REVIEW: Natural Compounds: Role in Reversal of Epigenetic Changes
Ruchi Aggarwal1#, Meenakshi Jha2#, Anju Shrivastava2, and Abhimanyu Kumar Jha1*
1Department of Biotechnology, IMS Engineering College, U. P. 201009, India; E-mail: abhimanyu2006@gmail.com2Department of Zoology, Delhi University, Delhi 110007, India
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received January 5, 2015; Revision received February 16, 2015
The hallmarks of carcinogenesis are characterized by alterations in the expression of multiple genes that occur via genetic and epigenetic alterations, leading to genome rearrangements and instability. The reversible process of epigenetic regulation, which includes changes in DNA methylation, histone modifications, and alteration in microRNA (miRNA) expression that alter phenotype without any change in the DNA sequence, is recognized as a key mechanism in cancer cell metabolism. Recent advancements in the rapidly evolving field of cancer epigenetics have shown the anticarcinogenic potential of natural compounds targeting epigenetic mechanism as a common molecular approach for cancer treatment. This review summarizes the potential of natural chemopreventive agents to reverse cancer-related epigenetic aberrations by regulating the activity of histone deacetylases, histone acetyltransferases, DNA methyltransferase I, and miRNAs. Furthermore, there is impetus for determining novel and effective chemopreventive strategies, either alone or in combination with other anticancer agents that exhibit similar properties, for improving the therapeutic aspects of cancer.
KEY WORDS: epigenetic, histone, DNA methylation, cancerDOI: 10.1134/S0006297915080027
Cancer is the second most common disease worldwide. According to Globocan 2012 (http://globocan.iarc.fr), 14.1 million new cancer cases, 8.2 million cancer deaths occurred, and 32.6 million people were living with cancer (within 5 years of diagnosis) worldwide. The majority of the cancer cases occurred in the less developed regions with an estimate of 57% (8 million) new cancer cases and 65% (5.3 million) cancer deaths. Cancer starts and progresses by synchronized genetic and epigenetic alterations, which cause changes in expression of multiple genes or leading to either activation of oncogenes or silencing of tumor suppressor genes [1]. Epigenetics is defined as heritable changes in gene expression caused by chemical modification of genes that do not involve changes in primary DNA sequence. These epigenetic modifications are considered as potential initiating events that occur during early carcinogenesis. Epigenetic mechanisms including chromatin remodeling in an appropriate manner via recruitment of a wide array of chromatin remodeling complex coregulators, effectors, and transcription factors, synthesis of non-coding microRNAs (miRNAs), DNA methylation, as well as covalent modification of histones such as acetylation, deacetylation, phosphorylation, ubiquitination, and sumoylation are considered as important determinants for regulation of gene expression [2].
EPIGENETIC MECHANISMS
DNA Methylation
DNA methylation is the covalent modification of eukaryotic DNA mediated by DNA methyltransferases (DNMT) that transfer methyl groups from S-adenosyl-L-methionine (SAM) to the 5th position of the cytosine pyrimidine ring located predominantly within CpG sequences [3]. The human genome contains CpG-rich regions, known as CpG islands, located in regulatory regions of housekeeping genes, tissue-specific genes, and tumor suppressors, are usually unmethylated in normal cells with the exception of about 6-8% of CGIs methylated in a tissue-specific manner [4].
By contrast, the hypermethylation of repetitive genomic sequences such as ribosomal DNA repeats, satellite repeats, or centromeric repeats prevents chromosomal instability, translocations, and gene disruption by limiting accessibility to the transcription machinery [5]. These methylation markers are added to DNA by enzymes of the DNMT family. DNMT1, a ubiquitous enzyme, is considered the major methyltransferase that maintains DNA methylation responsible for copying DNA methylation patterns to daughter strands during DNA replication [6]. The other two DNA-methylating enzymes (DNMT3A and DNMT3B) serve as de novo methyltransferases that set up DNA methylation patterns early in development [7].
At the same time, the genome of cancer cells undergoes global hypomethylation at repetitive genomic sequences and is associated with genomic instability and chromosomal aberrations [8, 9]. DNA methylation has critical roles in various physiological processes such as X chromosome inactivation, imprinting, and the silencing of germline-specific genes and may affect the transcription of genes by preventing the binding of key transcriptional factors [10]. DNA hypermethylation of promoter CpG islands of tumor suppressor genes leads to transcriptional silencing, which contributes to many of the hallmarks of cancer cells [11].
Histone Modifications
Within the nucleus, DNA is wrapped around a histone octamer formed by four histone partners – an H3-H4 tetramer and two H2A-H2B dimers [12]. Posttranslational modifications at the N-terminal of histones including acetylation, methylation, ubiquitination, phosphorylation, biotinylation, sumoylation, ADP ribosylation, and proline isomerization contribute to genomic stability, DNA damage response, and cell cycle checkpoints integrity affecting gene transcription and DNA repair [13]. These modifications are catalyzed by a variety of histone-modifying enzymes that add or remove specific groups such as histone methyltransferases (HMTs), histone demethylases (HDMs), histone acetyltransferases (HATs), and histone deacetylases (HDACs) [14].
Histone acetylation. Histone acetylation is a highly dynamic process maintained by the interplay of histone acetyltransferases (HATs) and histone deacetylases (HDACs) that acts in opposite manner. HATs use acetyl-CoA as a cofactor and catalyze the transfer of an acetyl group to the ε-amino group of lysine (K) residues in histone tails, neutralizing positive charge on lysine and resulting in a more relaxed chromatin structure, enabling the recruitment of the transcriptional machinery, whereas HDACs reverse the acetylation of lysine residues by catalyzing their transfer to coenzyme A (CoA) to restore their positive charge and stabilize the local chromatin architecture. Hyperacetylation of histone proteins results in activation of normally repressed genes, whereas hypoacetylation leads to silencing of gene transcription [15]. Various regulatory proteins and transcription factors such as p53, E2F, and nuclear factor-κB (NF-κB) involved in stress response, inflammation, and apoptosis are regulated by acetylation [16].
