Expression of Neuronal and Signaling Proteins in Penumbra around a Photothrombotic Infarction Core in Rat Cerebral Cortex
S. V. Demyanenko1, S. N. Panchenko2, and A. B. Uzdensky1*
1Academy of Biology and Biotechnology, Southern Federal University, 344090 Rostov-on-Don, Russia; fax: +7 (863) 223-0837; E-mail: auzd@yandex.ru2Rostov State Medical University, 344022 Rostov-on-Don, Russia; E-mail: pansvn@mail.ru
* To whom correspondence should be addressed.
Received January 12, 2015; Revision received March 11, 2015
Photodynamic impact on animal cerebral cortex using water-soluble Bengal Rose as a photosensitizer, which does not cross the blood-brain barrier and remains in blood vessels, induces platelet aggregation, vessel occlusion, and brain tissue infarction. This reproduces ischemic stroke. Irreversible cell damage within the infarction core propagates to adjacent tissue and forms a transition zone – the penumbra. Tissue necrosis in the infarction core is too fast (minutes) to be prevented, but much slower penumbral injury (hours) can be limited. We studied the changes in morphology and protein expression profile in penumbra 1 h after local photothrombotic infarction induced by laser irradiation of the cerebral cortex after Bengal Rose administration. Morphological study using standard hematoxylin/eosin staining showed a 3-mm infarct core surrounded by 1.5-2.0 mm penumbra. Morphological changes in the penumbra were lesser and decreased towards its periphery. Antibody microarrays against 224 neuronal and signaling proteins were used for proteomic study. The observed upregulation of penumbra proteins involved in maintaining neurite integrity and guidance (NAV3, MAP1, CRMP2, PMP22); intercellular interactions (N-cadherin); synaptic transmission (glutamate decarboxylase, tryptophan hydroxylase, Munc-18-1, Munc-18-3, and synphilin-1); mitochondria quality control and mitophagy (PINK1 and Parkin); ubiquitin-mediated proteolysis and tissue clearance (UCHL1, PINK1, Parkin, synphilin-1); and signaling proteins (PKBα and ERK5) could be associated with tissue recovery. Downregulation of PKC, PKCβ1/2, and TDP-43 could also reduce tissue injury. These changes in expression of some neuronal proteins were directed mainly to protection and tissue recovery in the penumbra. Some upregulated proteins might serve as markers of protection processes in a penumbra.
KEY WORDS: stroke, neurodegeneration, penumbra, proteomicsDOI: 10.1134/S0006297915060152
Abbreviations: CRMP2, collapsin response mediator protein 2; DYRK1A, dual-specificity tyrosine-phosphorylated regulated kinase 1A; ERK5, extracellular regulated kinase 2; GABA, γ-butyric acid; MAP1, microtubule-associated protein 1; NAV3, neuron navigator 3 protein; PINK1, PTEN-induced mitochondrial protein kinase; PKBα, protein kinase Bα; PKC, protein kinase C; PKCβ1, protein kinase C isoform β1; PMP22, peripheral myelin protein 22; PTI, photothrombotic infarction; SIRT1, NAD+-dependent deacetylase sirtuin-1; TDP-43, transactivation response DNA-binding protein; UCHL1, ubiquitin C-terminal hydrolase L1.
Ischemic stroke is one of the major factors of human disability and
death. Acute focal ischemia caused by occlusion of blood vessels leads
very quickly, within a few minutes, to ATP depletion, generation of
reactive oxygen species, membrane injury, loss of ion gradients,
depolarization, excitotoxicity, cell death, and tissue edema. These
injurious processes propagate from the infarction core to surrounding
tissue and kill cells in the adjacent transition zone (penumbra) [1, 2]. Acute cell necrosis in the
infarct core cannot be prevented. However, tissue damage in the
penumbra develops slower, during hours and days, and this
“therapeutic window” provides time for neuroprotection and
decrease in neuropathological consequences [1-3]. However, most current neuroprotective drugs are
not sufficiently effective. For example, detailed clinical study of
cerebrolysin, suggested previously as a promising neuroprotector, did
not show a significant difference in lethality of patients and side
effects between this drug and placebo [4].
Therefore, the comprehensive investigation of biochemical mechanisms
that regulate neurodegeneration and neuroprotection in penumbra is
necessary for development of new approaches to treatment of stroke
consequences.
Ligation-induced occlusion of the middle cerebral artery or insertion of a silicone-coated nylon thread are the most popular experimental models of ischemic stroke. Thrombotic occlusion can be also created by injection of thrombin or blood clots. But these methods are not always well controlled and reproducible. Photothrombotic infarction (PTI) induced by photodynamic impact and local laser irradiation create more reliable and reproducible cerebral infarction [5-11].
Photodynamic effect on stained cells is based on energy transfer of the photoexcitation energy from a photosensitizer molecule to oxygen, which is thereby transformed to highly toxic singlet form. Singlet oxygen and other reactive oxygen species induce oxidative stress and cell death. Photodynamic therapy is currently used in oncology for tumor destruction [12]. Photodynamic induction of local thrombosis in the animal brain is a non-traditional application of the photodynamic effect. With this method, local laser irradiation after administration of the water-soluble and cell-impermeable photosensitizer Bengal Rose, which does not cross the blood–brain barrier and remains in the blood vessels, causes photosensitization and local oxidative damage of the endothelium and basal lamina, platelet aggregation, and occlusion of microvessels in the animal brain. This quickly, in some minutes, induces local PTI of the brain tissue. This stroke model is noninvasive. The injury location, size, and degree are controlled and reproducible [5-11]. Brain microinfarction with occlusion of small brain vessels can induce cognitive impairment, dementia, and other neurological disorders. The mechanisms of ischemic injury of the nervous tissue and its recovery after microinfarction, in particular the involvement of diverse neurospecific and signaling proteins in the tissue responses are insufficiently studied [13-15].
