at 25°C. The CD instrument was routinely calibrated with D-10-camphorsulfonic acid.
Intrinsic fluorescence measurements. Fluorescence spectra of the protein samples were measured in a Perkin Elmer LS 55 spectrofluorimeter in a 3-mm quartz cell with both excitation and emission slits set at 10 nm. Protein concentration for all the experiments was 10 µM. For intrinsic fluorescence measurements, the excitation wavelength was 268 nm, while the emission spectra were recorded from 290-400 nm at 25°C. All measured spectra have been subtracted for the contribution of blanks. For the 1-anilino-8-naphthalene sulfonate (ANS) binding experiments, the excitation wavelength was 360 nm, and emission spectra were recorded from 400 to 600 nm. ANS concentration was kept at 16-fold that of the protein concentration. The concentration of ANS was determined experimentally using ε, the molar absorption coefficient value of 5000 M–1·cm–1 at 350 nm [19, 20] and was filtered before use to remove insoluble particles. Spectra were recorded in a 5-mm quartz cell at 25°C with excitation and emission slits of 10 nm at scanning speed of 100 nm/min in a Perkin-Elmer LS-55 spectroluminometer. Blanks were subtracted against corresponding samples.
RESULTS
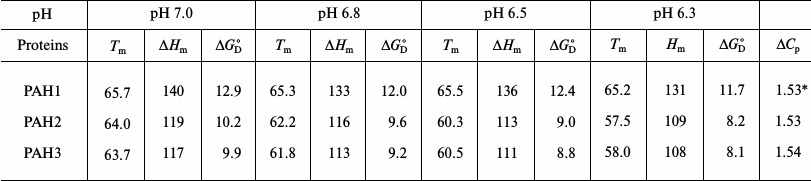
To investigate the possibility for differences in the thermodynamic stability of PAH domains, we first performed heat-induced denaturation of the proteins at different pH values (6.3, 6.5, 6.8, and 7.0) by following changes at θ222, the difference in CD signal at wavelength 222 nm, for all the three PAH proteins. We chose these pH values as the pH of the nucleus is slightly below 7.0 but higher than 6.0. In this study, pH 7.0 represents the optimum physiological pH condition. All denaturation curves were reversible. Figure 1 shows heat-induced denaturation profiles of PAH1, PAH2, and PAH3 at different pH values. Each denaturation curve of a protein at a given pH was analyzed for ΔHm and Tm using a nonlinear least squares method that involves fitting the entire data of the transition curve to Eq. (1) with all eight free parameters (aN, bN, cN, aD, bD, cD, ΔHm, and Tm). The thermodynamic parameters (ΔHm and Tm) for all the three proteins obtained at different pH values are given in Table 1. Values of ΔCp of the proteins were determined by plotting ΔHm and Tm values generated at the different pH values. The values of ΔCp evaluated in this manner were 1.53 and 1.54, respectively, for PAH2 and PAH3 (see Table 1). We could not evaluate the ΔCp of PAH1, as there was no significant change in the ΔHm and Tm values with change in pH. Since the estimated ΔCp values of PAH2 and PAH3 were identical, we used the same ΔCp for the estimation of ΔGDo of PAH1. Values of ΔGDo (the value of ΔGD at 25°C), estimated for the different PAH domains using Eq. (2), are also given in Table 1. Figure 2 shows plots of ΔTm (Tm of the protein at pH 7.0 – Tm at other pH values) versus pH (a-c) and ΔΔGDo (ΔGDo of the protein at pH 7.0 – ΔGDo at other pH values) versus pH (d-f). As evident from this figure, lowering the pH does not affect the stability of PAH1 but decreases the stability of PAH2 and PAH3 in terms of Tm and ΔGDo.
Table 1. Thermodynamic parameters of the PAH
domains at different pH values
Notes: The units of Tm, Hm,
ΔGDo, and
ΔCp are °C, kcal/mol, kcal/mol,
and kcal/mol, respectively. Errors in Tm,
Hm, ΔGDo, and
ΔCp are 0.2-1, 3-6, 7-9, and 5-7%,
respectively.