Histone methylation. Histone methylation is a process by which methyl groups are transferred from S-adenosylmethionine (SAM) to lysine and arginine residues of histone proteins of chromosomes, associated with either transcriptional activation, inactivation, or silencing of genomic regions [17]. Histone methylation is mediated by SAM-dependent histone methyl transferases. The effect of histone methylation on gene function and chromatin state is dependent on the methylated lysine residue (e.g. K4, K9, K27, K36, or K79 in H3), the methylation status (mono-, di-, or tri-methylation), and the location (interaction with promoter vs. gene coding regions) [18].
Histone phosphorylation. The process of histone phosphorylation takes place on serine, threonine, and tyrosine residues and is regulated by protein kinases (PKs) and protein phosphatases (PPs) that add and remove the modification, respectively [19]. The modification by histone kinases adds negative charge of the phosphate group from ATP to the histones, which results in repulsive force between them and is therefore associated with transcriptional activation. Phosphorylation of the core histones has been found to be associated with different cellular responses including transcriptional regulation, mitosis, DNA repair, developmental gene regulation, cell cycle progression, chromosome condensation, and apoptosis [20].
Histone ubiquitination and sumoylation. Histone ubiquitination is an enzymatic, posttranslational modification process in which the 76-amino acid protein ubiquitin is attached to a lysine residue of the histone core proteins H2A and H2B via the sequential action of three enzymes, E1 ubiquitin-activating, E2 ubiquitin-conjugating, and E3 ubiquitin-ligase enzymes. The enzyme complex is specific for a substrate and determines the degree of ubiquitination, i.e. either mono- or poly-ubiquitination. Ubiquitination of histones H2A and H2B is associated with different consequences on gene transcription. H2A mono-ubiquitination is considered as a repressive mark, whereas H2B ubiquitination plays an important role in transcription activation and repression. H2B ubiquitination is considered as prerequisite for H3 methylation, but H2A results in inhibition of this methylation [21, 22]. SUMO is a small ubiquitin-related modifier protein of length ∼100 amino acids and is attached to the targeted lysine residues of histone protein through an enzyme cascade similar to ubiquitination. Sumoylation of proteins results in a variety of effects including protein stability or enzymatic activity, altered localization, modulation of protein–protein or protein–DNA interactions, or antagonizing other lysine modifications such as ubiquitination, transcription regulation and included transcription factors, transcription machinery associated proteins, and components that modify chromatin structure [23].
Sumoylation of transcriptional activators has been found to be associated with transcriptional repression because of its interaction with other repressor complexes and due to its prevention of histone acetylation, which can be reversed by the action of desumoylating enzymes [24].
Interplay of DNA Methylation and Histone Modifications
In addition to performing their individual roles, interaction of histone modifications and DNA methylation with each other is medicated by a group of proteins with methyl-DNA-binding activity, including methyl-CpG-binding protein 2 (MeCP2), methyl-CpG-binding domain protein 1 (MBD1), and Kaiso, also known as ZBTB 33 (zinc finger and BTB domain-containing protein 33). Localization of these proteins to the promoter of the methylated DNA leads to the recruitment of a protein complex containing histone deacetylases (HDACs) and histone methyltransferases and thus determines gene expression status, chromatin organization, and cellular identity [25]. It has been shown that DNA methylation via several HMTs including G9a, SUV39H1, and PRMT5 to their specific genomic targets led to gene silencing by directly recruiting DNMTs [26-28]. In addition, HMTs and demethylases have also been associated with the regulation of the stability of DNMT proteins, which in turn recruit HDACs and methyl-binding proteins to result in gene silencing and chromatin condensation [29].
miRNA
MicroRNAs (miRNAs) are small non-coding RNAs of 20-22 nucleotides that function in RNA silencing and post transcriptional regulation of gene expression. MicroRNAs are involved in the regulation of biological processes such as cell cycle control, apoptosis, and several developmental and physiological processes including differentiation and development and are altered in cancer development [30]. The involvement of miRNA in suppressing gene expression can arise through numerous mechanisms, including genomic abnormalities, transcriptional regulation, and processing of miRNAs [31]. In addition, controlling the expression of DNMTs and other enzymes associated with epigenetic modifications as well as binding imperfectly to the 3′-untranslated region of the target mRNA leads to aberrant expression, which can affect mRNA translation and stability [32, 33].
Epigenetic Regulation of Gene Expression
Transcriptional deregulation can result in activation of transcription factors associated with oncogenes or silencing of genes responsible for tumor suppression leading to inappropriate gene expression [34]. The characteristic epigenetic markers associated with cancer cells include global hypomethylation, promoter-specific hypermethylation, posttranslational histone modifications, nucleosome remodeling with associated core proteins, global downregulation of miRNA, and upregulation of epigenetic machinery that regulates gene expression [35].
DNA methylation-mediated transcriptional silencing is thought to occur by interfering with the DNA binding of transcriptional factors such as CREB (cAMP response element binding protein), E2F (elongation 2 factor), NF-κB, and AP-2 (activator protein-2) that are unable to recognize specific sequences when they are methylated and by recruitment of methyl-CpG-binding transcriptional repressors, which in turn results in a condensed chromatin state [36]. In addition, methylated DNA can be bound by methyl-CpG-binding domain (MBD) proteins, which are essential for binding to 5-methylcytosine and hinders the binding of transcriptional factors to regulatory elements within promoters and enhancers [37]. These proteins thus help in the recruitment of other proteins such as histone deacetylases that are able to modify the methylation status of histones binding to some specific genetic locus, thereby causing transcriptional silencing of gene expressions.
Histone methylation is associated with different effects of gene activity. Methylation of lysine residue results in either activation or repression of gene activity, whereas arginine methylation leads to silencing of gene transcription. Enrichment of histone methylation at H3K4, H3K36, or H3K79 is generally associated with transcriptional activation. On the other hand, transcriptionally inactive regions are enriched with histone methylation at H3K9, H3K20, or H4K27me [38].
DNA methylation can also direct H3K9 methylation through effector proteins such as MeCP2, thereby establishing a repressive chromatin state [39]. The interactions between DNA methylation machinery and histone modifying enzymes further enhance the complexity of epigenetic regulation of gene expression, which determines and maintains cellular identity and function.
