REVIEW: Negative Feedback of Glycolysis and Oxidative Phosphorylation: Mechanisms of and Reasons for It
S. S. Sokolov1, A. V. Balakireva2, O. V. Markova1, and F. F. Severin1*
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia; fax: +7 (495) 939-0338; E-mail: severin@belozersky.msu.ru2Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia; fax: +7 (495) 939-4195
* To whom correspondence should be addressed.
Received December 25, 2014; Revision received January 26, 2015
There are two main pathways of ATP biosynthesis: glycolysis and oxidative phosphorylation. As a rule, the two pathways are not fully active in a single cell. In this review, we discuss mechanisms of glycolytic inhibition of respiration (Warburg and Crabtree effects). What are the reasons for the existence of this negative feedback? It is known that maximal activation of both processes can cause generation of reactive oxygen species. Oxidative phosphorylation is more efficient from the energy point of view, while glycolysis is safer and favors biomass synthesis. This might be the reason why quiescent cells are mainly using oxidative phosphorylation, while the quickly proliferating ones – glycolysis.
KEY WORDS: glycolysis, oxidative phosphorylation, mitochondria, Crabtree effectDOI: 10.1134/S0006297915050065
Abbreviations: FCCP, carbonyl cyanide 4-(fluoromethoxy)phenylhydrazone; ROS, reactive oxygen species; VDAC, voltage-dependent anion channel; YP, yeast extract-peptone.
During glycolysis, one molecule of glucose is converted into two
molecules of pyruvate, and this is accompanied by reduction of two
molecules of NAD+ and phosphorylation of two ADP molecules
[1]. Oxidation of pyruvate and NADH in the Krebs
cycle results in the formation of additional 28-30 ATP molecules. This
means that from the energy point of view under aerobic conditions, the
main function of glycolysis is not the energetic one but the conversion
of glucose and other hexoses into substrates of Krebs and oxidative
phosphorylation [2]. The velocity and the direction
of glycolytic flow are determined mainly by the regulation of enzymes
responsible for the irreversible steps: hexokinase,
phosphofructokinase-1, and pyruvate kinase. The excess of Krebs cycle
substrates such as acetyl-CoA or citrate, and ATP also, carry on the
negative feedback regulation of glycolysis by inhibiting the activity
of phosphofructokinase-1 to adjust its activity to match the one of the
respiratory chain [3].
At the same time, most of the cells possess mechanisms disturbing such regulation, i.e. Crabtree and Warburg effects. Crabtree effect was discovered by Herbert Crabtree [4] and describes a reversible phenomenon of yeast Saccharomyces cerevisiae. The phenomenon is that under aerobic conditions during glycolysis S. cerevisiae (unlike, for example, Kluyveromyces yeast) cells produce ethanol and do not channel the carbon flow through the Krebs cycle. The Crabtree effect is typical not only for S. cerevisiae but also for proliferating cells, in particular, cancerous ones, and cells infected with certain viruses. A similar phenomenon, described for cancer cells, was called the Warburg effect. It was noticed that even under aerobic conditions cancer cells repress respiration and produce lactate, which is synthesized during anaerobic glycolysis, indicating that the energy metabolism of cancer cells is based on glycolysis [5].
Therefore, glycolysis can inhibit respiration, or respiration can inhibit glycolysis. Thus, the question arises: why do the cells not prefer to use both pathways simultaneously at full power? We think that the most likely explanation is the following. Active respiration under the conditions of high ATP/ADP ratio (when glycolysis is active) can cause mitochondrial hyper-polarization as in such case the membrane potential is not being consumed for ATP production. As a result, the formation of reactive oxygen species (ROS) in the respiratory chain is strongly increased [6]. Interestingly, a number of cancerous cell lines that need ROS for proliferation use this mechanism for ROS production. Nevertheless, even in this case there is no simultaneous activation of glycolysis and respiration. Instead of raising ATP/ADP ratio, such cells inhibit mitochondrial ATP synthase and in this way prevent the dissipation of mitochondrial membrane potential and, as a result, cause mitochondrial hyperpolarization [7].