* The value has not been experimentally measured but based on
presumptions.
Fig. 1. Heat induced denaturation profile of PAH1 (a), PAH2 (b), and PAH3 (c) at different pH values as indicated in the figure.
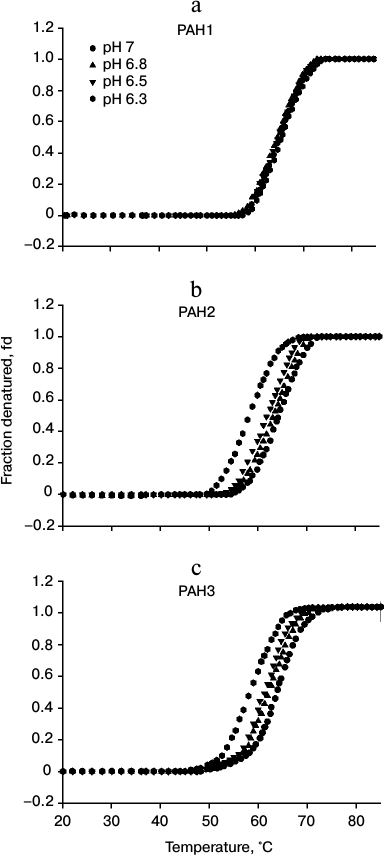
Fig. 2. pH dependence of protein stability. Plots of ΔTm versus pH (a-c) and ΔΔGDo versus pH (d-f) of PAH1, PAH2, and PAH3 at pH 7.0, 6.8, 6.5, and 6.3.
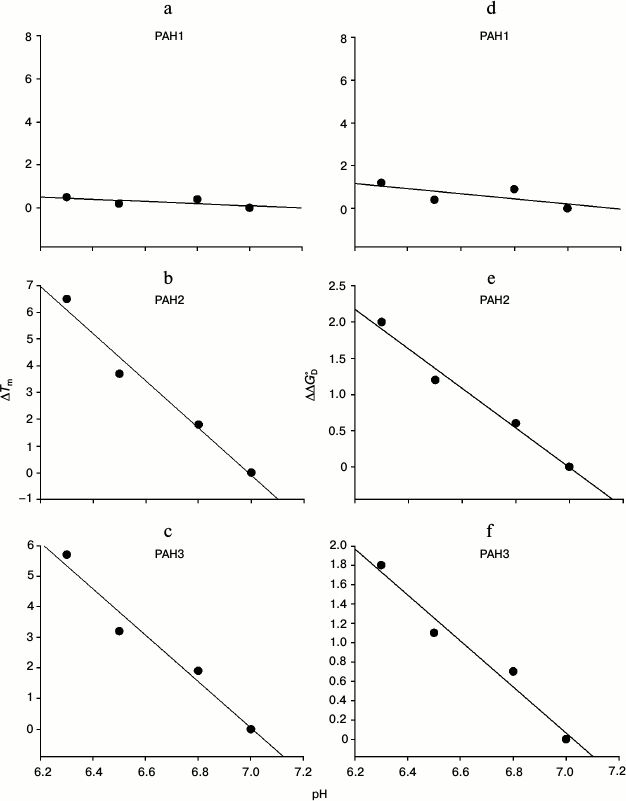
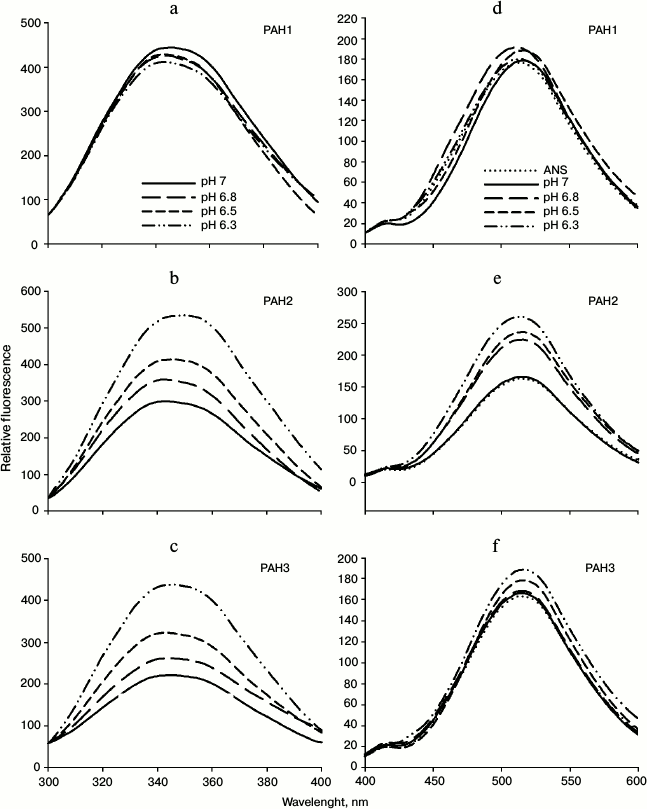
We then investigated the structural variations of the different PAH domains and evaluated how the native-state structural integrity is altered due to change in the pH. For this, we first measured far UV CD (secondary structure) of each of the PAH domains of hSin3B at different pH values (pH 6.3, 6.5, 6.8, and 7.0). It is seen in Fig. 3 that there is a decrease in the secondary structure of PAH2 and PAH3 in a pH-dependent manner, while the structure of PAH1 is not significantly affected by change in pH. Figure 4 (a-c) shows the effect of the pH on the tertiary structure of the different PAH domains in terms of the environment of tyrosine residues. It is also seen in this figure that there is an increase in the relative fluorescence intensity in the case of PAH2 and PAH3. However, there is no significant change in the relative fluorescence of PAH1 due to low pH. However, note that the peak maxima was at ~345 nm, which is the ideal peak for tryptophan, and not at ~310 nm, which should be the characteristic emission peak for tyrosine. None of the PAH domains have any tryptophan residues based on our sequencing report