REVIEW: Exploiting Natural Anti-carbohydrate Antibodies for Therapeutic Purposes
D. Bello-Gil1 and R. Manez1,2*
1Bellvitge Biomedical Research Institute (IDIBELL), Gran Via de l’Hospitalet 199, 08908 Hospitalet de Llobregat, Spain2Bellvitge University Hospital, Feixa Llarga s/n 08907, Hospitalet de Llobregat, Spain; E-mail: rmanez@bellvitgehospital.cat
* To whom correspondence should be addressed.
Received March 4, 2015; Revision received April 13, 2015
Natural anti-carbohydrate antibodies (NAbC) are antibodies that target glycans and are continuously produced without apparent external antigen stimulation. Clinically, NAbC are recognized by the adverse reactions to ABO mismatched blood transfusions or organ transplantation and the rejection of xenografts. These clinical effects do not reflect the biological functions of NAbC. However, they launch the possibility of using NAbC for boosting immunity in different clinical settings by means of: 1) expression of glycan antigens in elements that do not hold them to allow the binding and reactivity of existing NAbC; 2) removal of existing NAbC; 3) manipulation of the glycosylation pattern of NAbC.
KEY WORDS: natural antibodies, anti-carbohydrate antibodies, anti-blood group antibodies, anti-galactose α1,3 galactose antibodiesDOI: 10.1134/S0006297915070044
Abbreviations: ADCC, antibody-dependent cell cytotoxicity; ADE, antibody-dependent enhancement; CDC, complement-dependent cytotoxicity; Fab, antigen-binding fragment; Fc, constant fragment; HAPAb, hemolytic anti-pig antibodies; NAb, natural antibodies; NAbC, natural anti-carbohydrate antibodies; TAA, tumor-associated antigens.
NATURAL ANTI-CARBOHYDRATE ANTIBODIES
Natural antibodies (NAb) are defined as those immunoglobulins (mainly IgM isotype) continuously secreted by B-1 type lymphocytes without any previous external antigen stimulation [1, 2]. IgM NAb not only recognize and take part in the elimination of pathogens through the activation of complement, but also have an important role in apoptotic cell clearance, tissue homeostasis, and immune modulation [3, 4]. Less clear is the role of natural IgG antibodies considered on occasions as incompetent to identify antigens and nonreactive. However, recent studies demonstrated that they collaborate with lectins for rapid and effective removal of pathogens, suggesting that might be a part of the bridge between innate and adaptive immunity [5].
A broad range of NAb target carbohydrates (NAbC) and can be divided into three different groups: 1) conservative NAbC that are practically the same regarding specificity and blood levels in all healthy individuals; 2) allo-antibodies to blood group antigens and xenoantibodies to antigens from other species; 3) plastic antibodies, which include antibodies from the two previous groups but with variable levels during certain conditions, such as pregnancy or inflammation [6]. Plastic antibodies also include NAbC reacting to tumor-associated carbohydrate antigens, such as Galβ1-3GalNAcα (Thomsen–Friedenreich antigen) or GalNAcα (Tn antigen). These antibodies are not specific for tumors, but they show changes in cancer and other diseases [7]. In addition, they involve those antibodies reacting to Galα1-3Galβ (αGal) and particularly to Galα1-3Galβ1-4GlcNAcβ. For many years, they were considered the most abundant antibody in humans (up to 2% of total immunoglobulins), although recent studies have estimated this proportion as only 0.1%, as for many other NAbC [6]. The αGal epitope is expressed in various microorganisms including viruses, bacteria, and protozoa. This led to propose the continuous production of anti-αGal antibodies in humans, apes, and Old World monkeys as the result of antigenic stimulation by normal gut bacterial flora [8, 9]. In humans, anti-αGal antibodies react to red blood cells of patients with β-thalassemia or sickle cell anemia, and to normal senescent red blood cells [10]. Increased levels of anti-αGal antibodies have been described in patients with autoimmune diseases such as Grave’s disease, Henoch-Schönlein purpura, and Crohn’s disease [11]. Augmented levels of anti-αGal antibodies are also associated with higher mortality in patients undergoing renal replacement therapy and peritoneal dialysis-related enteric peritonitis [12].
Unfortunately, changes in anti-αGal antibodies and other NAbC evidenced a low sensitivity and specificity for the diagnosis of particular diseases or conditions. This may change in the near future with the great progress achieved in recent years in carbohydrate synthesis and microarray technology. To date, NAbC are clinically recognized by adverse reactions to ABO-mismatched blood transfusions or organ transplantation, and the rejection of xenografts expressing αGal epitopes. These clinical effects do not reflect the biological functions of NAbC. However, they unveil the possibility of using NAbC for boosting immunity at clinical level by means of: 1) expression of glycan antigens in elements that do not hold them to allow the binding and reactivity of existing NAbC; 2) removal of existing NAbC; 3) manipulation of the glycosylation pattern of NAbC.
ENHANCEMENT OF IMMUNITY THROUGH EXPOSURE TO CARBOHYDRATE
ANTIGENS
There is no evidence of any attempt to express ABO-incompatible antigens on cells to generate an immunological response against the cells. The clinical interest in this carbohydrate antigen–antibody reaction relies on the manipulation of antibodies to allow the transfusion or transplantation of ABO-incompatible blood or grafts, both on an intentional basis and when it occurs accidentally. GalNAcα1-3(Fucα1-2)Gal and Galα1-3(Fucα1-2)Gal were the classical antigenic determinants of blood group A and B, respectively. However, GalNAcα1-3(Fucα1-2)Galβ1-4GlcNAc and Galα1-3(Fucα1-2)Galβ1-4GlcNAc emerged recently as antigens for blood A and B, respectively [13]. Anti-blood group A antibodies, which generally show a low level, include both NAb against tetra- and trisaccharide moieties. However, only antibodies targeting the tetrasaccharide form can agglutinate red blood cells since anti-trisaccharide antibodies are present in both blood group B and A individuals. Interestingly, there is evidence that a large proportion of the elicited antibodies against the incompatible ABO antigens consists of anti-αGal antibodies, which interact with the αGal epitope in the blood group A or B core structure Galα1-3Galβ1-4GlcNAcβ [14]. These data, along with the classical consideration that anti-αGal antibodies constitute the largest quantity of immunoglobulins in humans, is probably the reason for the consideration of the αGal epitope as the carbohydrate antigen to target in the generation of an immunological response at clinical level.
One of the first potential applications of anti-αGal antibodies was to boost immunity against tumor cells. As we described previously, NAbC include plastic antibodies targeting tumor-associated antigens (TAA), which are not specific for tumor cells because they can be observed in normal tissues, although at different levels. An immune response that can be generated against TAA can be associated, in some cases, with an improved prognosis of cancer [15]. However, in most patients the tumor escapes this immune response. The immunogenicity of tumors can be increased by the expression of αGal epitopes after intratumoral injection of αGal glycolipids, as evidenced in the α1,3-galactosyltransferase knockout (Gal-KO) mouse model [16]. Anti-αGal antibodies bind αGal on glycolipids activating complement-mediated cytotoxicity and antibody-dependent cell-mediated cytotoxicity against tumor cells [16, 17]. This strategy has been evaluated in a phase I clinical trial including patients with advanced cancers. Intratumoral injection of αGal glycolipids was safe, with an apparent prolongation of survival in some patients [18].