The energy metabolism of resting cells differs significantly from the one of proliferating cells. Cancerous and other actively dividing (e.g. embryonic [8]) cells frequently demonstrate active glycolysis even under the physiological concentration of oxygen [5, 9]. Possibly, the glycolytic bias of proliferating cells can be explained by their needs for biomass production: inhibition of respiration directs carbon flow into lactate production instead of carbon dioxide [10]. Another possible explanation is that glycolytic bias of the proliferating cells is a precaution against a sudden hypoxia: a drop of oxygen in a dividing cell can cause a sharp decrease of ATP/ADP ratio leading to mitotic catastrophe. Apart from this, damage to actively respiring mitochondria can cause a sharp decrease of ROS production. Possibly, repression of mitochondrial activity is a precaution against their malfunctioning [11, 12].
Similar to proliferating cells, the S. cerevisiae yeast when growing on glucose produce ethanol and grow much faster than when they grow on non-fermentable carbon sources. What are the molecular mechanisms of respiration-to-glycolysis switch in proliferating cells? As aforementioned, high ATP/ADP ratio was shown to inhibit glycolysis [13, 14]. Therefore, a decrease in cytoplasmic [ATP] is a necessary step for switching to glycolysis. Otto Warburg suggested that the damage to mitochondria in cancer cells is the primary reason for such decrease [5]. Experimental evidence did not support this idea: the mitochondria isolated from cancer cells are capable of conducting oxidative phosphorylation like the ones isolated from the normal cells [15, 16]. One way to repress mitochondrial activity in cancer cells is reversible closure of the voltage-dependent anion channels (VDAC) by tubulin molecules [17]. VDAC is the major channel in the outer mitochondrial membrane responsible for metabolite transport. It was shown that the elevated concentration of free tubulin (which is typical for cancer cells) is able to block VDAC and in this way to slow mitochondrial activity and in this way to decrease cytoplasmic ATP/ADP ratio [18].
An increase of free tubulin concentration in the proliferating cells is because in mitosis microtubules are much more dynamic than in the interphase: fast growth and shrinking are necessary to establish the correct chromosomal centromeric regions [19]. Switching from respiration to glycolysis renders cancer cells independent of oxygen concentration and helps them avoid apoptosis [20]. There are multiple ways to execute this switching. To increase the rates of glycolysis, cancer cells can overexpress the high-affinity glucose transporters Glut1 and Glut3 [21].
In line with this, it was shown that high levels of glucose induced by diabetes result in p90RSK (p90 14 ribosomal S6 kinase)-dependent blockage of mitochondrial response to high glucose concentration. This signaling module is a part of the glucose → MEK5 → ERK5 → p90RSK cascade. Importantly, MEK5 and ERK5 kinases are typically activated in proliferating, including cancerous, cells [22]. It was also shown that p90RSK targets p53, one of the general factors affecting mitochondrial functioning. Apart from this, there is a link between the Crabtree effect and mitochondrial morphology, i.e. mitochondrial fragmentation and generation of ROS in response to high glucose concentration [23]. Possibly, mitochondrial fragmentation is an intermediate step between p90RSK activation and inhibition of respiration [24].
The comparison of transcriptomes of S. cerevisiae strains growing on glucose and galactose showed that transcription factors Bas1p, Pho2p, and Gcn4p play the central roles in the regulation of the respiration-to-glycolysis switch. Bas1p and Pho2p are responsible for the distribution of metabolic flows that accompany the switch from galactose to glucose consumption. When glucose concentration is high, the expression of Bas1p is also high. Apart from this, mutations in RAS2 gene decrease the levels of Crabtree effects and the rate of growth on glucose by decreasing of Gcn4p activity [25].
The Warburg effect in cancer cells and the Crabtree effect in yeast display several similarities: suppression of oxidative metabolism and active fermentation even in the presence of available oxygen. Moreover, both types of cells overexpress glycolytic enzymes in the presence of glucose. The only difference is that the cancer cells typically overexpress cytoplasmic lactate dehydrogenase, while high glucose in yeast causes overexpression of pyruvate dehydrogenase, causing accumulation of acetaldehyde that is then transformed into ethanol by alcohol dehydrogenase [26].