Modern proteomic techniques provide information on expression of hundreds of proteins in diverse biological samples [16-19]. To characterize changes in expression of various neuronal and signaling proteins in the penumbra after PTI, we used proteomic microarrays that provide simultaneous study of the difference between expression of 224 signaling and neuronal proteins in photosensitized rat cerebral cortex as compared with the control contralateral brain cortex.
MATERIALS AND METHODS
The experiments were performed on adult male Wistar rats (200-250 g). The animal holding room was maintained at temperature 22-26°C, 12-h light/dark schedule, and air exchange rate of 18 changes per hour. The animal care protocol corresponded to the institutional guidelines. All experimental procedures were performed according to the European Union guidelines 86/609/EEC for use of experimental animals.
Unilateral focal PTI in the somatosensory rat cortex was induced according to a modified method [7]. The rats were anaesthetized with chloral hydrate dissolved in 0.9% NaCl saline (300 mg/kg, i.p.). After the longitudinal incision of the skull skin, the periosteum was removed. Bengal Rose (Sigma-Aldrich Co, USA; 20 mg/kg) was injected in the v. subclavia. The rats were fixed in stereotaxis and unilateral irradiation of the somatosensory cortex (fields: FL, forelimb sensorimotor cortex; HL, hindlimb sensorimotor cortex; Par1, parietal primary somatosensory cortex [20]) was performed through the cranial bone using 532 nm laser beam (64 mW/cm2, 3 mm diameter, 30 min). The body temperature was maintained at 36.7-37.5°C.
One hour after the laser irradiation, the animals were euthanized with chloral hydrate overdose (600 mg/kg, i.p.) and decapitated. The infarction core was visualized by 15 min staining with 1% 3-phenyl-tetrazolium chloride (Sigma-Aldrich) solution at 37°C. For histological study, the rats were subjected to transcardial perfusion with 10% buffered formalin (pH 7.2) at 1 h after irradiation under chloral hydrate anesthesia. The extracted brains were post-fixed with formalin. The paraffin-embedded tissue samples were cross-sectioned by 6-8 µm slices and mounted on slides. After deparaffination, the sections were stained with hematoxylin and eosin, dehydrated by ethanol, cleared by xylene, mounted in Canada balsam, and examined under the microscope. All observed large pyramidal neurons were divided into three categories: normal, reversibly damaged (hypochromic or hyperchromic), and pyknotic (Table 1). Their numbers were counted using the ocular grid at 400-fold magnification in six randomized visual fields on three slices from irradiated and untreated contralateral hemisphere (control) in four rats. The average number of neurons per visual field at 1 h after PTI was compared to that in the control group using ANOVA test (Statistica 6.1).
In the proteomic study, we used the Panorama Antibody Array – Neurobiology kit (NBAA5; Sigma-Aldrich) that contains two identical microarrays, which are the nitrocellulose-coated glass slides containing 448 microdroplets with immobilized antibodies against 224 neuronal and signaling proteins. Each of 32 microdroplet sub-arrays has duplicate spots of seven antibodies plus a single spot with non-labeled bovine serum albumin (BSA) as a negative control and a single spot with Cy3- and Cy5-conjugated BSA as a positive control.
At 1 h after PTI, the cortical infarction region was excised from the extracted rat cortex using a 3-mm diameter circular knife. Then the surrounding 2-mm width ring-shape cortex area around the PTI core that comprised the penumbra was cut out with another 7-mm diameter circular knife. The similar piece from the non-irradiated contralateral somatosensory cortex was used as control. These samples were quickly frozen in liquid nitrogen and then transferred into a freezer (–85°C). To obtain sufficient mass of the experimental material, tissues from six rats were combined. Symmetrical samples of the untreated contralateral cortex of the same six rats were collected similarly and served as a control. These cortex pieces were weighed and homogenized on ice in extraction/labeling buffer supplemented with proteinase and phosphatase inhibitor cocktails and nuclease benzonase (components of NBAA5). Then the control and experimental lysates were centrifuged 2 min at 10,000 rpm in a cooled centrifuge at 4°C. The supernatants were collected, and the protein content was determined using the Bradford reagent. Then, both experimental and control samples were diluted to 1 mg/ml protein content and incubated 30 min with Cy3 or Cy5, respectively, at a room temperature in darkness. In another set, these samples were stained oppositely, by Cy5 and Cy3, respectively. The unbound dye was removed by centrifugation (4000 rpm, 4 min) in SigmaSpin Columns (NBAA5 components) filled with 200 µl of the labeled protein samples. The eluates were collected, and protein concentration was determined again. The mixture of the control and experimental samples (10 µg/ml protein content) labeled with Cy3 and Cy5, respectively, was added into a tube with 5 ml of the array incubation buffer (NBAA5 component) and incubated 40 min at room temperature on a rocking shaker. The same procedure was performed with the oppositely labeled samples: Cy5 and Cy3, respectively. Such swapped staining verifies results and compensates the potential bias in binding of Cy3 or Cy5 dyes to protein samples. This provides a double test and full control of the experiment. After following triple washing of the microarrays in the washing buffer (NBAA5 component) and triple washing in pure water, the microarray slides were air dried overnight in darkness. The microarrays were scanned using the GenePix 4100A Microarray Scanner (Molecular Devices, USA) at 532 and 635 nm (fluorescence maximums of Cy3 and Cy5, respectively). The integrated fluorescence intensity in each antibody spot was proportional to the quantity of the bound protein. Therefore, the comparison of the fluorescence levels in the experimental and control samples provides information on the changes in the protein expression induced by the studied impact. The fluorescence images of the antibody microarrays were analyzed and normalized using the GenePix Pro 6.0 software. Local fluorescence values in the rings around each spot (background) were subtracted. The median fluorescence value determined over all pixels was used for estimation of the protein content in each spot. The background fluorescence in the rings around each spot was subtracted. To estimate the ratio of experimental/control protein levels, the median values of the ratios of fluorescence intensity Cy5/Cy3 (or Cy3/Cy5 in swapped stained samples) were determined in each spot. Two samples labeled independently and reversely in duplicate in two microarrays provided four values of the ratio of the experimental/control median intensities of each protein fluorescence. Six experimental and six control rats were used in each experiment. The experiment was repeated twice, so eight ratio values were averaged. Mean and SD values are displayed in Table 2. Only >30% differences in the protein levels between the irradiated and control animals (the cutoff level) and significantly differed from 1 (p < 0.05) are displayed in Table 2 and discussed.