Aberrant miRNA regulation associated with cancer can be influenced by DNA methylation and histone covalent modifications [40]. On the other hand, miRNA targets enzymes of the epigenetic machinery. DNMT1 and DNMT3B are targets of miR-148a in cholangiocarcinoma and cervical cancer, respectively. HDAC1 is targeted by miR-449a and -b in prostate cancer, and HDAC4 by miR-1 in hepatocellular carcinoma [41]. In fact, DNA methylation and miRNA signal transcriptional silencing of associated genes, whereas specific histone modifications and their associated proteins can be characteristic of either gene silencing or gene expression. This review encompasses the various epigenetic changes and the role of natural compounds in their reversal.
ROLE OF NATURAL COMPOUNDS IN EPIGENETIC REVERSAL
The multistage nature of cancer development could potentially be modulated by chemicals that effect cellular enzyme systems, gene expression, signal transduction pathways, differentiation, or interactions with surrounding cells and extracellular matrices. Hence, rationally designed drugs that target a single gene product are unlikely to be of help in preventing or treating cancer. Moreover, theses targeted drugs can cause life-threatening side effects or therapy resistance [42]. When a complex system starts to dysfunction, it is generally best to fix it early, as prevention is better than cure. Often, cancers have a long latency period – 20 years or more. By the time they are clinically detectable, the system has degenerated into a disorganized, chaotic state at which point it may be beyond repair [43]. This has created an urgent need for safe and effective chemopreventive multifunctional drugs that act on entire networks in the body rather than single targets [44].
The basic idea of cancer chemoprevention is to arrest or reverse the progression of premalignant cells towards full malignancy using physiological mechanisms that do not kill healthy cells, but attenuate cancer inflammation [45]. The global demand for more affordable therapeutics and concerns about side effects of commonly used drugs has focused recent research in phytochemicals and traditional medicines that allow chronic use [46]. Natural compounds have been presumed to be safer than synthetic compounds due to their presence in the diet, wide availability, and tolerability. Natural dietary agents including fruits, vegetables, and spices can regulate gene expression via epigenetic mechanisms [47]; they have gained considerable importance owing to their demonstrated ability to suppress cancers. Epidemiological studies have shown that consumption of food rich in fruits, vegetables, and spices can effectively lower the incidence of cancers, indicating correlation between increased intake of dietary antioxidants and associated lower incidence of cancer mortality and morbidity [48, 49]. The reversible nature of epigenetic markers has led to great demand for the identification and development of natural compounds as epigenetic modulators for the prevention of cancer to restore “normal epigenome” [50, 51].
Natural Compounds Affecting the Epigenome
Accumulated evidence shows that natural compounds are associated with numerous health benefits along with their high potential for application in oncology owing to their anticarcinogenic properties [52]. Studies on natural bioactive compounds include plant secondary metabolites such as phytochemicals extracted as natural products from fruits, vegetables, spices and traditional medicinal herbs are associated with the regulation of multiple cancer-inflammation pathways, and epigenetic cofactors can be used in anticancer therapy because of its wide availability, low toxicity, and cost effective measures [53].
A variety of botanical species and plant parts are known for their chemopreventive potential, as they possess antiinflammatory, antioxidant, antiallergic, antiviral, and anticancer activities [54]. It has been shown that dietary agents and non-nutritional components of fruits and vegetables can regulate epigenetic-related mechanisms via initiation of apoptosis, repression of cancer-related genes, reactivation of tumor suppressor genes, cell cycle regulation, and the activation of cell survival protein in different cancers [55, 56].
The interference of multistage carcinogenesis by natural compounds may include their effects on detoxifying and antioxidant enzyme system, induction of antimetastasis response by targeting specific key transcription factors such as activation protein (AP-1), nuclear factor erythroid 2-related factor (NRF2), hypoxia inducible factor-1 (HIF-1) and NF-κB, inhibition of the mitogen-activated proteins (MAPKs)-, protein kinases and growth-factor receptor-mediated pathways, cell cycle arrest, induction of apoptosis, epigenetic cofactors, and microRNAs associated with tumor development and progression [57, 58]. Several natural compounds have been found to play an important role in the reversal of epigenetic changes.
Quercetin. Quercetin is strong natural antioxidant flavonol ubiquitously present in dietary plant sources such as fruits and vegetables, particularly citrus fruits, apples, onions, parsley, sage, tea, red wine, leaves, and grains. The properties underlying the antioxidant activity of quercetin may include oxygen radical scavenging, xanthine oxidase inhibition, protection against lipid peroxidation in vitro, and formation of inert complexes by chelating metal ions that cannot take part in the conversion of superoxide radicals and hydrogen peroxide into hydroxyl radicals [59]. The anticancer efficacy of quercetin has been linked to the modulation of mitogenic signaling, cell-cycle regulation, survival/apoptotic signaling, angiogenic, and metastatic events in cancer cells, with limited or no toxicity against normal cells.
Quercetin has been shown to have effect on histone acetylation. Quercetin decreases the level of COX-2 (cyclooxygenase-2) protein by blocking the binding of different transcription activators such as CREB2, NF-κB, p300, and c-Jun to the promoter of proinflammatory gene COX2, which accounts for its anticancer activity [60].
It has been reported that quercetin increases deacetylase activity of recombinant SIRT1, but on the other hand, it inhibits SIRT1 activity at the cellular level [61]. This biphasic effect on the activity of class III histone deacetylase SIRTs can be explained by metabolic transformation of quercetin. Ingestion of quercetin-3-O-glucuronide, a quercetin metabolite, leads to inhibitory function on recombinant SIRT-1. SIRT1-induced apoptosis in response to TNF-α (tumor necrosis factor alpha) suppresses transcription of NF-κB-regulated genes by deacetylating RelA/p65 at lysine 310 [62].
Quercetin induces FasL-dependent apoptosis through promotion of acetylation of histone H3 in leukemia HL60 cells by activating HAT and inhibiting HDAC, which results in activation of ERK (extracellular signal-regulated kinase) and JNK (jun N-terminus kinase) signaling pathways. Quercetin in human leukemia HL-60 cells induces significant histone hyperacetylation, indicating that its in vitro anticancer activity is linked to histone hyperacetylation [63]. Quercetin also activates signaling pathways by induction of the phosphorylation of ATM and H2AX [64].