The molecular mechanism of Crabtree effect is still not known. Originally, it was thought to be due to the competition between ATP/ADP antiporter and glycolytic enzymes for ADP [27], but later studies did not confirm this: the Michaelis constant of the antiporter for ADP is approximately 100 times lower than the one of the glycolytic enzymes [28]. Because in several cancer cell lines the Crabtree effect can be inhibited by the addition of extracellular phosphate, it was suggested that phosphate per se is the inhibitor [29]. This was in agreement with the fact that in cancer cells the addition of glucose decreases the levels of intracellular phosphate [30]. It was suggested that the value of thermodynamic phosphate potential, ATP/ADP·P, which affects the majority of metabolic processes in the cells [28], also regulates the onset of the Crabtree effect [31].
Calcium ions were also suggested to be a mediator of Crabtree effect [32]. In Ehrlich tumor cells, glucose addition stably increases cytosolic [Ca2+], while in these cells even micromolar [Ca2+] completely inhibits mitochondrial ATPase activity. It appeared that calcium binds the inhibitor protein of mitochondrial ATPase, thus repressing respiration and ATP synthesis [33].
Oxygen concentration also affects the Crabtree effect. It was shown that an increase of [O2] in the media from 1.2 to 2.7 µM induces the fermentative-to-mixed (fermentative–oxidative) metabolic switch in yeast. This is accompanied by activations of the Krebs cycle and NADH transport from mitochondria to cytoplasm. Thus, high concentrations of oxygen stimulate respiration even in the presence of high concentration of glucose. Nevertheless, under such conditions the enzymatic production of ethanol is still the main pathway of oxidation of intracellular NADH. For these reasons, the authors of this study suggest that a decrease in the rate of mitochondrial NADH oxidation is the main mechanism behind the Crabtree effect [34].
The yeast Pichia guilliermondii lacks the Crabtree effect and does not show mixed metabolism under aerobic conditions. Nevertheless, it was shown that a deleterious mutation in the CAT8 gene coding for a global activator of transcription causes a 20-fold increase in the production of ethanol. It appeared that the expression of the Krebs cycle enzymes, respiratory complexes, and activators of respiration (e.g. Hap4) was repressed in the mutant, while the genes responsible for fermentation were overexpressed. Thus, a simple genetic manipulation can activate the Crabtree effect [35].
It was also suggested that an intermediate product of glycolysis under certain conditions could inhibit respiration and serve as a mediator of the Crabtree effect. One of the candidates is fructose-1,6-diphosphate. Under physiological conditions, fructose-1,6-diphosphate decreases the activity of mitochondrial complexes III and IV [36]. It was shown that the Crabtree effect can be modeled on mitochondria isolated from rat liver by incubating them with fructose-1,6-diphosphate at concentrations similar to the ones present in hepatoma cells [36]. These results also demonstrate that the activation of the Crabtree effect does not require any permanent damage to mitochondria, but that its induction is reversible.
How does fructose-1,6-diphosphate interact with mitochondria? It was shown that both glucose-6-phosphate and fructose-1,6-diphosphate interact with mitochondrial unspecific channel (ScMUC): glucose-6-phosphate binds the channel, opens it partly causing proton leakage, uncoupling respiration and oxidative phosphorylation and increasing oxygen consumption. On the contrary, fructose-1,6-diphosphate addition closes the channel and decreases the rate of respiration. It appeared that fructose-1,6-bisphosphate reverses the effect of glucose-6-phosphate, which suggests that fructose-1,6-bisphosphate is the key molecule regulating the activity of ScMUC and the Crabtree effect itself [37].
We decided to test the hypothesis that fructose-1,6-diphosphate represses mitochondrial activity. To do that we used yeast strains with inactivated phosphoglycerate mutase (Δgpm1). The enzyme belongs to the glycolytic machinery; it catalyzes the reversible transfer of a phosphate group from C-2 to C-3 glycerol group forming 2-phosphoglycerate (Fig. 1). The Δgpm1 strain can grow in the presence of a mixture of glycerol and acetate (or ethanol) as carbon sources. In this case acetate (or ethanol) is metabolized downstream of phosphoglycerate mutase and produces substrates for the Krebs cycle, while glycerol is consumed by the enzymes upstream of phosphoglycerate mutase and serves as a substrate for gluconeogenesis [38]. It was shown that addition of glucose inhibits growth of Δgpm1 cells [39]. The authors suggested that glucose repression is the reason for the inhibition [39]. It is known that in the presence of glucose, S. cerevisiae yeast cells repress mitochondrial biogenesis at the level of transcription, and this phenomenon was called glucose repression. Indeed, repression of mitochondrial energy function must be lethal for Δgpm1 cells because the active fragment of the glycolytic pathway is unable to generate ATP when the cells are growing on the mixture of glycerol and acetate (Fig. 1). We confirmed the data of Papini et al. [38] and showed that, similar to glucose, galactose inhibits the growth of Δgpm1 cells (Fig. 2). As galactose does not activate glucose repression [40], one can speculate that the inhibition was due to the Crabtree effect. Indeed, as in Δgpm1 cells the metabolism of six-carbon sugars is blocked at the level of phosphoglycerate, an addition of such substrates is expected to induce accumulation of fructose-1,6-diphosphate (Fig. 1).