and other sequence information available in PubMed. Therefore, the observed peak at ~345 nm purely originates from tyrosine. Previous reports suggest that many proteins that lack tryptophan but have only tyrosine(s) exhibit peak maxima at ~345 nm due to the formation of tyrosinates (the conjugate phenolic base of tyrosine). Tyrosinates are likely formed via an intermolecular proton transfer from the excited state of one or more tyrosine residues to a suitable proton acceptor in the polypeptide. Aspartic and glutamic acid residues are the appropriate proton acceptors [20-23]. PAH1 and PAH3 have only one tyrosine residue, whereas PAH2 has two tyrosine residues. Interestingly, all tyrosine residues observed in the respective polypeptide sequence have either an adjacent or nearby aspartate residue. Figure 4 (d-f) clearly shows that the PAH1 and PAH3 domains do not bind to ANS, as evident from no significant increase in ANS fluorescence intensity and shift in λmax. However, PAH2 shows increase in relative ANS fluorescence but no shift in the λmax, indicating that there may be subtle binding of ANS. These results indicate that the different PAH domains have conformational variations.
Fig. 3. Effect of pH on the secondary structures of different PAH domains. Far-UV CD spectra (at 37°C) of PAH1 (a), PAH2 (b), and PAH3 (c) incubated overnight at the respective pH values (indicated in the figure).
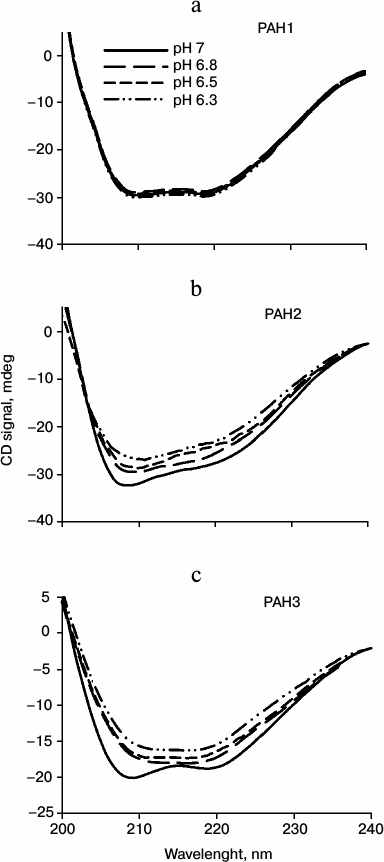
Fig. 4. Effect of pH on the tertiary structure of PAH domains. Intrinsic fluorescence spectra (a-c) and ANS fluorescence spectra (d-f) of PAH1, PAH2, and PAH3.