Quercetin showed a concentration-dependent effect on hypermethylation of p16INK4a, a tumor suppressor gene, in human colon cancer cell line RKO. It resulted in the reversal of hypermethylation after 120 h of treatment, expressing the influence of quercetin on DNA as well as protein methylation levels [65].
A study has revealed inhibition activity of quercetin against demethylase LSD1. This may result in controlling the gene transcription associated with cell growth and differentiation and is of potential therapeutic value [66]. Quercetin and other catechol polyphenols can inhibit DNMTs and thus DNA methylation indirectly through changing concentrations of SAM and SAH (S-adenosyl-L-homocysteine) intracellularly in a specific manner [67].
EGCG. Epigallocatechin gallate (EGCG), also known as epigallocatechin-3-gallate, is the ester of epigallocatechin and gallic acid and is the most abundant catechin in green tea, accounting for more than 50% of the active compounds present in green tea. Studies have shown that polyphenolic compounds, green tea catechins, can reduce the risks of diseases associated with cancer. Other major green tea catechins include (−)-epicatechin-3-gallate (ECG), (−)-epigallocatechin (EGC), and (−)-epicatechin (EC) [68]. EGCG is a powerful radical scavengers and acts as a pro- and antioxidant, triggers signal-transduction pathways, and induces the expression of phase II detoxifying and antioxidant enzymes such as glutathione peroxidase (GPx), glutathione, and many other enzymes. It is known to inhibit carcinogenesis by modulating some signal transduction pathways including JAK/STAT, Wnt, and Notch [69].
Studies have shown EGCG to have various properties that could give it anticancer activity that may include several different mechanisms based on modifications of epigenetic processes, particularly in cancer cells, which can be altered by the epigenetic machinery. They include apoptosis induction, regulation of signal transduction pathway, inhibition of oxidative stress and angiogenesis, cell cycle arrest, and reduction of cancer cell proliferation [70, 71].
EGCG showed a direct inhibitory effect on the activity of the DNMT1 molecule through the formation of hydrogen bonds on binding with different residues in the catalytic pocket of DNMT stabilized by Mg2+ [72]. An indirect inhibitory effect of EGCG on DNMT1 is due to that compounds with a catechol group are excellent substrates for methylation mediated by catechol-O-methyltransferase (COMT), which results in depletion of the methyl donor SAM and formation of SAH, a potent feedback inhibitor of DNA methylation considered as a potential demethylating agent.
EGCG effects at physiologically attainable concentrations have been shown to be associated with demethylation of specific promoters of genes including p16, MGMT, hMLH1 (human mutL homolog 1), GSTP1 (glutathione S-transferase Pi), and/or RARβ in various cancer cell lines [73]. Treating LNCaP cells with EGCG decreases transcriptional gene activity coding for HDACs 1-3 and thus decreases protein levels that are associated with increased acetylation level of lysines 9 and 18 on H3 histone and H4 histone related amino acid residues as well [74].
Recent studies involving human epidermoid carcinoma cells A431 revealed that treatment of these cells with EGCG results in re-expression of silenced tumor suppressor gene p16(INK4a) and p21/Cip1 mRNA and proteins through decreasing DNA methylation level of lysine 9 on histone 3 (H3-K9) and also associated with their effect on DNMT activity. On treating cells with EGCG at different concentrations (5-20 µM), decreased levels of 5-methyl-cytosine, DNMT activity, mRNA and protein levels of DNMT1, DNMT3a, and DNMT3b was expected, but treatment with these concentration showed increased acetylation of lysines 9 and 14 on H3 histone (H3-K9 and -K14) and acetylated lysine 5, 12, and 16 on H4 histone [75].
Treating AsPC-1 pancreatic metastatic adenocarcinoma cells with EGCG also inhibits HDAC activity, which in turn induces Raf kinase inhibitor protein (RKIP) via repressing ERK activation and upregulated E-cadherin expression. This leads to increase in histone H3 expression and inhibition of NF-κB nuclear translocation, Snail (zinc finger protein SNAI1) expression, and MMP-2 and-9 activities contributing to decreased metastatic potential of AsPC-1 cells [76].
EGCG treatment on human colon carcinoma HT29 cells showed a significant dose-dependent inhibition of HDAC activity and HDAC1 protein expression. In addition, this treatment also reversed the aberrant hypermethylation status of tumor suppressor gene RECK and thus enhanced mRNA expression significantly [77].
Experiments conducted with EGCG on prostate cancer cell lines LNCaP and PC-3 showed dose- and time-dependent inhibition of class I HDACs associated with increased histone H3 and p53 acetylation. Acetylation of p53 at Lys373 and Lys382 resulted in p53 accumulation in the G0/G1 cell cycle phase due to block of its MDM-2-mediated ubiquitination, which then binds with the p21 and BAX gene promoters leading to apoptosis induction, suggesting proteasomal degradation of class I HDACs by EGCG [78].
The inhibitory influences on HATs by EGCG at 50 µM may ultimately suppress agonist-dependent androgen receptor (AR) activation by downregulation of its acetylation, and it is thus considered beneficial to hormone-dependent prostate cancer. AR remains “locked” in the cytoplasm, which provides transcriptional silencing of AR-related genes. Acetylation of AR by p300/CBP and PCAF (HAT KAT2B)/TIP60 (HAT KAT5) supports the understanding that HAT coactivators compete with HDAC corepressors for binding to promoter regions and/or protein substrates and determine the level of transcription [79].
In addition to study the influence of EGCG on HDACs, it has also been identified as a HAT inhibitor with global specificity for the majority of HAT enzymes. Inhibitory effect of 90% on HAT activity was observed on treating HeLa cell nuclear extract with 100 µM EGCG, with no change in HDAC, SIRT1, and histone methyltransferase activities. Hypoacetylation of RelA (p65) on inhibition of NF-κB activation by EGCG resulted in increased level of cytosolic IκBα, which in turn prevents p65 translocation into the nucleus, thus interrupting the TNFα-induced cascade of events [80]. In addition, EGCG has been found to demethylate the promoter of the Wnt oncogene in lung cells [81].