Fig. 1. A scheme of glycolysis.
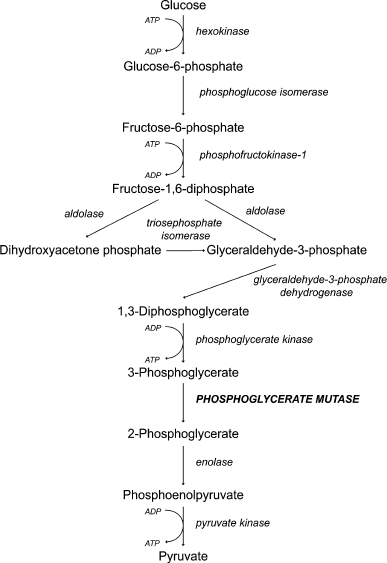
Fig. 2. Growth of the wild type (wt) and Δgpm1 cells on solid media containing ethanol and glycerol; ethanol, glycerol and galactose, or ethanol, glycerol and glucose as carbon sources. The strains were grown on solid YP/ethanol/glycerol medium, resuspended in water at 107 cells/ml, and then plated (10-µl samples) in serial 10-fold dilutions as shown by the figure. The colonies were photographed after 3 days of incubation at 30°C. Ethanol, glycerol, and the sugars were used at 1% concentration.
Unexpectedly, it appeared that neither galactose (data not shown) nor glucose suppresses the oxygen consumption rate by Δgpm1 cells (table). At the same time, when glucose is added to wild-type cells, activation of respirations takes place very rapidly – within seconds (data not shown). These data argue against the role of fructose-1,6-diphosphate as an intermediate of the Crabtree effect. It is important to mention that relatively slow mechanisms of the Crabtree effect have also been described, e.g. the ones relying on transcriptional changes [41]. Possibly, something similar takes place in our experimental system. We are currently screening for genes whose deletions allow Δgpm1 cells to grow in the presence of galactose. Hopefully, the results of this work will help to understand the mechanisms of mitochondrial repressions during activation of glycolysis.
Rates of oxygen consumption (nmol O2/min ×
2·107) by wild-type and Δgpm1 cells
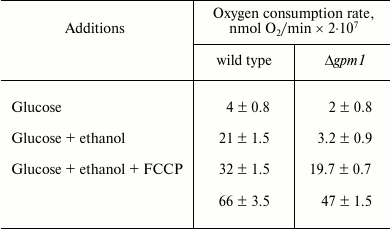
Note: Measuring of respiration of intact cells. Glucose and ethanol were
used at 0.2% concentrations, FCCP at 2 µM. The measurements
were performed as described in [34] with the
following modifications. The cells were grown in YP/ethanol/glycerol
medium. To deplete the intracellular respiration substrate prior to the
measurements, the cells were incubated with 2 mM dinitrophenol at
room temperature for 45 min.
This work was supported by the Russian Science Foundation grant 14-24-00107.
REFERENCES
1.Barer, A. P. (1931) A study of glycolysis, J.
Clin. Invest., 10, 507-520.
2.Vitols, E., and Linnane, A. W. (1961) Studies on
the oxidative metabolism of Saccharomyces cerevisiae. II.
Morphology and oxidative phosphorylation capacity of mitochondria and
derived particles from baker’s yeast, J. Biophys. Biochem.
Cytol., 9, 701-710.
3.Van den Brink, J., Canelas, A. B., van Gulik, W.
M., Pronk, J. T., Heijnen, J. J., de Winde, J. H., and Daran-Lapujade,
P. (2008) Dynamics of glycolytic regulation during adaptation of
Saccharomyces cerevisiae to fermentative metabolism, Appl.