RESULTS
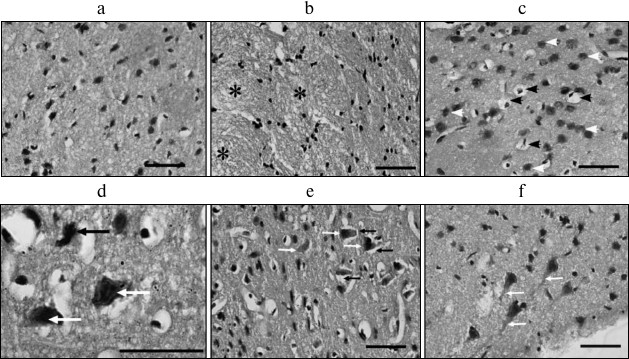
The 3-mm PTI core was surrounded by a penumbra about 1.5-2.0-mm in width (Fig. 1). The number of pyramidal neurons (these were mainly observed in the histological samples) with pathological alterations in the sensomotor cortex of the control contralateral brain hemisphere was insignificant (Fig. 2a and Table 1). Morphological changes characteristic for necrosis within the infarction core at 1 h after light exposure (Fig. 2) included local neuropil vacuolization (Fig. 2b), swelling of glial cells, and perineuronal edema (Fig. 2c). Some neurons had hyperchromic or pyknotic nuclei (Fig. 2d). The average number of hyperchromic and pyknotic neurons per visual field in the infarction core increased from 3 ± 1 in the contralateral cortex (control) to 18 ± 4 (p < 0.05) and from 0 to 3 ± 1 (p < 0.05), respectively (Table 1). Tissue alterations in the penumbra gradually decreased from the regions neighboring the PTI core to the periphery (at a distance of 2 mm), where most neurons looked undamaged (Fig. 2, e and f). The mean number of hyperchromic neurons per visual field increased to 12 ± 2 as compared with control (p < 0.05); however, it was lesser than inside the PTI core (Table 1). Pyknotic neurons were not observed. In some places, perineuronal edema (Fig. 2e) and swelling of neuronal bodies and neuritis (Fig. 2f) was observed.
Table 1. Number of altered neurons in the
photothrombotic infarction core and penumbra 1 h after local
photodynamic treatment of rat cerebral cortex

Note: Mean cell numbers are averaged over six randomized visual fields
(magnification 400×) in three slices in four rats (M
± SEM; * p < 0.05).
Fig. 1. Local photothrombotic infarction in rat cerebral cortex induced by photodynamic treatment. a) Infarction core in the sensomotor cortex. The ring-shape penumbra is shown by the white arrowhead. Scale bar, 3 mm. b) Frontal section through the infarction core and penumbra. Infarct is represented as a zone of pale-staining cortex, surrounded by a vacuolated edematous neuropil, beyond which the cortex retained normal staining (hematoxylin–eosin). Scale bar, 100 µm.
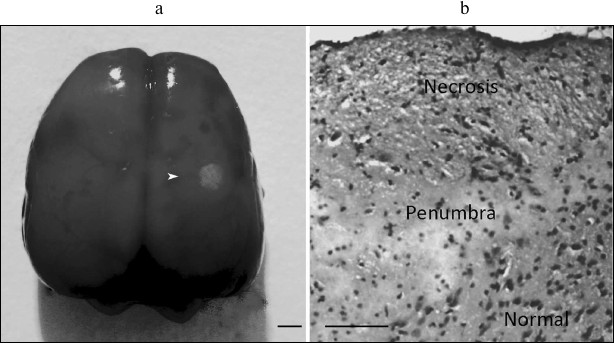
Fig. 2. Morphological changes in rat cerebral cortex 1 h after PTI. a) Control cortical tissue in the non-treated hemisphere; b) infarction core; * vacuolated neuropil; c) infarction core; black arrows – pericellular edema; d) infarction core; black arrows – pyknotic neurons; white arrows – hyperchromic neurons; e) penumbra; black arrows – perineuronal edema; white arrows – hyperchromic neurons; f) penumbra; white arrows – swollen dendrites of pyramidal neurons. Scale bars, 50 µm (a-c, e, f); 20 µm (d).