Other potential clinical application of carbohydrate antigens is related to the enhancement of immunogenicity in vaccines against infectious diseases. Synthetic β1-6-D-glucosamine and β1-6-D-N-acetylglucosamine oligosaccharides produce conjugated vaccines protecting against a broad range of pathogens. Many bacteria generate the surface polysaccharide β1-6-poly-N-acetyl-D-glucosamine (PNAG), and production of antibodies against this epitope can generate immunity to a large number of germs [19]. Furthermore, expression of αGal epitopes in inactivated influenza virus vaccine and in gp120 vaccine of HIV produces a prominent increase of the humoral and cellular immune responses generated by the two vaccines [20, 21]. Additionally, polyanhydride nanoparticles carrying αGal-transformed antigens induce higher antibody responses with broader epitope recognition compared to other adjuvants [22]. Combination of αGal-modified antigens with nanoparticle technology could be a novel approach to design the next generation of vaccines against emerging and re-emerging pathogens.
Anti-αGal antibodies can also accelerate wound and burn healing. The idea is based on a local recruitment and activation of macrophages to promote the regeneration of damaged tissue. Topical application of liposomes carrying αGal epitopes improve wound healing time, avoiding fibrosis and scar formation in both Gal-KO mice and pigs [23-25]. The capacity to restore and revitalize skin wounds achieved by αGal glycoconjugates led to propose the use of these particles for the regeneration of internal tissues such as myocardium after infarction and injured nerves [26].
DEPLETION OF ANTI-CARBOHYDRATE ANTIBODIES TO BOOST IMMUNITY
AGAINST MICROORGANISMS
Antibody-dependent enhancement of infection. Antibodies play a crucial role in host defense against infections. However, there is evidence that in some conditions antibodies can enhance the infective potential of microorganisms instead of protecting the host. Antibody-dependent enhancement (ADE) of infection was initially described in viral diseases, especially in virus from the family of Flaviviridae [27]. Thus, the severity of Dengue disease seems associated with preexisting maternal anti-Dengue virus antibodies or a previous exposure to one of the four Dengue virus serotypes before a secondary infection. Similarly, the presence of non-neutralizing antibodies in HIV infection, besides failing to generate a protective immune response, can enhance viral infectivity [28]. The list of human and animal viruses that may exploit ADE is long and includes influenza A, Coxsackievirus, respiratory syncytial virus, Ebola, and others [29]. After vaccination, ADE of viral infections can also occur, and the induction of non-neutralizing antibodies appears as a primary cause for incapacity to develop vaccines against some viruses, including HIV [30].
The same ADE phenomenon appears to be present during the development of bacterial infections. The specific antibody response to capsular polysaccharides of Streptococcus pneumoniae or Bacillus anthracis and Staphylococcus aureus toxins can enhance infections caused by these bacteria [31]. In addition, natural antibodies to specific polysaccharide of S. aureus such as poly-N-acetyl glucosamine interfere with the protective antibodies induced by different vaccines against this microorganism, causing the failure of immunization [32]. The case of anti-αGal antibodies is particularly paradoxical. These antibodies appear to be produced in response to enterobacteria of the intestinal microbiota [8]. We were able to strengthen this hypothesis by showing a reduction of anti-αGal IgG antibodies after the removal of these microorganisms from the bowel in non-human primates [9]. However, there is also evidence that anti-αGal antibodies facilitate the survival of Enterobacteriaceae or Neisseria meningitidis by enhancing the infectivity and disease caused by these bacteria [33, 34].
Several factors appear to be associated with ADE of infections. These include the strain and load of the microorganism, along with the concentration, class, and epitope specificity of the antibody [29]. Complement is also important in controlling the biological activity of antibodies and seems to have a particular role in the ADE of bacterial infections. The lack of alternative complement pathway activation is a mechanism described to explain the deleterious effect of anti-αGal antibodies on Enterobacteriaceae and N. meningitidis infections [33, 34]. The failure of complement-mediated bacterial killing can result from the binding of IgG2 antibodies to the O-linked glycans of the lipopolysaccharide (LPS) of Gram-negative bacteria. This type of response is the cause for an increased severity of lung infection by Pseudomonas aeruginosa in patients with bronchiectasis [35]. The amount of alternative complement pathway amplification required for optimal bactericidal activity depends on the extent of classical complement activation, which in turns is dependent on the quantity of antigen expression [36]. This high epitope density is also crucial for efficient activation of complement by IgG2 antibodies, which are the principal IgG antibodies in immunity against carbohydrate antigens. Accordingly, binding of IgG2 antibodies to a low density of carbohydrate-antigens can lead to deficient alternative complement pathway activation. Removal of these antibodies, by passing the serum in vitro through an affinity column coated with a monoclonal anti-IgG2 antibody, restores complement activation and serum bactericidal activity [35]. We also have evidence that this strategy works in vivo. Depletion of anti-αGal antibodies, which includes IgG2 subclass, prevents mortality by Enterobacteriaceae sepsis in Gal-KO mice after a cecal ligation puncture (CLP) procedure [37]. Therefore, the removal of existing non-neutralizing anti-carbohydrate antibodies might be an innovative tool to enhance immunity for the prevention or treatment of infectious diseases. The potential impact of this strategy might be similar to that achieved by the production of new antibodies by vaccination.
Methods for depletion of anti-carbohydrate antibodies. Extracorporeal absorption of antibodies, obtained by plasmapheresis or adsorption through columns carrying specific antigens, is the method usually employed both clinically and experimentally for the depletion of antibodies. Patients can undergo these techniques on a daily basis, demonstrating the safety of the procedures. However, it also requires a sophisticated technology that makes the application of these methods feasible only for limited periods. Thus, a soluble inhibitor that can bind and clear antibodies systemically is certainly a much better strategy to remove antibodies of the bloodstream. Such an inhibitor should bind antibodies at least as tightly as the natural antigen; it should be non-immunogenic; and the resulting immune complexes should be rapidly cleared from the circulation to prevent diseases related to immune complexes. Another feature to take in consideration in the particular case of anti-carbohydrate antibodies is their high avidity for multivalent antigens, glycoproteins, and glycolipids, but low affinity for single oligosaccharides [38]. Thus, polymers were selected to provide flexible backbones for the multivalent presentation of αGal and maximize avidity to anti-αGal antibodies [39]. These antibodies emerged for many years as the most important hurdle for the transplantation of pig grafts in humans (xenoantibodies), requiring a continuous depletion that was not attainable by extracorporeal adsorptions [40].