Environ. Microbiol., 74, 5710-5723.
4.Crabtree, H. G. (1928) The carbohydrate metabolism
of certain pathological overgrowths, Biochem. J., 22,
1289-1298.
5.Warburg, O. (1956) On the origin of cancer cells,
Science, 123, 309-314.
6.Korshunov, S. S., Skulachev, V. P., and Starkov, A.
A. (1997) High protonic potential actuates a mechanism of production of
reactive oxygen species in mitochondria, FEBS Lett., 416,
15-18.
7.Martinez-Reyes, I., and Cuezva, J. M. (2014) The
H+-ATP synthase: a gate to ROS-mediated cell death or cell
survival, Biochim. Biophys. Acta, 1837, 1099-1112.
8.Krisher, R. L., and Prather, R. S. (2012) A role
for the Warburg effect in preimplantation embryo development: metabolic
modification to support rapid cell proliferation, Mol. Reprod.
Dev., 79, 311-320.
9.Harvey, A. J., Kind, K. L., and Thompson, J. G.
(2002) REDOX regulation of early embryo development,
Reproduction, 123, 479-486.
10.Vander Heiden, M. G., Cantley, L. C., and
Thompson, C. B. (2009) Understanding the Warburg effect: the metabolic
requirements of cell proliferation, Science, 324,
1029-1033.
11.Zorov, D. B., Juhaszova, M., and Sollott, S. J.
(2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release, Physiol. Rev., 94, 909-950.
12.Slavov, N., Macinskas, J., Caudy, A., and
Botstein, D. (2011) Metabolic cycling without cell division cycling in
respiring yeast, Proc. Natl. Acad. Sci. USA, 108,
19090-19095.
13.Hers, H. G., and Van Schaftingen, E. (1982)
Fructose 2,6-bisphosphate 2 years after its discovery, Biochem.
J., 206, 1-12.
14.Mor, I., Cheung, E. C., and Vousden, K. H. (2011)
Control of glycolysis through regulation of PFK1: old friends and
recent additions, Cold. Spring Harb. Symp. Quant. Biol.,
76, 211-216.
15.Nakashima, R. A., Paggi, M. G., and Pedersen, P.
L. (1984) Contributions of glycolysis and oxidative phosphorylation to
adenosine 5′-triphosphate production in AS-30D hepatoma cells,
Cancer Res., 44, 5702-5706.
16.Singleterry, J., Sreedhar, A., and Zhao, Y.
(2014) Components of cancer metabolism and therapeutic interventions,
Mitochondrion, 17, 50-55.
17.Maldonado, E. N., Patnaik, J., Mullins, M. R.,
and Lemasters, J. J. (2010) Free tubulin modulates mitochondrial
membrane potential in cancer cells, Cancer Res., 70,
10192-10201.
18.Rostovtseva, T. K., and Bezrukov, S. M. (2008)
VDAC regulation: role of cytosolic proteins and mitochondrial lipids,
J. Bioenerg. Biomembr., 40, 163-170.
19.Mitchison, T. J. (1988) Microtubule dynamics and
kinetochore function in mitosis, Annu. Rev. Cell Biol.,
4, 527-549.
20.Schlisio, S. (2009) Neuronal apoptosis by prolyl
hydroxylation: implication in nervous system tumours and the Warburg
conundrum, J. Cell Mol. Med., 13, 4104-4112.
21.Macheda, M. L., Rogers, S., and Best, J. D.
(2005) Molecular and cellular regulation of glucose transporter (GLUT)
proteins in cancer, J. Cell Physiol., 202, 654-662.
22.Zhou, G., Bao, Z. Q., and Dixon, J. E. (1995)
Components of a new human protein kinase signal transduction pathway,
J. Biol. Chem., 270, 12665-12669.
23.Yu, T., Sheu, S. S., Robotham, J. L., and Yoon,
Y. (2008) Mitochondrial fission mediates high glucose-induced cell
death through elevated production of reactive oxygen species,
Cardiovasc. Res., 79, 341-351.
24.Redman, E. K., Brookes, P. S., and Karcz, M. K.
(2013) Role of p90(RSK) in regulating the Crabtree effect: implications
for cancer, Biochem. Soc. Trans., 41, 124-126.