EGCG alone or in combination other epigenetic-modifying compounds such as HDAC inhibitors can be considered as effective anticancer agents and have led to great attention to the development of an epigenetic diet for cancer prevention [82].
Curcumin. The phytochemical curcumin (1,7-bis(4-hydroxy-3-methoxy-phenyl)-1,6-heptadiene-3,5-dione) is a biologically active phenolic component of Curcuma longa (turmeric). It has long been used in traditional Indian Ayurvedic medicine [83]. Curcumin has many pharmacological effects and possesses antioxidative, antiinflammatory, antiproliferative, antiangiogenic, and anticancer properties, thus being used as a therapeutic anticancer agent [84].
It is considered as nontoxic and has been shown to suppress tumor growth through multiple signaling pathways by regulation of expression of various proteins including NF-κB, Akt, MAPK, p53, Nrf2, Notch-1, JAK/STAT, β-catenin, and AMPK (5′-AMP-activated protein kinase). The anticancer efficacy of curcumin has been linked to the modulation of mitogenic signaling, cell-cycle regulation, survival/apoptotic signaling, angiogenic, and metastatic events in cancer cells [85]. Pharmacokinetic analyses have shown that human plasma contains a low concentration of curcumin compared with measured concentration in experimental cell culture systems, which might be related to its biological effect through remodeling of the epigenome network. Curcumin is considered a strong epigenome modulator and functions as a histone-modifying compound, thereby acting as an inhibitor of HDAC and HAT [86].
In 2004, curcumin, in its physiological range, was reported to be a specific inhibitor of p300/CREB-binding protein (CBP) HAT activity in HeLa cervical cancer cell line, while there is no change in p300/CBP-associated factors, histone deacetylase HDAC1, and histone methyltransferases. As a consequence, acetylation of histones H3 and H4 is inhibited with the most potent inhibitory IC50 concentration of 25 µM, which in turn inhibited p300-mediated acetylation of p53 [87]. It also promotes proteasomal degradation of p300 and CBP with no effects on the HATs PCAF or GCN5 [88].
It has been shown that curcumin induces cell cycle arrest and apoptosis of numerous cancer cell lines, which is mainly attributed to the NF-κB and PI3K/AKT signaling pathway [89]. NF-κB, which is known as a proinflammatory transcription factor, contributed to the activation of different genes involved in various cellular processes as it undergoes acetylation with p300/CBP acetyltransferases at multiple lysine residues on histone. It has been reported that treating cancer cells with curcumin could significantly reduce HAT activity, p300 levels, and acetylated CBP/p300 gene expression, and consequently suppressed NF-κB binding, thus contributing to its potent NF-κB inhibitory activity [90].
The ability of HDAC inhibitors to alter various cellular functions that are deregulated in cancer cells enables them to be used as cancer therapeutic compounds [91]. Various studies have revealed the influence of curcumin on HDAC expression. Curcumin is found to be a more potent HDAC inhibitor than sodium butyrate and valproic acid, which are well-known HDAC inhibitors. It has been reported that curcumin treatment can lead to significant decrease in protein levels of HDAC 1, 3, and 8, thereby increasing acetylation level of histone H4 [92].
The contradicting effect of curcumin on different subtypes of HDAC enzymes requires better understanding of the mechanism involved in HDAC expression on curcumin treatment [93]. Curcumin also showed potent activity as an inhibitor of HDAC in medulloblastoma cells, and it directly inhibited HDAC4 transcription [94].
Numerous animal studies have shown in vitro inhibition of HATs and HDACs in different cancer models. Evidences suggesting the inhibition of both HATs and HDACs together might provide a novel strategy to prevent cancer [95]. Curcumin induces epigenetic modifications via modulating DNA methylation and is considered as an effective hypomethylating agent decreasing DNMT activity, thus facilitating inactive prometastatic and proto-oncogene expressions. Curcumin inhibits DNMT activity by interacting directly with SssI methyltransferase, a homolog of DNMT1, and covalently blocks the catalytic thiolate of C1226 of DNMT1, which inhibits DNMT1 catalytic activity [86, 96].
In a breast cancer cell line at 10 µM concentration, curcumin induced significant decrease in protein levels of DNMT1, HDAC1, and MeCP2 as well as in the transcriptional levels of all three DNMTs, thereby reversing epigenetic modifications through preventing the binding of NF-κB/Sp1 binding to the promoter region of DNMT1 [97].
Curcumin showed epigenomic effects by inducing global DNA hypomethylation in MV4-11 leukemia cells and decreasing (by 15-20%) DNMT1 expression in an acute myeloid leukemia (AML) cell line MV4-11 model with IC50 of 3 µM after 72 h treatment. This effect may be associated with reactivation of tumor suppressor gene p15INK4B (p15) through promoter hypomethylation, G1 cell cycle arrest, and in vitro induction of apoptosis [98]. Curcumin has been found to demethylate and reactivate tumor suppressor genes RARβ2 and WIF-1 in human cervical and non-small-cell lung cancer cells, respectively [99, 100]. In addition, it may lead to NEUROG1 and NRF1 promoter demethylation and re-expression in human prostate cancer cells [101, 102].
Treating selected colorectal cancer cell lines HCT116, HT29, and RKO with curcumin changed methylation pattern at selected partially methylated loci rather than completely methylated CpG sites [103]. It has been reported to selectively induce NRF2 and RARβ2 gene promoter demethylation [104]. In MDA-MB-435 cells, curcumin has been found to downregulate EZH2 expression and thus reduced methylation of H3K27me3 [105]. Studies also showed induction of antiinflammatory effects of curcumin and suggest correlation between inflammation and epigenetic alterations that occurs during tumorigenesis [106].
Curcumin has been found to alter miRNA expression in BxPC-3 pancreatic cancer cell line via upregulation of tumor suppressor miR-22 and downregulation of oncogenic miR-199a, while the upregulation of miR-22 is associated with suppression of the target gene Sp1 and Era expressions. In contrast, curcumin has also been found to suppress miR-21 expression and thus induce tumor suppressor PTEN [107]. Exposure of MCF-7 breast cancer cells to curcumin can lead to increase in the levels of proapoptotic miR-15a and miR-16 accompanied by suppression of the expression of their target gene Bcl-2 [108].