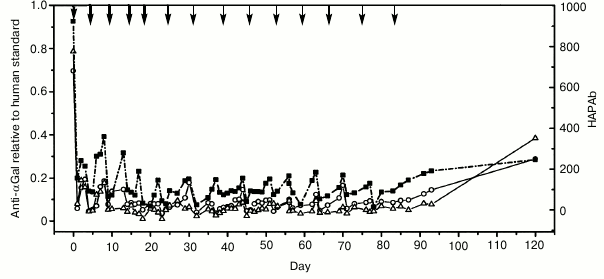
Maximal increase in avidity of anti-αGal antibodies relative to αGal monomer was achieved with polyethylene glycol polymers and a linear polylysine backbone with an average length of 1000 lysines and with 25% of side chains conjugated to αGal trisaccharide (GAS914; Fig. 1) [41, 42]. Administration of GAS914 to non-human primates intravenously or subcutaneously led to a rapid and long-lasting removal of anti-αGal antibodies from circulation with minimal complement and B cell activation (Fig. 2). GAS914 accumulated in the lymphoid organs. This may cause an antigen-specific B cell unresponsiveness that can explain the suppression of anti-αGal antibodies for periods far in excess of the period that the glycoconjugated was measurable in the blood (Fig. 2). The ability of small doses of GAS914 to efficiently eliminate anti-αGal antibodies, along with the possibility of synthesizing large amounts with restricted physicochemical variability, generated many expectations regarding the possibility of offering a relevant therapeutic option for pig graft xenotransplantation. Unfortunately, continuous removal of anti-αGal antibodies using GAS914, as the use of pig organs from Gal-KO pigs that do not express αGal, could not be clinically applied. The reason is the xenograft rejection observed in non-human primate models as result of the emergence of antibodies targeting non-αGal antigens [43, 44].
Fig. 1. Structure of GAS914 [42]. a) Chemical structure of random copolymer with: n, average degree of polymerization; x, fraction of glycosylated monomer; (1 – x), fraction of thioglycerol-capped monomer. Arrows indicate the positions labeled with 14C for pharmacokinetic studies. b) Atomic force microscopy of GAS914 shows a chain-like molecule with an average chain diameter of 4.0 ± 0.2 nm and average chain length of 79 ± 24 nm.
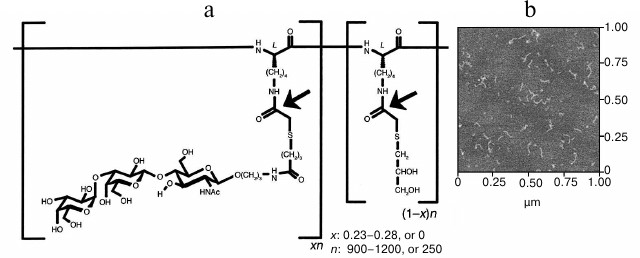
Fig. 2. Inhibition of αGal IgG, IgM, and hemolytic anti-pig Ab (HAPAb) by GAS914 [42]. Baboons treated with intravenous injections of GAS914 at the times indicated by arrows. Thick arrows indicate injection of 5 mg/kg and thin arrows indicate injection of 1 mg/kg. Anti-αGal IgM (circles), IgG (triangles), and HAPAb (squares) titers are relative to a human standard serum (1 for anti-αGal antibodies and 1000 for HAPAb).
The availability of GAS914 allowed us to assess in vivo the impact of anti-αGal antibody removal in infections caused by enteric bacteria with the positive results previously described. The enhancement of immunity achieved by the depletion of anti-αGal antibodies might have applications in the prevention or treatment of several infections. These include most Gram-negative bacterial infections that now cause the majority of hospital-acquired infections, involving in many cases microorganisms resistant to multiple antibiotics and producing severe diseases such as sepsis. Severe sepsis is a common, expensive, and frequently fatal condition, with as many deaths annually as those caused by acute myocardial infarction [45]. Case-fatality rate depends on the setting and severity of disease, but it can reach up to 30% for sepsis, 50% for severe sepsis, and 80% for septic shock. If sepsis is identified and treated earlier, mortality can be reduced producing cost-effective benefits in terms of life years/quality-adjusted life years gained [46]. With αGal-glycoconjugates such as GAS914 or similar, we might have for first time created a tool to prevent sepsis, a disease that now has no specific treatment. Finally, the αGal-glycoconjugates can also have an impact on the treatment of protozoan parasitic infections such as malaria and Chagas disease. Both are associated with boosts of anti-αGal antibodies that cannot prevent the progress of the infection to a chronic state [47, 48].
Another strategy that may be used to systemically remove anti-carbohydrate antibodies involves the insertion of synthetic glycolipids on red blood cells (KODE technology), generating what is defined as kodecytes [49]. Kodecytes were originally designed with the purpose of creating quality control systems in blood transfusions, to identify and quantify special antibodies, or for a more precise analysis of antibody responses through the reactivity to cells expressing single antigens [49]. However, they have also the potential to be used clinically in humans to determine cell survival [50], and to deplete anti-blood group antibodies or any other antibody targeting carbohydrate antigens that can be conjugated to function-spacer lipids [51]. Undoubtedly, the availability of methods for systemically eliminating antibodies, with a more than acceptable toxicity profile in animal models, could be a major clinical advance to treat disorders associated with excess of particular antibodies (autoimmune diseases) or modulate antibody-mediated immune responses.
MODULATION OF ANTIBODY GLYCOSYLATION
Antibody glycosylation. Antibodies are serum glycoproteins essential for the humoral immune response that can be categorized, according to the heavy chain, into five main classes: IgG, IgM, IgA, IgE, and IgD [52, 53]. In serum, the most abundant class is IgG comprising 75% of antibodies in the circulation [54]. This immunoglobulin also has the longest half-life and can be subdivided into four different subclasses: IgG1, IgG2, IgG3, and IgG4 [55, 56]. IgM and IgA are the following most abundant immunoglobulins (10 and 15%, respectively) and have markedly shorter serum half-lives [54, 57]. In general, IgG comprises two identical heavy chains and two identical light polypeptide chains (Fig. 3). Each heavy chain features one variable domain (VH) and three constant domains (CH1, CH2, and CH3). In addition, each light chain features one variable domain (VL) and one constant domain (CL) [52, 58, 59]. The two heavy chains are linked to each other and to a light chain by disulfide bonds [60]. The resulting tetrameric-quaternary structure (“Y”-shaped) has three functional units, two antigen-binding fragments (Fab), and one constant fragment (Fc) [54, 61].
Fig. 3. Schematic representation of general architecture of an IgG antibody. Ag, antigen; VH, variable heavy chain; VL, variable light chain; CL, constant light chain. CH1, CH2, and CH3 are constant domains of heavy chains. Enlarged representation of the Asn297-linked oligosaccharide complex is also shown (Fuc, fucose; Man, mannose; GlcNAc, N acetylglucosamine; Gal, galactose; Sia, sialic acid). The carbohydrate chain can contain 0, 1, or 2 galactose residues, defining G0, G1, and G2 glycoforms, respectively.