Using NBAA5 protein microarrays, we compared the averaged levels of 224 neuronal and signaling proteins within the 2 mm cerebral cortex ring around the PTI core (penumbra) with that in the untreated contralateral cortical tissue 1 h after PTI. We found more than 30% overexpression of some proteins in these penumbra samples (Table 2). These included: ubiquitin-1 and ubiquitin C-terminal hydrolase L1 (UCHL1) involved in ubiquitin-mediated proteolysis (+51 and +78%, respectively); PTEN-induced mitochondrial protein kinase PINK1 and Parkin (+75 and +41%, respectively) involved in mitochondrial quality control, fission, and mitophagy; myelin protein PMP22 (+72%); protein kinase PKBα regulating cell proliferation and apoptosis (+58%); N-cadherin participating in cell–cell adhesion (+57%); glutamic acid decarboxylase that converts L-glutamate into γ-butyric acid (GABA) (+51%); tryptophan hydroxylase that synthesizes serotonin (+48%); collapsin response mediator protein 2 (CRMP2), a component of the collapsin/semaphorin pathway that regulates axon guidance (+49%); dual-specificity tyrosine-phosphorylated regulated kinase 1A (DYRK1A) that phosphorylates various transcription factors and other proteins (+46%); neuron navigator 3 protein (NAV3) (+45%); MAP kinase ERK5 that mediates oxidative stress signals and protects cells from apoptosis (+40%); synaptic proteins Munc-18-1 and Munc-18-3 (+33 and +53%), synphilin-1 that participates in synaptic function and ubiquitin-mediated protein degradation (+38%), and microtubule-associated protein MAP1 (+36%). At the same time, some proteins were downregulated: protein kinase C (6.3-fold) and its Cβ1 isoform (–65%); TDP-43 (transactivation response DNA-binding protein) (2-fold), synaptic proteins synaptophysin and syntaxin (–33 and –71%), and neurofilament 68, the cytoskeleton component (–51%).
Table 2. Relative changes in the expression
of neuronal and signaling proteins in the penumbra surrounding the
local photothrombotic infarction in rat somatosensory cortex compared
with the contralateral cortex tissue 1 h after light exposure
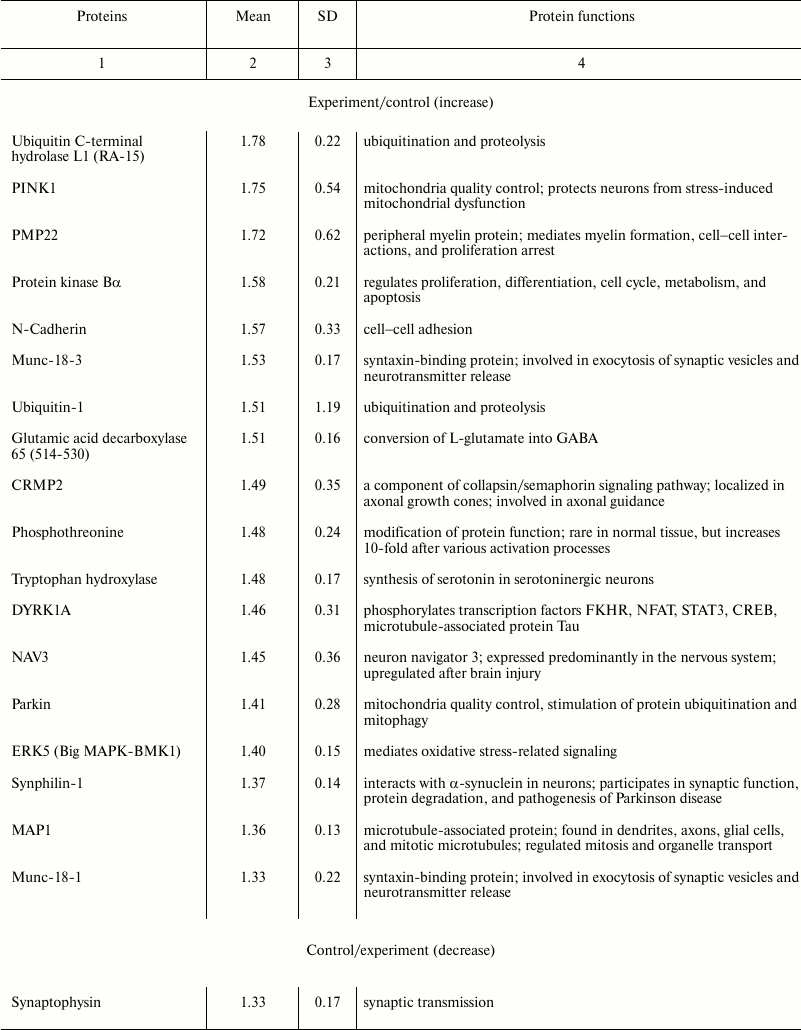

Note: Mean ratios of median fluorescence values of experimental and
control samples, or, oppositely, control and experimental values in the
case of protein level decrease, and SD are given.
DISCUSSION
The photothrombotic infarct used in the present study as a stroke model reproduced well the morphological alterations in the cerebral cortex after occlusion of small blood vessels. Local photodynamic impact induced typical ischemic alterations in the infarction core: edema, massive tissue vacuolization, and degenerative changes in neurons, glia, and blood vessels. Alterations in the penumbra were similar to those in the PTI core but lesser. They were more profound near the infarction core and were reduced at the penumbra periphery.
Stroke-induced neurodegeneration and neuroprotection are controlled by the complex intracellular signaling system consisting of hundreds and thousands signaling proteins. Its components, internal interactions, and role in cell death and survival are insufficiently studied. Proteomic methods provide simultaneous study of changes in expression of hundreds of cellular proteins. This is a novel approach to study complex, multifactor processes such as stroke and consequent organism responses. Previous proteomic studies of stroke mechanisms revealed changes in the expression of some bioenergetic [21]; antiapoptotic, inflammatory, antioxidant, and mitochondria heat shock proteins [22]; protein kinases [23], and extracellular proteins [18] in the penumbra.