Low bioavailability of curcumin due to its insolubility and instability in water may lead to an issue of concern of use of curcumin as a bioactive agent in chemotherapy. It can be enhanced by utilizing properties of dietary factors such as rubusoside, found in Chinese blackberry extract, as well as compounds such as phosphatidylcholine, found in soy and egg yolks, thus increasing its therapeutic potential in cancer prevention [109].
Genistein. Genistein (4′,5,7-trihydroxyisoflavone) is a phytoestrogen belonging to a class of polyphenolic flavonoids primarily abundant in soybeans [110]. This polyphenol has been shown to act as chemopreventive agent against several cancer types, particularly hormone-responsive breast and prostate malignancies attributed to the hormonal activity mediated by binding to estrogen receptor, ER-α, and ER-β, as well as androgen receptor downregulation in androgen-dependent prostate cancer cell lines such as LNCaP [111, 112]. Exposure to moderate doses of genistein induced inhibitory effects on cervical, prostate, colon, and esophageal cancers with the associated mechanism contributing to its anticancer properties including carcinogen bioactivation, cell-signaling, cell cycle regulation, angiogenesis, oxidative stress, inflammation, and gene transcription regulation by affecting epigenetic mechanisms that are altered in cancer cells [113, 114].
Genistein is only a moderately strong radical scavenger, thus exerting its antioxidant effects at low concentrations corresponding to the relevant physiological concentrations in plasma genistein shown to have inhibitory effect on cellular growth followed by apoptosis in various hematological cancer cell lines and cell lines of solid tumor origin (e.g. HCT-116 and SW-480) [115]. Its effect on cell cycle is commonly associated with induction of G2/M cell cycle arrest in breast, colon, malignant glioma, and prostate cancer cell lines [116, 117].
Genistein is also a potent modulator of epigenetic markers (including DNA methylation and/or covalent histone modifications), acting either directly or through a steroid receptor-dependent process. Treatment with genistein (10-25 µM) on prostate cell lines LNCaP and DuPro promotes hyperacetylation of histones H3 and H4 associated with increased H3K4 acetylation at tumor suppressor genes p16INK4a and p21WAF1/CIP1 transcription start sites, resulting in increased expression of transcriptional activating HATs and thus subsequent re-expression [118].
In vivo experiments on a murine orthotopic breast cancer model supplemented with genistein-enriched diet showed inhibition of cancer growth with considerable lower net weight of tumor and significantly less proliferating cell nuclear antigen-positive cells compared with tumors developed in animals fed with a genistein-free diet. Genistein effects the epigenome at mRNA level via increasing tumor suppressor genes p16 and p21 expression and downregulating oncogenes BMI1 (polycomb complex protein BMI-1) and c-MYC. It also increased acetylation of histone H3 and H3K4me3. On the other side, formation of suppressive chromatin markers H3K9me3 and H3K27me3 was decreased. The more prominent changes were seen in precancerous breast cell line, whereas breast cancer cells exhibited mild changes [119].
Treatment of ER-α positive, MCF-7 and ER-α negative MDA-MB 231 breast cancer cell line with 18.5 µM concentration of genistein showed decrease in trimethylated markers at H3K27, H3K9, and H3K4 on six selected genes including SRC3 (steroid receptor coactivator protein 3 that displays HAT activity), BRCA1, ER-α, ER-β, EZH2 (histone-lysine N-methyltransferase that adds methyl group to H3K27), and p300 [120].
When genistein is added to HT29 cell line, it showed significant inhibitory effects on HDAC activity with potent inhibitory IC50 concentration of 97 ± 18 µM [77]. However, treatment of HT29 cells with high concentration (200 µM) of genistein was associated with downregulation of HDAC1 protein. Likewise, treatment of KYSE510 esophageal squamous carcinoma cells with 5 and 100 µM genistein induced HDAC activity inhibition of 13.2 and 33%, respectively, resulting in reversal of promoter hypermethylation of p16, RARβ2, and MGMT (O6-methylguanine methyltransferase) and thus gene re-expression [121].
Genistein showed significant reversal of promoter hypermethylation and reactivation of tumor suppressor gene BTG3 in prostate carcinoma cell lines LNCaP and PC3 as well as in renal cell carcinoma cell lines A498, ACHN, and HEK-293, associated with increased level of histone acetylation and binding of RNA polymerase II with the promoter of BTG3 gene resulting in the formation of active chromatin, while in squamous cervical cancer cell line SiHa it was found to be associated with promoter demethylation of RARβ2, a tumor suppressor gene, and thus led to gene reactivation [122].
In addition to the effect of genistein on histone modifications, particularly histone acetylation and methylation, it also influences DNMTs in vitro at protein expression level and by inhibiting DNMT1 activity as well. It downregulates methylation status specifically at tumor suppressor genes p21 and p16 promoter, thereby affecting survival of cancerous cells [123].
Genistein can reactivate the gene silenced by methylation in breast cancers not only by DNMT1 activity inhibition, but also via downregulating DNMT1 protein level. In MCF-7 and MDAMB-231 cell lines, it was shown to significantly inhibit HDAC1 but also to decrease DNMT1 and MeCP2 protein levels associated with decreased transcription expression of DNMT1, DNMT3a, and DNMT3b. In addition, treatment of breast cancer with a low concentration (3.125 µM) of genistein resulted in demethylation at the GSTP1 gene promoter.
The intrinsic synergistic/additive effect by genistein may be associated with its inhibitory effects on DNMTs and HDACs together. Genistein-mediated hypomethylation and hyperacetylation associated with reactivation of silenced PTEN tumor suppressor gene expression in prostate cancer cells. In addition, genistein has also been shown to regulate the expression of miRNA, an epigenetic marker, and their targets in several cancer types [124, 125]. Other natural compounds that have been found to be of importance in the reversal of epigenetic changes are mentioned in the table.