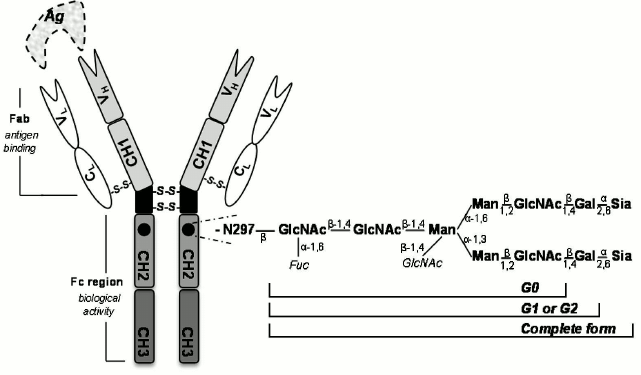
Immunoglobulins elicit their effector functions (biological activity) through interactions of the Fc portion with distinct target molecules and receptors [62, 63]. Glycosylation of the antibody Fc region is a versatile enzyme-directed site-specific process, which occurs as a posttranslational modification in the endoplasmic reticulum and in the Golgi complex of the cell [59, 63]. The Fc region of IgG contains one highly conserved N-linked glycosylation site at N297 (Fig. 3) in the CH2 domain of each heavy chain [64, 65]. The N-linked glycans influence the steric hindrance between the two heavy chains, maintaining the IgG Fc in an opened conformation. These glycans are essential for initiation of many IgG effector functions [54]. Removal of glycans results in collapse of the heavy chains [66], ablating binding to Fc receptors and C1q [52, 54]. In addition, roughly 10-20% of the Fab has N-glycosylation sites in the binding region [53].
The core structure of the IgG-Fc glycans comprises N-acetylglucosamine (GlcNAc) and mannose residues (Fig. 3). This can be further extended by variable additions of terminal and branching sugar residues such as galactose, sialic acid, fucose, and bisected GlcNAc [53, 54]. The majority of human IgG-Fc glycans contain a core-fucose (>92%) [67]. The most variable residue of the Fc sugar core is the galactose (Fig. 3), with a variability that is age-dependent [65]. Depending on the absence or presence of galactose on one or both branches of the sugar moiety, three glycoforms called IgG-G0 (no galactose), IgG-G1 (galactose on one arm), and IgG-G2 (galactose on both arms) have been defined [54, 62].
Antibody-mediated effector functions are crucially dependent on the interaction of its constant region with the complement system, initiated through binding and subsequent activation of C1q (complement-dependent cytotoxicity, CDC), as well as with cellular Fc receptors (antibody-dependent cell cytotoxicity, ADCC, and phagocytosis, ADCP) [65]. Fc-mediated effector functions are determined by the antibody isotype and the presence and type of oligosaccharides attached to the Fc region [53, 62, 63]. Glycosylation of antibodies, particularly in the Fc region of IgG, has been extensively studied in disease and health [64, 68, 69], detecting more than 30 distinct glycan patterns in the circulating IgG antibodies of healthy individuals [59, 67, 70].
Variations in the composition of the sugar moiety can dramatically influence antibody activity. Increased levels of antibodies lacking terminal sialic acid and galactose residues (IgG-G0) are associated with autoimmune diseases like rheumatoid arthritis, systemic lupus erythematosus, and Crohn’s disease [54, 65]. The absence of these terminal sugar residues exposes the high mannose core heptasaccharide, which can now be recognized by mannose-binding lectin (MBL) in vitro [71]. The increased MBL-binding in agalactosylated IgG, however, did not translate into increased CDC in vivo [64]. The significance of an elevated presence of G0 glycoforms is still unclear [54, 65]. However, recent evidence suggests that agalactosylated IgG might have a role in the lack of antibody effector functions observed in some bacterial infections, in chronic HIV infections, and on anti-αGal IgG of hepatitis C virus-infected individuals with fibrosis and cirrhosis [68, 72-74]. IgG galactose deficiency was also correlated with the severity of liver necro-inflammation and fibrosis in chronic hepatitis B [69]. It is remarkable that the presence or absence of a single sugar on IgG Fc glycans so significantly influence the antibody effector functions. Thus, the possibility of modulating such phenomena is a very appealing way to have impact on the effector functions of antibodies.
Controlling antibody glycosylation. The potential for glycosylation is genetically determined; thus, the glycan profile is dependent on host production. The structure of the resulting glycans depends on the glycosyltransferases and modification enzymes present in the cell. The glycosylation pattern of human serum IgG can vary markedly depending on the physiological status of the individual [54]. Thus, levels of galactosylated and sialylated serum IgG increases during pregnancy and decreases postpartum [75]. IgG glycosylation is also altered in several other conditions. As mentioned before, compared to healthy individuals, there is a marked increase of agalactosylated and asialylated serum IgG in individuals with autoimmune diseases and infectious diseases. However, it remains to be elucidated how the disease affects the pattern of antibody glycosylation [54].
So far, little is known about the factors that could drive the composition of Fc-coupled carbohydrate structures of secreted IgG during the activation and differentiation of B lymphocytes [62]. It is speculated that the regulation of expression of glycosyltransferases and glycosidases and their activity is differentially regulated in response to stimulation of environmental factors. Both environmental factors, such as all-trans retinoic acid, as well as factors stimulating the innate immune system (i.e. CpG oligodeoxynucleotide, a ligand for toll-like receptor-9) or coming from the adaptive immune system (i.e. interleukin-21, a T-cell derived cytokine), can modulate IgG1 Fc-glycosylation [62]. There are several examples where antibody glycosylation has been modified in order to achieve a better immunological response against disorders such as breast, colon, and hematological cancers [76]. Among the antibodies, human IgG1 is the main isotype employed as a therapeutic antibody because has the longest half-life in blood and strongest effector functions (CDC, ADCC) compared to other antibody classes and subclasses. Modification of glycosylation pattern of these antibodies can have great therapeutic value. Thus, the removal of α1,6 fucose from oligosaccharides attached to Asn297 of human IgG1 Fc greatly increased ADCC, producing a stronger effect than any other structural change in the Fc oligosaccharides [77, 78]. After the discovery of ADCC enhancement by defucosylation, almost 20 glycoengineered antibodies have entered clinical trials using this modification [76].