Using proteomic antibody microarrays, we found changes in expression of some neuronal and signaling proteins in penumbra 1 h after PTI in comparison with the untreated contralateral cortex of the same rats (Table 2). More than 6-fold downregulation of protein kinase C (PKC) was most profound. PKC regulates numerous cellular processes from proliferation to apoptosis. It is known to be rapidly upregulated after ischemia, and its inhibitors protect brain cells from anoxic [24] and excitotoxic injury [25]. This suggests the involvement of PKC in the neurotoxic processes. The inhibition and downregulation of PKC during ischemic neurodegeneration [26] are in agreement with our data. It could be a result of tissue destruction and proteolysis. PKC isoforms γ, δ, and ε are known to be involved in ischemic brain injury [26, 27]. In our experiments, they were slightly upregulated in the penumbra (+16-23%) (data not shown). The observed downregulation of PKC could be rather associated with 65 and 27% decrease in the levels of β1 and β2 isoforms (Table 2). PKCβ1/2 mediates glutamate excitotoxicity, and its downregulation protects neurons from excitotoxic and ischemic injury [28]. The observed downregulation of PKC β1 and β2 could contribute to neuroprotective processes in the penumbra [28]. On the other hand, PKCβ1 inhibition is known to exacerbate the injury of astrocytes under oxygen and glucose deprivation [29]. One can suggest the simultaneous involvement of PKCβ1 in ischemic neuron injury and astroglia protection in the penumbra tissue.
The upregulation of protein kinase Bα (+58%) and protein kinase ERK5 (+40%) could contribute to protection of the penumbra tissue after PTI. In fact, PKBα can mediate protection of neurons from stroke-induced apoptosis [30]. ERK5 is involved in the cell response to stress and plays antiapoptotic and neuroprotective roles [31].
The level of DYRK1A that phosphorylates a variety of transcription factors (FKHR, NFAT, STAT3, CREB, etc.), microtubule-associated protein tau, and other proteins was increased by 46%. The literature data on its role in signaling pathways that regulate cell survival or death are controversial. It can play the antiapoptotic role due to phosphorylation and inactivation of caspase-9 [32]. DYRK1A can also stimulate the proapoptotic ASK1-JNK signaling pathway [33]. Alternatively, it phosphorylates and activates SIRT1, a NAD+-dependent protein deacetylase essential for energy metabolism and cell survival [34].
The upregulation of PTEN-induced mitochondrial kinase PINK1 and Parkin that control mitochondrial “quality” and induce mitophagy (a form of autophagy that eliminates damaged mitochondria and thereby protects cells from stress-induced mitochondrial dysfunction) may be attributed to compensatory changes in the penumbra [35, 36].
Many damaged proteins are accumulated in nervous tissue under oxidative stress, and the ubiquitin-proteasome system eliminates them [37]. The level of ubiquitin-1 and ubiquitin C-terminal hydrolase L1 (UCHL1) involved in ubiquitin-mediated proteolysis and clearance of cells from damaged proteins was increased in penumbra by 51 and 78%. Overexpression of UCHL1 serves as a marker of human brain injury [38]. Its upregulation correlated with 38% upregulation of synphilin-1, which participates in ubiquitin-mediated protein degradation in neurons and also in synaptic function. The complexes of synphilin-1 with proteins PINK1, UCHL1, SIAH, and Parkin are implicated in the pathogenesis of Parkinson’s disease [39]. The upregulation of UCHL1 and its interaction with PKCβ2 in the mouse brain was possibly involved in neuroprotection induced by ischemic preconditioning [23].
Some proteins involved in maintaining the integrity and growth of neurites were also overexpressed in the penumbra. We observed upregulation of neuronal navigator NAV3, which mediates recovery after neurodegeneration [40]; microtubule-associated protein MAP1, which fills the axons and dendrites and regulates organelle transport along neurites [41]; and CRMP2, a component of the collapsin/semaphorin signaling pathway, which regulates growth of axons and formation of interneuron contacts during neurogenesis. CRMP2 is known to be upregulated in the ischemic mouse cerebral cortex [42]. It also modulates axon integrity under ischemic and glutamate neurotoxicity [43]. The PKCβ2–CRMP2 complex is involved in neuroprotection induced by ischemic preconditioning [23]. We also observed 72% upregulation of peripheral myelin protein PMP22, which mediates formation of myelin sheaths and neuroglial interactions. PMP22 mutations lead to demyelination [44]. The upregulation of PMP22 in penumbra is possibly needed to repair myelin disorganization.
We also observed upregulation of some synaptic proteins in the penumbra: synphilin-1, tryptophan hydroxylase that mediates biosynthesis of serotonin, and glutamic acid decarboxylase that converts L-glutamate into GABA was possibly aimed to recover synaptic functions. The level of syntaxin-binding proteins Munc-18-1 and Munc-18-3, the components of the SNARE complex involved in exocytosis of synaptic vesicles and neurotransmitter release [45], increased by 32 and 53%, respectively. However, the levels of syntaxin and synaptophysin deceased by 71 and 33%, respectively, which could be a result of their release from the disrupted Munc-18–syntaxin complex. These controversial changes could be due to simultaneous synapse destruction and the efforts of brain cells to repair synaptic structures.
Two-fold downregulation of TDP-43 in the penumbra could indicate compensatory processes in this zone. Apparently, TDP-43 is normally involved in transcription, mRNA splicing, transport, and stability. However, modified TDP-43 (phosphorylated, ubiquitinated, or truncated) can mediate formation of inclusions in neurons involved in neurodegeneration in amyotrophic lateral sclerosis, frontal-temporal lobar degeneration, and Alzheimer’s disease [46]. Overexpression of TDP-43 was observed in the brain infarction core after acute ischemic stroke in rats [47]. The downregulation of neurofilament 68, a marker of cytoskeleton disorganization, also indicates the recovery tendency in the penumbra. N-cadherin mediates cell–cell adhesion [48]. Its 57% upregulation in the penumbra is evidently associated with maintaining of tissue integrity.