DISCUSSION
We have investigated the difference in the thermodynamic stability and structural variations of the different PAH domains of hSin3B and how alteration in pH affect the PAH domains. Thermodynamic stabilities of the three PAH domains were determined by measuring the heat-induced denaturation of the proteins, for which change in θ222, was monitored. At a given pH, ΔGDo was estimated using Eq. (2) with known values of ΔHm, Tm, and ΔCp. However, this estimation requires a large extrapolation. Hence, a large error may be associated with ΔGDo determination due to errors in the estimations of ΔHm, Tm, and ΔCp. We used Becktel and Schellman’s procedure [24] to determine the maximum and minimum errors associated with the ΔGDo determination at a given solvent condition. This procedure involves the estimation of ΔGDo of proteins using the maximum and minimum fitting parameter errors of ΔHm and ΔCp (one with maximum error in ΔHm and minimum error in ΔCp and the other with minimum error in ΔHm and maximum error in ΔCp) obtained from the analysis of individual denaturation curves to yield two different ΔGDo values (one minimum and one maximum). Because there were three independent measurements of ΔHm and Tm of a protein at the given pH, we obtained six values of ΔGDo (three maximum and three minimum values). All of these six values were used to determine the average ΔGDo and the mean error. It was observed that the mean error associated with the ΔGDo estimation was in the range 7-9% for all the proteins.
It is seen in Table 1 that PAH1 is more stable than PAH2 and PAH3 at physiological pH in terms of Tm and ΔGDo of the proteins. However, it appears that the protein stability for each of the PAH domains is different at physiological and nuclear pH values since the Tm and ΔGDo values are different at pH 7.0 and lower pH values (Table 1) indicating that change in pH might regulate the structure and stability of the PAH domains. Therefore, we investigated the effect of different pH values on the PAH domains. For this, we plotted ΔTm versus pH and ΔΔGDo versus pH (Fig. 2). As shown in this figure, for both PAH2 and PAH3 there is a linear relation between the protein stability and pH (in terms of ΔTm or ΔΔGDo), i.e. protein stability decreases with decrease in pH. However, the stability of PAH1 does not depend on pH. Thus, protein stability is not always a function of pH for the three PAH domains. The decrease in protein stability due to low pH in the case of PAH2 and PAH3 might mean that the structure should also be destabilized. To investigate this possibility, we measured secondary and tertiary structures of the proteins using far UV CD (Fig. 3) and tyrosine fluorescence (Fig. 4, a-c) as probes, respectively. It is seen in the figures that both the secondary and tertiary structures of PAH2 and PAH3 decrease on lowering pH. As expected, the structure of the PAH1 is not at all perturbed by change in pH. Thus, our thermodynamic and structural measurements are in agreement. The result clearly indicates that change in pH might help to regulate the function of hSin3B. In support to our conclusion, the presence of Sin3 has been reported not only in the nucleus, but also in cytoplasm and mitochondria [25-29]. In addition, there is different partitioning of hSin3B in both the nuclear and cytoplasmic compartments (unpublished results) under stress conditions. It has also been reported that difference in the pH of nucleus and cytoplasm also plays a role in intracellular localization and movement of various proteins between the nuclear and cytoplasmic compartments [30]. Therefore, structural and functional regulation of proteins by change in pH might be a general strategy for many proteins in cells.