Another possible way to modulate antibody glycosylation involves the control of extracellular metabolites (mainly glycans) and the enzymes related to their synthesis, transport, and in general, the glycosylation reactions. N-glycosylation of antibodies begins in the endoplasmic reticulum. Then, the glycoprotein is transported to the Golgi apparatus where the high-mannose glycans are enzymatically processed giving rise to large variations in glycoform depending on host cell line [79]. The local specific activities of enzymes and availability of the reaction substrates, like the nucleotide sugar donor (NSD) species [80], are key elements that determine type and grade of antibody glycosylation. The NSDs are synthesized in the cytoplasm and are then transported into the Golgi. Their synthesis is highly influenced by the presence of key nutrients during cell culture [81, 82], e.g. glucose and galactose. The addition of specific metabolic intermediates of the nucleotide-sugar synthesis pathways to the culture will drive metabolic flux towards the desired nucleotide sugar [83, 84]. This supports a novel approach for engineering of the glycoform by means of feeding strategies [79, 85]. As an example, the effects of nucleotide-sugar precursor feeding on intracellular glycosylation activities were analyzed in CHO cells producing recombinant human interferon-gamma (IFN-γ) [86]. Galactose (+/–uridine), glucosamine (+/–uridine), and N-acetylmannosamine (ManNAc) (+/–cytidine) feeding resulted in 12, 28, and 32% increase in IFN-γ sialylation as compared to the untreated control cultures. This could be directly attributed to increases in nucleotide-sugar substrates, UDP-Hex (approximately 20-fold), UDP-HexNAc (6- to 15-fold), and CMP-sialic acid (30- to 120-fold), respectively [86]. Recently, a modeling framework was developed to provide a link from the extracellular environment and its effect on intracellular metabolites to the distribution of glycans on the constant region of an antibody product. This work is focused on the mechanistic in silico reconstruction of the nucleotide sugar donor metabolic network. The result is a modeling platform that is able to describe the product glycoform based on extracellular conditions [80].
Finally, a nonenzymatic glycation of serum proteins, including Ig, has been described in diabetic patients and is associated with the impairment of immunoreactivity described in these patients [87]. The nonenzymatic IgG glycation observed when the extracellular glucose-fructose concentration is elevated suggests that exposure to other carbohydrates can also lead to new changes in antibody glycation. Thus, we postulate that after removing anti-αGal antibodies, continuing the exposure to αGal through glycoconjugates might provide a tool to modify IgG glycation. Exposure to αGal glycoconjugates could be particularly useful in pathological conditions associated with an increase in agalactosylated IgG antibodies such as observed in autoimmune and infectious diseases. This approach along with the expression of new carbohydrate antigens or the removal of non-neutralizing anti-carbohydrate antibodies might provide novel therapeutic applications for a broad number of disorders.
This work was supported by the “Fondo de Investigaciones Sanitarias” (FIS) grants PI10/01727 and PI13/01098 from Carlos III Health Institute, Spanish Ministry of Health, and European Commission’s Seventh Framework Program grant HEALTH-F4-2013-603049 TransLink Collaborative Project.
REFERENCES
1.Schwartz-Albiez, R., Monteiro, R. C., Rodriguez,
M., Binder, C. J., and Shoenfeld, Y. (2009) Natural antibodies,
intravenous immunoglobulin and their role in autoimmunity, cancer and
inflammation, Clin. Exp. Immunol., 158 (Suppl. 1),
43-50.
2.Holodick, N. E., Tumang, J. R., and Rothstein, T.
L. (2010) Immunoglobulin secretion by B1 cells: differential intensity
and IRF4-dependence of spontaneous IgM secretion by peritoneal and
splenic B1 cells, Eur. J. Immunol., 40, 3007-3016.
3.Sorman, A., Zhang, L., Ding, Z., and Heyman, B.
(2014) How antibodies use complement to regulate antibody responses,
Mol. Immunol., 61, 79-88.
4.Gronwall, C., and Silverman, G. J. (2014) Natural
IgM: beneficial autoantibodies for the control of inflammatory and
autoimmune disease, J. Clin. Immunol., 34 (Suppl. 1),
S12-21.
5.Panda, S., and Ding, J. L. (2015) Natural
antibodies bridge innate and adaptive immunity, J. Immunol.,
194, 13-20.
6.Bovin, N. V. (2013) Natural antibodies to glycans,
Biochemistry (Moscow), 78, 786-797.
7.Bovin, N., Obukhova, P., Shilova, N., Rapoport, E.,
Popova, I., Navakouski, M., Unverzagt, C., Vuskovic, M., and Huflejt,
M. (2012) Repertoire of human natural anti-glycan immunoglobulins. Do
we have auto-antibodies? Biochim. Biophys. Acta, 1820,
1373-1383.
8.Galili, U., Mandrell, R. E., Hamadeh, R. M.,
Shohet, S. B., and Griffis, J. M. (1988) Interaction between human
natural anti-α-galactosyl immunoglobulin G and bacteria of the
human flora, Infect. Immun., 56, 1730-1737.
9.Manez, R., Blanco, F. J., Diaz, I., Centeno, A.,
Lopez-Pelaez, E., Hermida, M., Davies, H. F., and Katopodis, A. (2001)
Removal of bowel aerobic gram-negative bacteria is more effective than
immunosuppression with cyclophosphamide and steroids to decrease
natural alpha-galactosyl IgG antibodies, Xenotransplantation,
8, 15-23.
10.Galili, U., Korkesh, A., Kahane, I., and
Rachmilewitz, E. A. (1983) Demonstration of a natural antigalactosyl
IgG antibody on thalassemic red blood cells, Blood, 61,
1258-1264.
11.D’Alessandro, M., Mariani, P., Lomanto, D.,
Bachetoni, A., and Speranza, V. (2002) Alterations in serum
anti-α-galactosyl antibodies in patients with Crohn’s and
ulcerative colitis, Clin. Immunol., 103, 63-68.
12.Fontan, M. P., Manez, R., Rodriguez-Carmona, A.,
Peteiro, J., Martinez, V., Garcia-Falcon, T., and Domenech, N. (2006)
Serum levels of anti-alpha galactosyl antibodies predict survival and
peritoneal dialysis-related enteric peritonitis rates in patients
undergoing renal replacement therapy, Am. J. Kidney Dis.,
48, 972-982.
13.Obukhova, P., Korchagina, E., Henry, S., and
Bovin, N. (2011) Natural anti-A and anti-B of the ABO system: allo- and
autoantibodies have different epitope specificity,
Transfusion, 52, 860-869.
14.Galili, U., Ishida, H., Tanabe, K, and Toma, H.
(2002) Anti-gal A/B, a novel anti-blood group antibody identified in
recipients of abo-incompatible kidney allografts,
Transplantation, 74, 1574-1580.
15.Galon, J., Costes, A., Sanchez-Cabo, F.,
Kirilovsky, A., Mlecnik, B., Lagorce-Pages, C., Tosolini, M., Camus,
M., Berger, A., Wind, P., Zinzindohoue, F., Bruneval, P., Cugnenc, P.
H., Trajanoski, Z., Fridman, W. H., and Pages, F. (2006) Type, density,
and location of immune cells within human colorectal tumors predict
clinical outcome, Science, 313, 1960-1964.
16.Galili, U., Wigglesworth, K., and Abdel-Motal, U.