The depletion of the oxygen supply and energetic resources in the infarction core is known to impair ion homeostasis and induces depolarization, water influx, edema, necrosis, and osmotic cell lysis. Glutamate release from damaged cells activates NMDA and AMPA receptors, which opens associated ion channels in neighboring cells. The following Ca2+ influx causes cell necrosis and apoptosis. Simultaneous K+ release induces sustained depolarization of neurons and glial cells outside the PTI core that further maintains glutamate and K+ release. Such self-developing excitotoxic process results in injury propagation and formation of the penumbra [49, 50]. The energy deficit induces generation of reactive oxygen species in mitochondria, which enhances oxidative tissue damage [51].
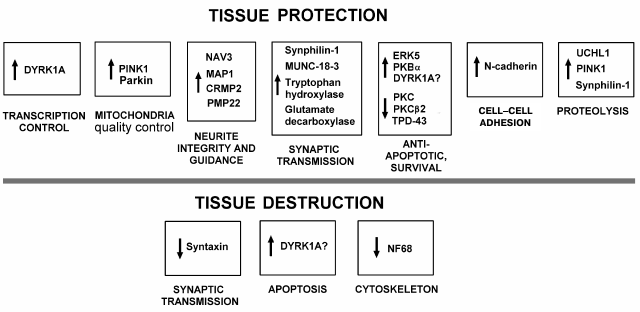
On the other hand, reparative processes occur simultaneously with injurious ones in damaged tissue. The present data have demonstrated neuroprotective processes in penumbra 1 h after PTI (Fig. 3). We observed the overexpression of proteins involved in maintaining the integrity, guidance, and growth of neurites (NAV3, MAP1, CRMP2, and PMP22); intercellular interactions (N-cadherin); structure and functions of mitochondria (PINK1 and Parkin) and synapses (glutamic acid decarboxylase, tryptophan hydroxylase, Munc-18-1 and Munc-18-3, synphilin-1); ubiquitination and proteolysis of damaged proteins that mediate tissue clearance (UCHL1, PINK1, and synphilin-1); some antiapoptotic proteins (PKBα, ERK5, and DYRK1A). Simultaneous downregulation of PKC and PKCβ1/2 could reduce the oxidative and excitotoxic tissue injury. The downregulation of TDP-43 could prevent accumulation of neurotoxic inclusions in neurons. The downregulation of neurofilament 68, a marker of cytoskeleton disorganization, also indicated the recovery tendency.
Fig. 3. Summary scheme of changes in expression of neuronal and signaling proteins in penumbra 1 h after local photothrombotic infarction in rat cerebral cortex.
Thus, in the present work we found a number of proteins that are so important for response of the penumbra tissue to PTI that they are overexpressed. Many other cellular proteins are expressed sufficiently and their level does not change. Under external impact or change in their functional state, these proteins are only activated via, for example, phosphorylation or limited proteolysis. Many of the proteins found in the present work are poorly studied, and their role in the ischemic brain is still unstudied. Our proteomic results are only the first step in a complex study. It should be kept in mind that proteomic data only provide a list of proteins potentially involved in infarction propagation or restriction. They do not disclose the mechanisms of expression changes, because the signaling pathways and transcription factors, which control expression of these proteins, remain unknown. Further mechanistic investigations should reveal the signaling pathways underlying these changes and determine the roles of these and other related proteins in development of tissue damage and reparation. Possibly, the consequences of stroke will be efficiently treated in future by a drug complex aimed to a range of tissue processes.
Another limitation of this method as well as other biochemical assays, which require some mass of biological material, is associated with the heterogeneity of the nervous tissue that consists of neurons, different glial cells (astroglia, oligodendroglia, microglia, etc.), and blood vessels. Different processes in these cell types can determine the observed variable tendencies. Moreover, all observed changes were averaged over the whole penumbra tissue (2 mm ring around the PTI core). But the penumbra is inhomogeneous. In penumbra, damaging processes dominate near the PTI core, whereas protective processes prevail at the penumbra periphery. The balance between damaging and protective tendencies forms the “death border”. This balance determines the cell fate, and the goal of the anti-stroke therapy is to restrict the propagation of this border due to inhibition of injurious processes and stimulation of tissue recovery. This can be reached based on deep knowledge of their biochemical mechanisms. The data also indicate potential markers and targets for diagnostics and treatment of infarction in the penumbra.
The authors used the equipment of the Center for Collective Use of Southern Federal University “High Technology” supported by the Ministry of Education and Science of Russian Federation (project RFMEFI59414X0002).
The work was supported by the Russian Scientific Foundation (grant 14-15-00068). A. B. Uzdensky’s work was supported by the Ministry of Education and Science of Russian Federation (grant “Science organization” #790).
REFERENCES
1.Meisel, A., Prass, K., Wolf, T., and Dirnagl, U.
(2004) Stroke, in Neuroprotection: Models, Mechanisms and Therapies
(Bahr, M., ed.) Wiley-Blackwell, Hoboken, NJ, pp. 9-43.
2.Iadecola, C., and Anrather, J. (2011) Stroke
research at a crossroad: asking the brain for directions, Nat.
Neurosci., 14, 1363-1368.
3.Moskowitz, M. A. (2010) Brain protection: maybe
yes, maybe no, Stroke, 41, S85-S86.
4.Zhiganshina, L. E., and Abakumova, T. R. (2013)
Cerebrolysin in a treatment of acute ischemic stroke, Vestnik
RAMN, 1, 21-29.
5.Watson, B. D., Dietrich, W. D., Busto, R., Wachtel,
M. S., and Ginsberg, M. D. (1985) Induction of reproducible brain
infarction by photochemically initiated thrombosis, Ann.
Neurol., 17, 497-504.
6.Dietrich, W. D., Watson, B. D., Busto, R.,
Ginsberg, M. D., and Bethea, J. R. (1987) Photochemically induced
cerebral infarction. I. Early microvascular alterations, Acta
Neuropathol., 72, 315-325.