Sin3B is a nuclear protein and therefore it is important to compare the structure and stability of the different PAH domains at each of nuclear pH value. ΔGDo values given in Table 1 at different nuclear pH values suggest that PAH1 is more stable than PAH2 and PAH3. Interestingly, the stability of both PAH2 and PAH3 are nearly identical at each of the nuclear pH values. We further investigated if the similarity in the thermodynamic stability results in similar structural properties in case of PAH2 and PAH3. Table 2 shows the structural comparison of the three PAH domains under nuclear pH conditions. It is seen in this table that the extent of decrease in secondary structure is higher for PAH2 than for PAH3, while there is no significant change in the secondary structure of PAH1 due to low pH. In agreement with this result, the tertiary structure of PAH2 (based on the relative tyrosine fluorescence) is also a little more disordered relative to PAH3, while the tertiary structure of PAH1 remains unaffected by change in pH. Thus, results on secondary and tertiary measurements indicate that there is subtle variation in the structure of the PAH domains. It has been reported that destabilization of the tertiary structure results in the exposure of hydrophobic clusters (that were buried in the interior) to the solvent [20]. We further examined the presence of hydrophobic clusters using ANS binding assay. As evident from Table 2, there is no binding of ANS to PAH1 and PAH3. However, in the case of PAH2 we observed an increase in ANS fluorescence intensity without having a shift in emission maxima, apparently indicating poor binding. All these data led us to believe that the conformation of the different PAH domains at each nuclear pH are different. PAH1 appears to have a very stable native structure, which with subtle variation in pH does not influence its structural integrity, while PAH2 and PAH3 might have relatively flexible structure. It was reported earlier that the structural flexibility and orientation of the PAH domains in case of mammalian Sin3B is crucial for having different binding partners for each of the PAH domains [5, 14, 15, 31]. Interestingly, human PAH1 that is conformationally stable (relative to PAH2 or PAH3) has so far been reported to have only one binding partner – HCF-1. Other than human, in lower organisms only two binding partners have been reported so far for PAH1, SMRTER in S. cerevisiae and Opi1 in Drosophila [31]. Thus, it seems that the evolutionarily lower conformational flexibility has restrained PAH1 not to interact with a large pool of transcription factors. Human PAH2 and PAH3 that are relatively flexible in structure have been reported to have a large number of binding partners [13]. Taken together, the results indicate that the difference in the conformation of native structure or flexibility of the different PAH domains could result in the recognition of different sets of binding partners.
Table 2. Effect of pH on the structure of
PAH domains of human Sin3B (hSin3B)
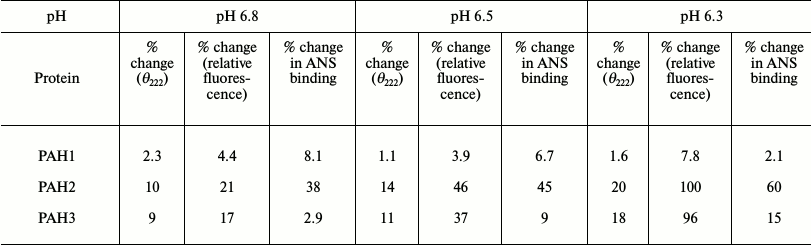
Note: Percentage change in θ222, relative fluorescence
change, and ANS binding has been evaluated relative to the observed
intensities at physiological pH.
In conclusion, our studies indicate at least two things: (i) the native state structures and stability of the different PAH domains is different at nuclear pH where Sin3 functions; (ii) PAH2 and PAH3 behave differently at both nuclear and physiological pH in terms of native state structure and stability, while the structure of PAH1 remains unaltered at both pH values. The study indicates the importance of structural heterogeneity in PAH domains in recruiting or recognizing a specific set of binding partners. Further research should focus on identifying the functional importance of PAH1 in hSin3B, as it represents a highly stable native structure.
This work was supported by grant from the Council of Scientific and Industrial Research (File No. 37 (1596/13/EMR-II)). L.R.S. and T.H. acknowledge Indian Council for Medical Research for the financial assistance provided in the form of research fellowship (File No. 3/1/3/JRF-2010/MBD-2 (33050)).