M. (2007) Intratumoral injection of α-gal glycolipids induces
xenograft-like destruction and conversion of lesions into endogenous
vaccines, J. Immunol., 178, 4676-4687.
17.Galili, U., Albertini, M. R., Sondel, P. M.,
Wigglesworth, K., Sullivan, M., and Whalen, G. F. (2010) In situ
conversion of melanoma lesions into autologous vaccine by intratumoral
injections of α-gal glycolipids, Cancers, 2,
773-793.
18.Whalen, G. F., Sullivan, M., Piperdi, B.,
Wasseff, W., and Galili, U. (2012) Cancer immunotherapy by intratumoral
injection of α-gal glycolipids, Anticancer Res.,
32, 3861-3868.
19.Gening, M. L., Maira-Litran, T., Kropec, A.,
Skurnik, D., Grout, M., Tsvetkov, Y. E., Nifantiev, N. E., and Pier, G.
B. (2010) Synthetic β-(1→6)-linked N-acetylated and
nonacetylated oligoglucosamines used to produce conjugate vaccines for
bacterial pathogens, Infect. Immun., 78, 764-772.
20.Abdel-Motal, U. M., Guay, H. M., Wigglesworth,
K., Welsh, R. M., and Galili, U. (2007) Immunogenicity of influenza
virus vaccine is increased by anti-gal-mediated targeting to
antigen-presenting cells, J. Virol., 81, 9131-9141.
21.Abdel-Motal, U., Wang, S., Lu, S., Wigglesworth,
K., and Galili, U. (2006) Increased immunogenicity of human
immunodeficiency virus gp120 engineered to express
Galα1-3Galβ1-4GlcNAc-R epitopes, J. Virol.,
80, 6943-6951.
22.Phanse, Y., Carrillo-Conde, B. R., Ramer-Tait, A.
E., Broderick, S., Kong, C. S., Rajan, K., Flick, R., Mandell, R. B.,
Narasimhan, B., and Wannemuehler, M. J. (2014) A systems approach to
designing next generation vaccines: combining α-galactose
modified antigens with nanoparticle platforms, Sci. Rep.,
4, 3775.
23.Wigglesworth, K. M., Racki, W. J., Mishra, R.,
Szomolanyi-Tsuda, E., Greiner, D. L., and Galili, U. (2011) Rapid
recruitment and activation of macrophages by anti-Gal/α-Gal
liposome interaction accelerates wound healing, J. Immunol.,
186, 4422-4432.
24.Galili, U., Wigglesworth, K., and Abdel-Motal, U.
M. (2010) Accelerated healing of skin burns by anti-Gal/α-gal
liposomes interaction, Burns, 36, 239-251.
25.Hurwitz, Z. M., Ignotz, R., Lalikos, J. F., and
Galili, U. (2012) Accelerated porcine wound healing after treatment
with α-gal nanoparticles, Plast. Reconstr. Surg.,
129, 242e-251e.
26.Galili, U. (2013) Anti-Gal: an abundant human
natural antibody of multiple pathogeneses and clinical benefits,
Immunology, 140, 1-11.
27.Ubol, S., Phuklia, W., Kalayanarooj, S., and
Modhiran, N. (2010) Mechanisms of immune evasion induced by a complex
of dengue virus and preexisting enhancing antibodies, J. Infect.
Dis., 201, 923-935.
28.Willey, S., Aasa-Chapman, M. M., O’Farrell,
S., Pellegrino, P., Williams, I., Weiss, R. A., and Neil, S. J. (2011)
Extensive complement-dependent enhancement of HIV-1 by autologous
non-neutralizing antibodies at early stages of infection,
Retrovirology, 8, 16.
29.Takada, A., and Kawaoka, Y. (2003)
Antibody-dependent enhancement of viral infection: molecular mechanisms
and in vivo implications, Rev. Med. Virol., 13,
387-398.
30.Huisman, W., Martina, B. E., Rimmelzwaan, G. F.,
Gruters, R. A., and Osterhaus, A. D. (2009) Vaccine-induced enhancement
of viral infections, Vaccine, 27, 505-512.
31.Mahalingam, S., and Lidbury, B. A. (2003)
Antibody-dependent enhancement of infection: bacteria do it too,
Trends Immunol., 24, 465-467.
32.Skurnik, D., Kropec, A., Roux, D., Theilacker,
C., Huebner, J., and Pier, G. B. (2012) Natural antibodies in normal
human serum inhibit Staphylococcus aureus capsular
polysaccharide vaccine efficacy, Clin. Infect. Dis., 55,
1188-1197.
33.Hamadeh, R. M., Jarvis, G. A., Galili, U.,
Mandrell, R. E., Zhou, P., and Griffiss, J. M. (1992) Human natural
anti-Gal IgG regulates alternative complement pathway activation on
bacterial surfaces, J. Clin. Invest., 89, 1223-1235.
34.Hamadeh, R. M., Estabrook, M. M., Zhou, P.,
Jarvis, G. A., and Griffiss, J. M. (1995) Anti-Gal binds to pili of
Neisseria meningitidis: the immunoglobulin A isotype blocks
complement-mediated killing, Infect. Immun., 63,
4900-4906.
35.Wells, T. J., Whitters, D., Sevastsyanovich, Y.
R., Heath, J. N., Pravin, J., Goodall, M., Browning, D. F.,
O’Shea, M. K., Cranston, A., De Soyza, A., Cunningham, A. F.,
MacLennan, C. A., Henderson, I. R., and Stockley, R. A. (2014)
Increased severity of respiratory infections associated with elevated
anti-LPS IgG2 which inhibits serum bactericidal killing, J. Exp.
Med., 211, 1893-1904.
36.Giuntini, S., Reason, D. C., and Granoff, D. M.
(2012) Combined roles of human IgG subclass, alternative complement
pathway activation, and epitope density in the bactericidal activity of
antibodies to meningococcal factor H binding protein, Infect.
Immun., 80, 187-194.
37.Manez, R., Perez-Cruz, M., Bello, D., Dominguez,
M. A., and Costa, C. (2014) Removal of natural anti-galactose
α1,3 galactose antibodies with GAS914 enhances humoral immunity
and prevents sepsis mortality in mice, Intens. Care Med. Exp.,
2 (Suppl. 1), P50.
38.Galili, U., and Matta, K. L. (1996) Inhibition of
anti-Gal IgG binding to porcine endothelial cells by synthetic
oligosaccharides, Transplantation, 62, 256-262.
39.Byrne, G. W., Schwarz, A., Fesi, J. R., Birch,
P., Nepomich, A., Bakaj, I., Velardo, M. A., Jiang, C., Manzi, A.,
Dintzis, H., Diamond, L. E., and Logan, J. S. (2002) Evaluation of
different alpha-galactosyl glycoconjugates for use in
xenotransplantation, Bioconj. Chem., 13, 571-581.