7.Pevsner, P. H., Eichenbaum, J. W., Miller, D. C.,
Pivawer, G., Eichenbaum, K. D., Stern, A., Zakian, K. L., and Koutcher,
J. A. (2001) A photothrombotic model of small early ischemic infarcts
in the rat brain with histologic and MRI correlation, J. Pharmacol.
Toxicol. Methods, 45, 227-233.
8.Shanina, E. V., Redecker, C., Reinecke, S.,
Schallert, T., and Witte, O. W. (2005) Long-term effects of sequential
cortical infarcts on scar size, brain volume and cognitive function,
Behav. Brain Res., 158, 69-77.
9.Schmidt, A., Hoppen, M., Strecker, J. K.,
Diederich, K., Schabitz, W. R., Schilling, M., and Minnerup, J. (2012)
Photochemically induced ischemic stroke in rats, Exp. Transl. Stroke
Med., 4, 13.
10.Romanova, G. A., Barskov, I. V., Ostrovskaya, R.
U., Gudasheva, T. A., and Viktorov, I. V. (1998) Behavioral and
morphological changes induced by bilateral photoinduced thrombosis of
cerebral vessels in the rat frontal cortex, Pathol. Physiol. Exp.
Ther., No. 2, 8-10.
11.Romanova, G. A., Shakova, F. M., Barskov, I. V.,
Stelmashuk, E. V., Genrihs, E. E., Cheremnyh, A. M., Kalinina, T. I.,
and Yurin, V. L. (2014) Neuroprotective and anti-amnestic effect of
erythropoietin derivatives at experimental ischemic damage to brain
cortex, Bull. Exp. Biol. Med., 158, 299-302.
12.Uzdensky, A. B. (2010) Cellular and Molecular
Mechanisms of Photodynamic Therapy [in Russian], Nauka, St.
Petersburg.
13.Brundel, M., de Bresser, J., van Dillen, J. J.,
Kappelle, L. J., and Biessels, G. J. (2012) Cerebral microinfarcts: a
systematic review of neuropathological studies, J. Cereb. Blood Flow
Metab., 32, 425-436.
14.Pantoni, L. (2010) Cerebral small vessel disease:
from pathogenesis and clinical characteristics to therapeutic
challenges, Lancet Neurol., 9, 689-701.
15.Del Zoppo, G. J., and Mabuchi, T. (2003) Cerebral
microvessel responses to focal ischemia, J. Cereb. Blood Flow
Metab., 23, 879-894.
16.Spisak, S., Tulassay, Z., Molnar, B., and
Guttman, A. (2007) Protein microchips in biomedicine and biomarker
discovery, Electrophoresis, 28, 4261-4273.
17.Wingren, C., and Borrebaeck, C. A. (2009)
Antibody-based microarrays, Methods Mol. Biol.,
509, 57-84.
18.Dayon, L., Turck, N., Garci-Berrocoso, T.,
Walter, N., Burkhard, P. R., Vilalta, A., Sahuquillo, J., Montaner, J.,
and Sanchez, J. C. (2011) Brain extracellular fluid protein changes in
acute stroke patients, J. Proteome Res., 10,
1043-1051.
19.Demyanenko, S. V., Uzdensky, A. B., Sharifulina,
S. A., Lapteva, T. O., and Polyakova, L. P. (2014) PDT-induced
epigenetic changes in the mouse cerebral cortex: a protein microarray
study, Biochim. Biophys. Acta, 1840, 262-270.
20.Zilles, K. (1985) The Cortex of the Rat: A
Stereotaxis Atlas, Springer-Verlag, Berlin.
21.Villa, R. F., Gorini, A., Ferrari, F., and Hoyer,
S. (2013) Energy metabolism of cerebral mitochondria during aging,
ischemia and post-ischemic recovery assessed by functional proteomics
of enzymes, Neurochem. Int., 63, 765-781.
22.Datta, A., Park, J. E., Li, X., Zhang, H., Ho, Z.
S., Heese, K., Lim, S. K., Tam, J. P., and Sze, S. K. (2010)
Phenotyping of an in vitro model of ischemic penumbra by
iTRAQ-based shotgun quantitative proteomics, J. Proteom.
Res., 9, 472-484.
23.Bu, X., Zhang, N., Yang, X., Liu, Y., Du, J.,
Liang, J., Xu, Q., and Li, J. (2011) Proteomic analysis of
PKCβII-interacting proteins involved in HPC-induced
neuroprotection against cerebral ischemia of mice, J.
Neurochem., 117, 346-356.
24.Hara, H., Onodera, H., Yoshidomi, M.,
Matsuda, Y., and Kogure, K. (1990) Staurosporine, a novel protein
kinase C inhibitor, prevents postischemic neuronal damage in the gerbil
and rat, J. Cereb. Blood Flow Metab., 10, 646-653.
25.Felipo, V., Minana, M. D., and Grisolia, S.
(1993) Inhibitors of protein kinase C prevent the toxicity of glutamate
in primary neuronal cultures, Brain Res., 604,
192-196.
26.Bright, R., and Mochly-Rosen, D. (2005) The role
of protein kinase C in cerebral ischemic and reperfusion injury,
Stroke, 36, 2781-2790.
27.Chou, W. H., and Messing, R. O. (2005) Protein
kinase C isozymes in stroke, Trends Cardiovasc. Med.,
15, 47-51.
28.Lee, B. K., Yoon, J. S., Lee, M. G., and Jung, Y.
S. (2014) Protein kinase C-β mediates neuronal activation of
Na+/H+ exchanger-1 during glutamate
excitotoxicity, Cell Signal., 26, 697-704.
29.Wang, J., Bright, R., Mochly-Rosen, D., and
Giffard, R. G. (2004) Cell-specific role for ε- and
βI-protein kinase C isozymes in protecting cortical neurons and
astrocytes from ischemia-like injury, Neuropharmacology,
47, 136-145.