REFERENCES
1.Grzenda, A., Lomberk, G., Zhang, J.-S., and
Urrutia, R. (2009) Sin3: master scaffold and transcriptional
corepressor, Biochim. Biophys. Acta (BBA)-Gene Regul.
Mechanisms, 1789, 443-450.
2.Kadamb, R., Mittal, S., Bansal, N., Batra, H., and
Saluja, D. (2013) Sin3: insight into its transcription regulatory
functions, Europ. J. Cell Biol., 92, 237-246.
3.McDonel, P., Costello, I., and Hendrich, B. (2009)
Keeping things quiet: roles of NuRD and Sin3 co-repressor complexes
during mammalian development, Int. J. Biochem. Cell Biol.,
41, 108-116.
4.Hassig, C. A., Fleischer, T. C., Billin, A. N.,
Schreiber, S. L., and Ayer, D. E. (1997) Histone deacetylase activity
is required for full transcriptional repression by mSin3A, Cell,
89, 341-347.
5.Le Guezennec, X., Vermeulen, M., and Stunnenberg,
H. G. (2006) Molecular characterization of Sin3 PAH-domain interactor
specificity and identification of PAH partners, Nucleic Acids
Res., 34, 3929-3937.
6.Sahu, S. C., Swanson, K. A., Kang, R. S., Huang,
K., Brubaker, K., Ratcliff, K., and Radhakrishnan, I. (2008) Conserved
themes in target recognition by the PAH1 and PAH2 domains of the Sin3
transcriptional corepressor, J. Mol. Biol., 375,
1444-1456.
7.Ayer, D. E., Lawrence, Q. A., and Eisenman, R. N.
(1995) Mad-Max transcriptional repression is mediated by ternary
complex formation with mammalian homologs of yeast repressor Sin3,
Cell, 80, 767-776.
8.Yang, Q., Kong, Y., Rothermel, B., Garry, D. J.,
Bassel-Duby, R., and Williams, R. S. (2000) The winged-helix/forkhead
protein myocyte nuclear factor beta (MNF-beta) forms a co-repressor
complex with mammalian sin3B, Biochem. J., 345,
335-343.
9.Alland, L., Muhle, R., Hou, H., Potes, J., Chin,
L., Schreiber-Agus, N., and DePinho, R. A. (1997) Role for N-CoR and
histone deacetylase in Sin3-mediated transcriptional repression,
Nature, 387, 49-55.
10.Rayman, J. B., Takahashi, Y., Indjeian, V. B.,
Dannenberg, J.-H., Catchpole, S., Watson, R. J., te Riele, H., and
Dynlacht, B. D. (2002) E2F mediates cell cycle-dependent
transcriptional repression in vivo by recruitment of an
HDAC1/mSin3B corepressor complex, Genes Devel., 16,
933-947.
11.Spronk, C. A. E. M., Tessari, M., Kaan, A. M.,
Jansen, J. F. A., Vermeulen, M., Stunnenberg, H. G., and Vuister, G. W.
(2000) The Mad1-Sin3B interaction involves a novel helical fold,
Nature Struct. Mol. Biol., 7, 1100-1104.
12.Olsson, A., Olsson, I., and Dhanda, R. S. (2008)
Transcriptional repression by leukemia-associated ETO family members
can be independent of oligomerization and coexpressed hSIN3B and N-CoR,
Biochim. Biophys. Acta (BBA)-Gene Regul. Mechanisms,
1779, 590-598.
13.Silverstein, R. A., and Ekwall, K. (2005) Sin3: a
flexible regulator of global gene expression and genome stability,
Curr. Genet., 47, 1-17.
14.Van Ingen, H., Baltussen, M. A. H., Aelen, J.,
and Vuister, G. W. (2006) Role of structural and dynamical plasticity
in Sin3: the free PAH2 domain is a folded module in mSin3B, J. Mol.
Biol., 358, 485-497.