40.Manez, R., Domenech, N., Centeno, A.,
Lopez-Pelaez, E., Crespo, F., Juffe, A., Duthaler, R. O., and
Katopodis, A. G. (2004) Failure to deplete anti-Gal α1-3Gal
antibodies after pig-to-baboon organ xenotransplantation by
immunoaffinity columns containing multiple Galα1-3Gal
oligosaccharides, Xenotransplantation, 11, 408-415.
41.Diamond, L. E., Byrne, G. W., Schwarz, A., Davis,
T. A., Adams, D. H., and Logan, J. S. (2002) Analysis of the control of
the anti-gal immune response in a non-human primate by galactose
α1-3 galactose trisaccharide-polyethylene glycol conjugate,
Transplantation, 73, 1780-1787.
42.Katopodis, A. G., Warner, R. G., Duthaler, R. O.,
Streiff, M. B., Bruelisauer, A., Kretz, O., Dorobek, B., Persohn, E.,
Andres, H., Schweitzer, A., Thoma, G., Kinzy, W., Quesniaux, V. F.,
Cozzi, E., Davies, H. F., Manez, R., and White, D. (2002) Removal of
anti-Galα1,3Gal xenoantibodies with an injectable polymer, J.
Clin. Invest., 110, 1869-1877.
43.Lam, T. T., Paniagua, R., Shivaram, G.,
Schuurman, H. J., Borie, D. C., and Morris, R. E. (2004) Anti-non-Gal
porcine endothelial cell antibodies in acute humoral xenograft
rejection of hDAF-transgenic porcine hearts in cynomolgus monkeys,
Xenotransplantation, 11, 531-535.
44.Chen, G., Qian, H., Starzl, T., Sun, H., Garcia,
B., Wang, X., Wise, Y., Liu, Y., Xiang, Y., Copeman, L., Liu, W.,
Jevnikar, A., Wall, W., Cooper, D. K., Murase, N., Dai, Y., Wang, W.,
Xiong, Y., White, D. J., and Zhong, R. (2005) Acute rejection is
associated with antibodies to non-Gal antigens in baboons using
Gal-knockout pig kidneys, Nat. Med., 11, 1295-1298.
45.Angus, D. C., Linde-Zwirble, W. T., Lidicker, J.,
Clermont, G., Carcillo, J., and Pinsky, M. R. (2001) Epidemiology of
severe sepsis in the United States: analysis of incidence, outcome, and
associated costs of care, Crit. Care Med., 29,
1303-1310.
46.McPherson, D., Griffiths, C., Williams, M.,
Baker, A., Klodawski, E., Jacobson, B., and Donaldson, L. (2013)
Sepsis-associated mortality in England: an analysis of multiple cause
of death data from 2001 to 2010, B. M. J. Open, 3,
e002586.
47.Almeida, I. C., Milani, S. R., Gorin, P. A., and
Travassos, L. R. (1991) Complement-mediated lysis of Trypanosoma
cruzi trypomastigotes by human anti-α-galactosyl antibodies,
J. Immunol., 146, 2394-2400.
48.Yilmaz, B., Portugal, S., Tran, T. M., Gozzelino,
R., Ramos, S., Gomes, J., Regalado, A., Cowan, P. J., d’Apice, A.
J., Chong, A. S., Doumbo, O. K., Traore, B., Crompton, P. D., Silveira,
H., and Soares, M. P. (2014) Gut microbiota elicits a protective immune
response against malaria transmission, Cell, 159,
1277-1289.
49.Frame, T., Carroll, T., Korchagina, E., Bovin,
N., and Henry, S. (2006) Synthetic glycolipid modification of red blood
cell membranes, Transfusion, 47, 876-882.
50.Oliver, C., Blake, D., and Henry, S. (2011)
Modeling transfusion reactions and predicting in vivo cell
survival with kodecytes, Transfusion, 51, 1723-1730.
51.Oliver, C., Blake, D., and Henry, S. (2011) In
vivo neutralization of anti-A and successful transfusion of A
antigen-incompatible red blood cells in an animal model,
Transfusion, 51, 2664-2675.
52.Jefferis, R. (1991) Structure–function
relationships in human immunoglobulins, Netherlands J. Med.,
39, 188-198.
53.Vidarsson, G., Dekkers, G., and Rispens, T.
(2014) IgG subclasses and allotypes: from structure to effector
functions, Front. Immunol., 5, 520.
54.Shade, K. T., and Anthony, R. M. (2013) Antibody
glycosylation and inflammation, Antibodies, 2,
392-414.
55.Schur, P. H. (1988) IgG subclasses. A historical
perspective, Monogr. Allergy, 23, 1-11.
56.Rispens, T., Davies, A. M., Ooijevaar-de Heer,
P., Absalah, S., Bende, O., Sutton, B. J., Vidarsson, G., and Aalberse,
R. C. (2014) Dynamics of inter-heavy chain interactions in human
immunoglobulin G (IgG) subclasses studied by kinetic Fab arm exchange,
J. Biol. Chem., 289, 6098-6109.
57.Schroeder, H. W., Jr., and Cavacini, L. (2010)
Structure and function of immunoglobulins, J. Allergy Clin.
Immunol., 125, S41-S52.
58.Edelman, G. M., Cunningham, B. A., Gall, W. E.,
Gottlieb, P. D., Rutishauser, U., and Waxdal, M. J. (1969) The covalent
structure of an entire gammaG immunoglobulin molecule, Proc. Natl.
Acad. Sci. USA, 63, 78-85.
59.Hristodorov, D., Fischer, R., and Linden, L.
(2013) With or without sugar? (A)glycosylation of therapeutic
antibodies, Mol. Biotechnol., 54, 1056-1068.
60.Grossberg, A. L., Stelos, P., and Pressman, D.
(1962) Structure of fragments of antibody molecules as revealed by
reduction of exposed disulfide bonds, Proc. Natl. Acad. Sci.
USA, 48, 1203-1209.
61.Huber, R. (1980) Spatial structure of
immunoglobulin molecules, Klin. Wochenschr., 58,
1217-1231.
62.Wang, J., Balog, C. I., Stavenhagen, K.,
Koeleman, C. A., Scherer, H. U., Selman, M. H., Deelder, A. M.,
Huizinga, T. W., Toes, R. E., and Wuhrer, M. (2011) Fc-glycosylation of
IgG1 is modulated by B-cell stimuli, Mol. Cell Proteom.,
10, M110.004655.
63.Hmiel, L. K., Brorson, K. A., and Boyne, M. T.
(2015) Post-translational structural modifications of immunoglobulin G
and their effect on biological activity, Anal. Bioanal. Chem.,
407, 79-94.
64.Nimmerjahn, F., Anthony, R. M., and Ravetch, J.
V. (2007) Agalactosylated IgG antibodies depend on cellular Fc
receptors for in vivo activity, Proc. Natl. Acad. Sci.
USA, 104, 8433-8437.
65.Abes, R., and Teillaud, J. L. (2010)
Glycosylation on effector functions of therapeutic IgG,
Pharmaceuticals, 3, 146-157.