25.Martinez, J. L., Bordel, S., Hong, K. K., and
Nielsen, J. (2014) Gcn4p and the Crabtree effect of yeast: drawing the
causal model of the Crabtree effect in Saccharomyces cerevisiae
and explaining evolutionary trade-offs of adaptation to galactose
through systems biology, FEMS Yeast Res., 14,
654-662.
26.Diaz-Ruiz, R., Rigoulet, M., and Devin, A. (2011)
The Warburg and Crabtree effects: on the origin of cancer cell energy
metabolism and of yeast glucose repression, Biochim. Biophys.
Acta, 1807, 568-576.
27.Gatt, S., and Racker, E. (1959) Regulatory
mechanisms in carbohydrate metabolism. I. Crabtree effect in
reconstructed systems, J. Biol. Chem., 234,
1015-1023.
28.Veech, R. L., Lawson, J. W., Cornell, N. W., and
Krebs, H. A. (1979) Cytosolic phosphorylation potential, J. Biol.
Chem., 254, 6538-6547.
29.Koobs, D. H. (1972) Phosphate mediation of the
Crabtree and Pasteur effects, Science, 178, 127-133.
30.Rodriguez-Enriquez, S., Juarez, O.,
Rodriguez-Zavala, J. S., and Moreno-Sanchez, R. (2001) Multisite
control of the Crabtree effect in ascites hepatoma cells, Eur. J.
Biochem., 268, 2512-2519.
31.Sussman, I., Erecinska, M., and Wilson, D. F.
(1980) Regulation of cellular energy metabolism: the Crabtree effect,
Biochim. Biophys. Acta, 591, 209-223.
32.Evtodienko, Iu. V., and Teplova, V. V. (1996)
Biological role and mechanisms of realization of the Crabtree effect in
rapidly proliferating cells. The role of Ca2+ ions,
Biochemistry (Moscow), 61, 1423-1431.
33.Wojtczak, L. (1996) The Crabtree effect: a new
look at the old problem, Acta Biochim. Pol., 43,
361-368.
34.Aceituno, F. F., Orellana, M., Torres, J.,
Mendoza, S., Slater, A. W., Melo, F., and Agosin, E. (2012) Oxygen
response of the wine yeast Saccharomyces cerevisiae EC1118 grown
under carbon-sufficient, nitrogen-limited enological conditions,
Appl. Environ. Microbiol., 78, 8340-8352.
35.Qi, K., Zhong, J. J., and Xia, X. X. (2014)
Triggering respirofermentative metabolism in the crabtree-negative
yeast Pichia guilliermondii by disrupting the CAT8 gene,
Appl. Environ. Microbiol., 80, 3879-3887.
36.Diaz-Ruiz, R., Averet, N., Araiza, D., Pinson,
B., Uribe-Carvajal, S., Devin, A., and Rigoulet, M. (2008)
Mitochondrial oxidative phosphorylation is regulated by fructose
1,6-bisphosphate. A possible role in Crabtree effect induction? J.
Biol. Chem., 283, 26948-26955.
37.Rosas-Lemus, M., Uribe-Alvarez, C.,
Chiquete-Felix, N., and Uribe-Carvajal, S. (2014) In Saccharomyces
cerevisiae fructose-1,6-bisphosphate contributes to the Crabtree
effect through closure of the mitochondrial unspecific channel,
Arch. Biochem. Biophys., 555/556, 66-70.
38.Papini, M., Nookaew, I., Scalcinati, G., Siewers,
V., and Nielsen, J. (2010) Phosphoglycerate mutase knock-out mutant
Saccharomyces cerevisiae: physiological investigation and
transcriptome analysis, Biotechnol. J., 5, 1016-1027.
39.Trumbly, R. J. (1992) Glucose repression in the
yeast Saccharomyces cerevisiae, Mol. Microbiol.,
6, 15-21.
40.Conrad, M., Schothorst, J., Kankipati, H. N., Van
Zeebroeck, G., Rubio-Texeira, M., and Thevelein, J. M. (2014) Nutrient
sensing and signaling in the yeast Saccharomyces cerevisiae,
FEMS Microbiol. Rev., 38, 254-299.
41.Swerdlow, R. H., Lezi, E., Aires, D., and Lu, J.
(2013) Glycolysis-respiration relationships in a neuroblastoma cell
line, Biochim. Biophys. Acta, 1830, 2891-2898.