15.Swanson, K. A., Knoepfler, P. S., Huang, K.,
Kang, R. S., Cowley, S. M., Laherty, C. D., Eisenman, R. N., and
Radhakrishnan, I. (2004) HBP1 and Mad1 repressors bind the Sin3
corepressor PAH2 domain with opposite helical orientations, Nature
Struct. Mol. Biol., 11, 738-746.
16.Kumar, G. S., Xie, T., Zhang, Y., and
Radhakrishnan, I. (2011) Solution structure of the mSin3A
PAH2–Pf1 SID1 complex: a Mad1/Mxd1-like interaction disrupted by
MRG15 in the Rpd3S/Sin3S complex, J. Mol. Biol.,
408, 987-1000.
17.Van Ingen, H., Lasonder, E., Jansen, J. F. A.,
Kaan, A. M., Spronk, C. A. E. M., Stunnenberg, H. G., and Vuister, G.
W. (2004) Extension of the binding motif of the Sin3 interacting domain
of the Mad family proteins, Biochemistry, 43,
46-54.
18.Saluta, M., and Bell, P. A. (1998)
Troubleshooting GST fusion protein expression in E. coli,
Life Sci. News, 1.
19.Marty, A., Boiret, M., and Deumie, M. (1986) How
to illustrate ligand–protein binding in a class experiment: an
elementary fluorescent assay, J. Chem. Ed., 63,
365.
20.Lakowicz, J. R. (2007) Principles of
Fluorescence Spectroscopy, Springer.
21.Szabo, A. G., Lynn, K., Krajcarski, D., and
Rayner, D. M. (1979) Tyrosine fluorescence at 345 nm in proteins
lacking tryptophan, J. Luminesc., 18, 582-586.
22.Szabo, A. G., Lynn, K. R., Krajcarski, D. T., and
Rayner, D. M. (1978) Tyrosinate fluorescence maxima at 345 nm in
proteins lacking tryptophan at pH 7, FEBS Lett.,
94, 249-252.
23.Ruan, K., Li, J., Liang, R., Xu, C., Yu, Y.,
Lange, R., and Balny, C. (2002) A rare protein fluorescence behavior
where the emission is dominated by tyrosine: case of the 33-kDa protein
from spinach photosystem II. Biochem. Biophys. Res. Commun.,
293, 593-597.
24.Becktel, W. J., and Schellman, J. A. (1987)
Protein stability curves, Biopolymers, 26,
1859-1877.
25.Barnes, V. L., Strunk, B. S., Lee, I., Huttemann,
M., and Pile, L. A. (2010) Loss of the SIN3 transcriptional corepressor
results in aberrant mitochondrial function, BMC Biochem.,
11, 26.
26.Backues, S. K., Lynch-Day, M. A., and Klionsky,
D. J. (2012) The Ume6-Sin3-Rpd3 complex regulates ATG8 transcription to
control autophagosome size, Autophagy, 8,
1835-1836.
27.Kong, Q., Zeng, W., Wu, J., Hu, W., Li, C., and
Mao, B. (2010) RNF220, an E3 ubiquitin ligase that targets Sin3B for
ubiquitination, Biochem. Biophys. Res. Commun.,
393, 708-713.
28.Khochbin, S., Verdel, A., Lemercier, C., and
Seigneurin-Berny, D. (2001) Functional significance of histone
deacetylase diversity, Curr. Opin. Genet. Devel.,
11, 162-166.
29.Vega, A. V., Avila, G., and Matthews, G. (2013)
Interaction between the transcriptional corepressor Sin3B and
voltage-gated sodium channels modulates functional channel expression,
Sci. Rep., 3.
30.Cunningham, J., Estrella, V., Lloyd, M., Gillies,
R., Frieden, B. R., and Gatenby, R. (2012) Intracellular electric field
and pH optimize protein localization and movement, PloS One,
7, e36894.
31.Nomura, M., Uda-Tochio, H., Murai, K., Mori, N.,
and Nishimura, Y. (2005) The neural repressor NRSF/REST binds the PAH1
domain of the Sin3 corepressor by using its distinct short hydrophobic
helix, J. Mol. Biol., 354, 903-915.