66.Feige, M. J., Nath, S., Catharino, S. R.,
Weinfurtner, D., Steinbacher, S., and Buchner, J. (2009) Structure of
the murine unglycosylated IgG1 Fc fragment, J. Mol. Biol.,
391, 599-608.
67.Zauner, G., Selman, M. H. J., Bondt, A.,
Rombouts, Y., Blank, D., Deelder, A. M., and Wuhrer, M. (2013)
Glycoproteomic analysis of antibodies, Mol. Cell. Proteom.,
12, 856-865.
68.Lamontagne, A., Long, R. E., Comunale, M. A.,
Hafner, J., Rodemich-Betesh, L., Wang, M., Marrero, J., Di Bisceglie,
A. M., Block, T., and Mehta, A. (2013) Altered functionality of
anti-bacterial antibodies in patients with chronic hepatitis C virus
infection, PLoS One, 8, e64992.
69.Ho, C. H., Chien, R. N., Cheng, P. N., Liu, J.
H., Liu, C. K., Su, C. S., Wu, I. C., Li, I. C., Tsai, H. W., Wu, S.
L., Liu, W. C., Chen, S. H., and Chang, T. T. (2015) Aberrant serum
immunoglobulin G glycosylation in chronic hepatitis B is associated
with histological liver damage and reversible by antiviral therapy,
J. Infect. Dis., 211, 115-124.
70.Kaneko, Y., Nimmerjahn, F., and Ravetch, J. V.
(2006) Antiinflammatory activity of immunoglobulin G resulting from Fc
sialylation, Science, 313, 670-673.
71.Malhotra, R., Wormald, M. R., Rudd, P. M.,
Fischer, P. B., Dwek, R. A., and Sim, R. B. (1995) Glycosylation
changes of IgG associated with rheumatoid arthritis can activate
complement via the mannose-binding protein, Nat. Med., 1,
237-243.
72.Moore, J. S., Wu, X., Kulhavy, R., Tomana, M.,
Novak, J., Moldoveanu, Z., Brown, R., Goepfert, P. A., and Mestecky, J.
(2005) Increased levels of galactose-deficient IgG in sera of
HIV-1-infected individuals, AIDS, 19, 381-389.
73.Ackerman, M. E., Crispin, M., Yu, X., Baruah, K.,
Boesch, A. W., Harvey, D. J., Dugast, A. S., Heizen, E. L., Ercan, A.,
Choi, I., Streeck, H., Nigrovic, P. A., Bailey-Kellogg, C., Scanlan,
C., and Alter, G. (2013) Natural variation in Fc glycosylation of
HIV-specific antibodies impacts antiviral activity, J. Clin.
Invest., 123, 2183-2192.
74.Mehta, A. S., Long, R. E., Comunale, M. A., Wang,
M., Rodemich, L., Krakover, J., Philip, R., Marrero, J. A., Dwek, R.
A., and Block, T. M. (2008) Increased levels of galactose-deficient
anti-Gal immunoglobulin G in the sera of hepatitis C virus-infected
individuals with fibrosis and cirrhosis, J. Virol., 82,
1259-1270.
75.Van de Geijn, F. E., Wuhrer, M., Selman, M. H.,
Willemsen, S. P., de Man, Y. A., Deelder, A. M., Hazes, J. M., and
Dolhain, R. J. (2009) Immunoglobulin G galactosylation and sialylation
are associated with pregnancy-induced improvement of rheumatoid
arthritis and the postpartum flare: results from a large prospective
cohort study, Arthritis Res. Ther., 11, R193-R193.
76.Niwa, R., and Satoh, M. (2015) The current status
and prospects of antibody engineering for therapeutic use: focus on
glycoengineering technology, J. Pharm. Sci., 104,
930-941.
77.Shields, R. L., Lai, J., Keck, R.,
O’Connell, L. Y., Hong, K., Meng, Y. G., Weikert, S. H., and
Presta, L. G. (2002) Lack of fucose on human IgG1 N-linked
oligosaccharide improves binding to human Fc-gamma RIII and antibody
dependent cellular toxicity, J. Biol. Chem., 277,
26733-26740.
78.Shinkawa, T., Nakamura, K., Yamane, N.,
Shoji-Hosaka, E., Kanda, Y., Sakurada, M., Uchida, K., Anazawa, H.,
Satoh, M., Yamasaki, M., Hanai, N., and Shitara, K. (2003) The absence
of fucose but not the presence of galactose or bisecting
N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows
the critical role of enhancing antibody-dependent cellular
cytotoxicity, J. Biol. Chem., 278, 3466-3473.
79.Jedrzejewski, P. M., del Val, I. J.,
Constantinou, A., Dell, A., Haslam, S. M., Polizzi, K. M., and
Kontoravdi, C. (2014) Towards controlling the glycoform: a model
framework linking extracellular metabolites to antibody glycosylation,
Int. J. Mol. Sci., 15, 4492-4522.
80.Del Val, I. J., Kontoravdi, C., and Nagy, J. M.
(2010) Towards the implementation of quality by design to the
production of therapeutic monoclonal antibodies with desired
glycosylation patterns, Biotechnol. Progr., 26,
1505-1527.
81.Murrell, M. P., Yarema, K. J., and Levchenko, A.
(2004) The systems biology of glycosylation, Chembiochem,
5, 1334-1347.
82.Sou, S. N., Sellick, C., Lee, K., Mason, A.,
Kyriakopoulos, S., Polizzi, K. M., and Kontoravdi, C. (2014) How does
mild hypothermia affect monoclonal antibody glycosylation?
Biotechnol. Bioeng., doi: 10.1002/bit.25524 [Epub ahead of
print].
83.Hills, A. E., Patel, A., Boyd, P., and James, D.
C. (2001) Metabolic control of recombinant monoclonal antibody
N-glycosylation in GS-NS0 cells, Biotechnol. Bioeng.,
75, 239-251.
84.Grainger, R. K., and James, D. C. (2013) CHO cell
line specific prediction and control of recombinant monoclonal antibody
N-glycosylation, Biotechnol. Bioeng., 110,
2970-2983.
85.Jedrzejewski, P. M., del Val, I. J., Polizzi, K.
M., and Kontoravdi, C. (2013) Applying quality by design to
glycoprotein therapeutics: experimental and computational efforts of
process control, Pharm. Bioprocess., 1, 51-69.
86.Wong, N. S., Wati, L., Nissom, P. M., Feng, H.
T., Lee, M. M., and Yap, M. G. (2010) An investigation of intracellular
glycosylation activities in CHO cells: effects of nucleotide sugar
precursor feeding, Biotechnol. Bioeng., 107,
321-336.
87.Kalia, K., Sharma, S., and Mistry, K. (2004)
Non-enzymatic glycosylation of immunoglobulins in diabetic nephropathy,
Clin. Chem. Acta, 347, 169-176.