30.Zhao, H., Sapolsky, R. M., and Steinberg, G.
K. (2006) Phosphoinositide-3-kinase/akt survival signal pathways are
implicated in neuronal survival after stroke, Mol.
Neurobiol., 34, 249-270.
31.Wang, R. M., Zhang, Q. G., Li, C. H., and
Zhang, G. Y. (2005) Activation of extracellular signal-regulated kinase
5 may play a neuroprotective role in hippocampal CA3/DG region after
cerebral ischemia, J. Neurosci. Res., 80, 391-399.
32.Laguna, A., Aranda, S., Barallobre, M. J.,
Barhoum, R., Fernandez, E., Fotaki, V., Delabar, J. M., de la
Luna, S., Villa, P., and Arbones, M. L. (2008) The protein kinase
DYRK1A regulates caspase-9-mediated apoptosis during retina
development, Dev. Cell, 15, 841-853.
33.Choi, H. K., and Chung, K. C. (2011) DYRK1A
positively stimulates ASK1-JNK signaling pathway during apoptotic cell
death, Exp. Neurobiol., 20, 35-44.
34.Guo, X., Williams, J. G., Schug, T. T., and Li,
X. (2010) DYRK1A and DYRK3 promote cell survival through
phosphorylation and activation of SIRT1, J. Biol. Chem.,
285, 3223-3232.
35.Trancikova, A., Tsika, E., and Moore, D. J.
(2012) Mitochondrial dysfunction in genetic animal models of
Parkinson’s disease, Antioxid. Redox Signal.,
16, 896-919.
36.De Vries, R. L., and Przedborski, S. (2013)
Mitophagy and Parkinson’s disease: be eaten to stay healthy,
Mol. Cell Neurosci., 55, 37-43.
37.Caldeira, M. V., Salazar, I. L., Curcio, M.,
Canzoniero, L. M., and Duarte, C. B. (2014) Role of the
ubiquitin–proteasome system in brain ischemia: friend or foe?
Prog. Neurobiol., 112, 50-69.
38.Yamauchi, T., Sakurai, M., Abe, K., Matsumiya,
G., and Sawa, Y. (2008) Ubiquitin-mediated stress response in the
spinal cord after transient ischemia, Stroke, 39,
1883-1889.
39.Kruger, R. (2004) The role of synphilin-1 in
synaptic function and protein degradation, Cell Tissue Res.,
318, 195-199.
40.Maes, T., Barcelo, A., and Buesa, C. (2002)
Neuron navigator: a human gene family with homology to unc-53, a cell
guidance gene from Caenorhabditis elegans, Genomics,
80, 21-30.
41.Halpain, S., and Dehmelt, L. (2006) The MAP1
family of microtubule-associated proteins, Genome Biol.,
7, 224.
42.Chen, A., Liao, W. P., Lu, Q., Wong, W. S., and
Wong, P. T. (2007) Up-regulation of dihydropyrimidinase-related protein
2, spectrin alpha II chain, heat shock cognate protein 70 pseudogene 1
and tropomodulin 2 after focal cerebral ischemia in rats – a
proteomics approach, Neurochem. Int., 50,
1078-1086.
43.Hou, S. T., Jiang, S. X., Aylsworth, A.,
Ferguson, G., Slinn, J., Hu, H., Leung, T., Kappler, J., and Kaibuchi,
K. (2009) CaMKII phosphorylates collapsin response mediator protein 2
and modulates axonal damage during glutamate excitotoxicity, J.
Neurochem., 111, 870-881.
44.Quarles, R. H. (2002) Myelin sheaths:
glycoproteins involved in their formation, maintenance and
degeneration, Cell. Mol. Life Sci., 59,
1851-1871.
45.Gallwitz, D., and Jahn, R. (2003) The riddle of
the Sec1/Munc-18 proteins – new twists added to their
interactions with SNAREs, Trends Biochem. Sci.,
28, 113-116.
46.Lee, E. B., Lee, V. M., and Trojanowski, J.
Q. (2011) Gains or losses: molecular mechanisms of TDP43-mediated
neurodegeneration, Nat. Rev. Neurosci., 13,
38-50.
47.Kanazawa, M., Kakita, A., Igarashi, H.,
Takahashi, T., Kawamura, K., Takahashi, H., Nakada, T., Nishizawa, M.,
and Shimohata, T. (2011) Biochemical and histopathological alterations
in TAR DNA-binding protein-43 after acute ischemic stroke in rats,
J. Neurochem., 116, 957-965.
48.Zechariah, A. E., Ali, A., Hagemann, N., Jin, F.,
Doeppner, T. R., Helfrich, I., Mies, G., and Hermann, D. M. (2013)
Hyperlipidemia attenuates vascular endothelial growth factor-induced
angiogenesis, impairs cerebral blood flow, and disturbs stroke recovery
via decreased pericyte coverage of brain endothelial cells,
Arterioscler. Thromb. Vasc. Biol., 33, 1561-1567.
49.Back, T., Ginsberg, M. D., Dietrich, W. D., and
Watson, B. D. (1996) Induction of spreading depression in the ischemic
hemisphere following experimental middle cerebral artery occlusion:
effect on infarct morphology, J. Cereb. Blood Flow Metab.,
16, 202-213.
50.Puyal, J., Ginet, V., and Clarke, P. G. (2013)
Multiple interacting cell death mechanisms in the mediation of
excitotoxicity and ischemic brain damage: a challenge for
neuroprotection, Prog. Neurobiol., 105, 24-48.
51.Sims, N. R., and Anderson, M. F. (2002)
Mitochondrial contributions to tissue damage in stroke, Neurochem.
Int., 40, 511-526.