Natural compounds playing important role in reversal of epigenetic
changes
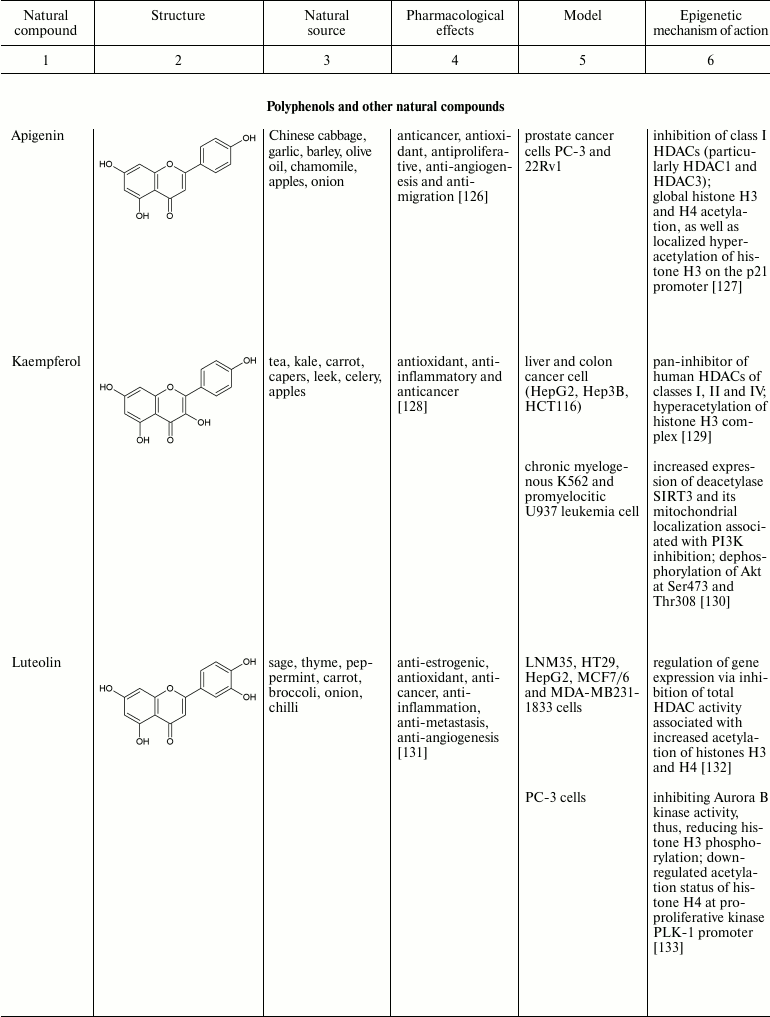
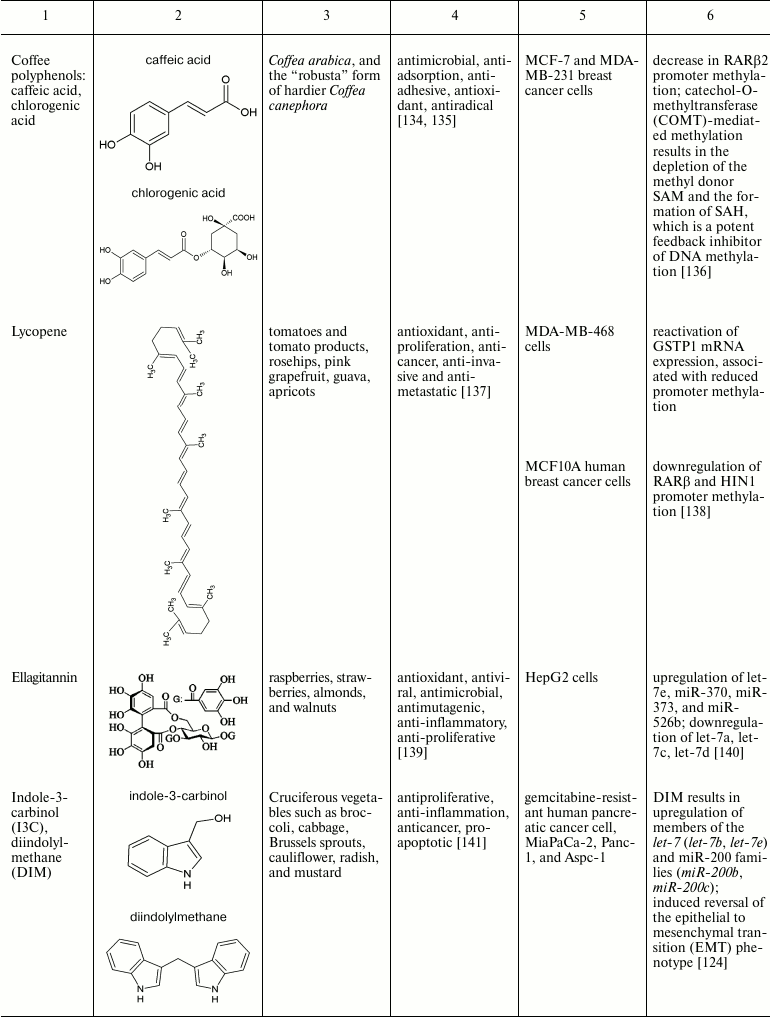
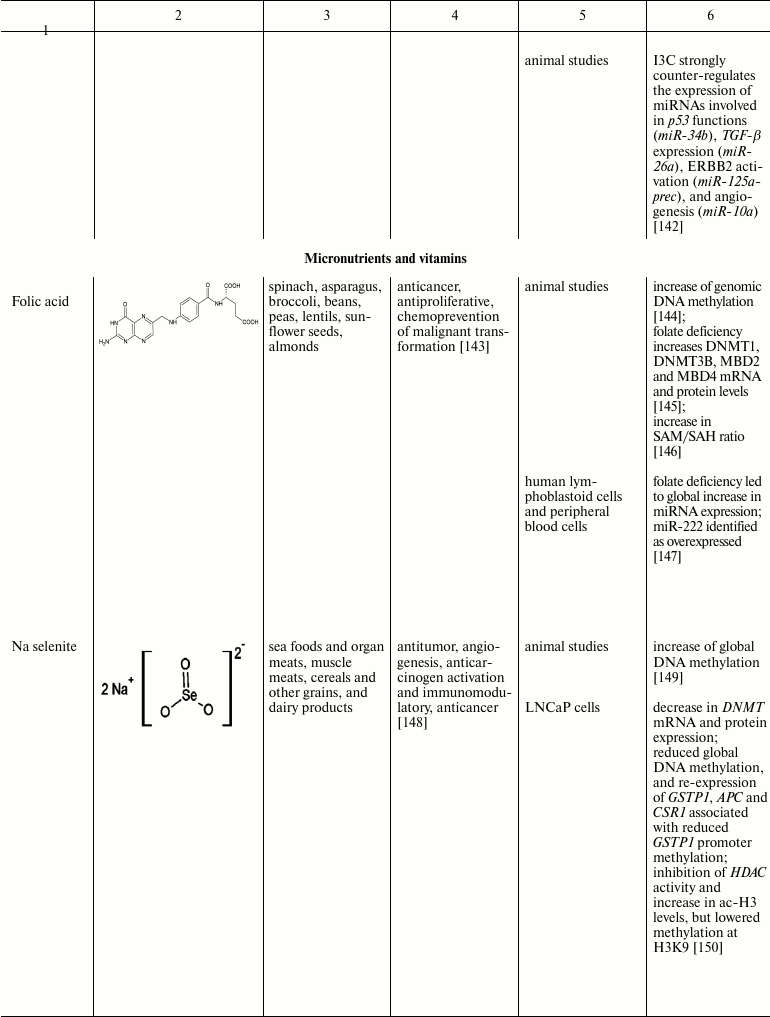
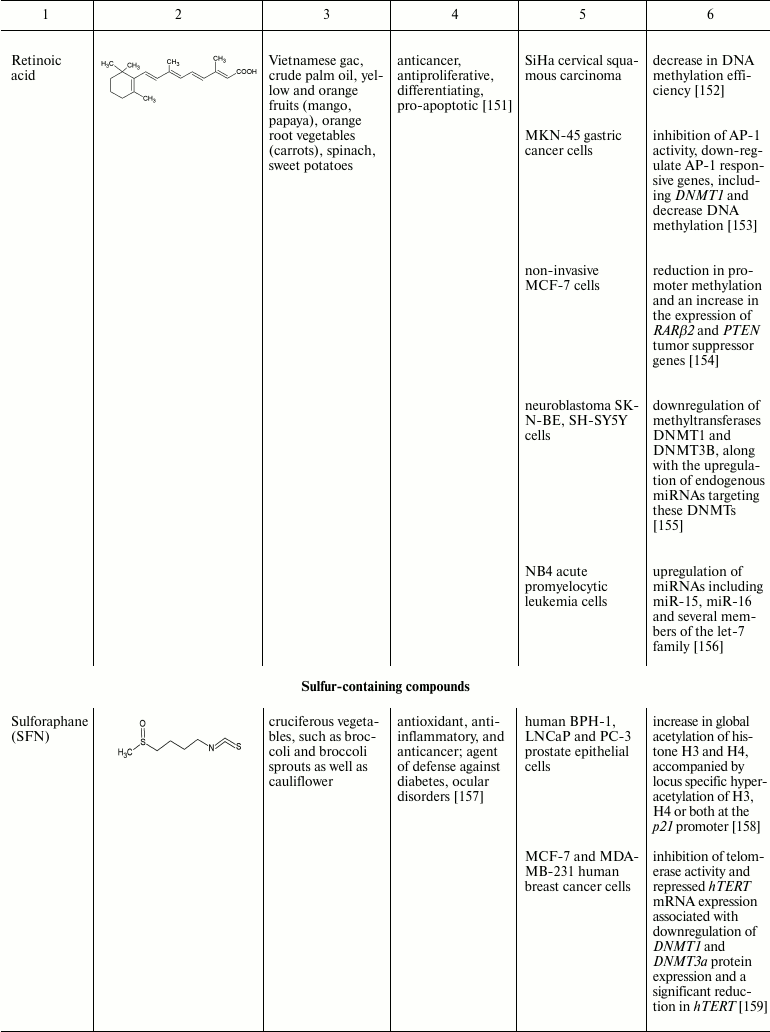
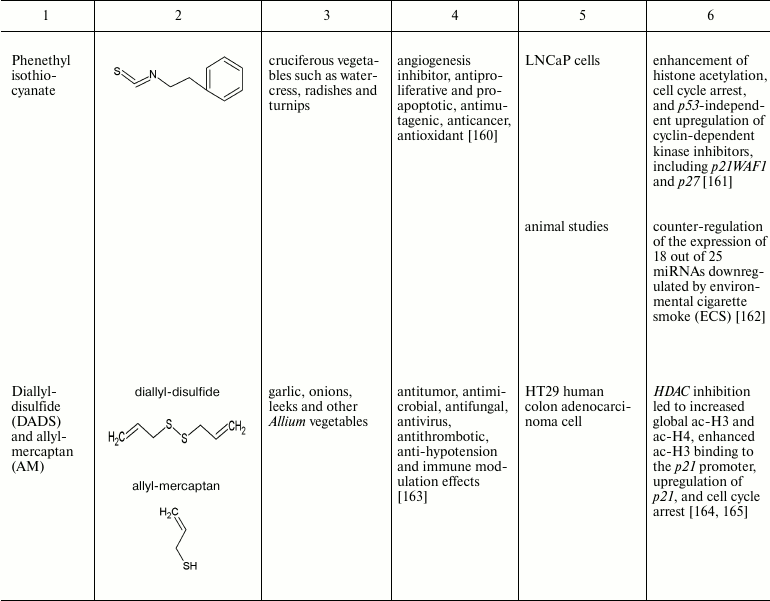
CONCLUSION AND PROSPECTS
Alterations in epigenetic modifications regulating essential cellular processes required for maintaining cellular identity have been found to be associated with cancer. Studies discussed in this review have shown that dietary chemopreventive agents can reverse these abnormal epigenetic alterations by affecting global DNA methylation accompanied by reactivation of tumor suppressor genes silenced by promoter hypermethylation, upregulating genes by altering histone covalent modifications as well as miRNA, and thus they could be considered as chemotherapeutic agents for cancer therapy. This characteristic has resulted in increasing enthusiasm for developing therapeutic strategies by targeting various epigenetic factors such as HDAC, HAT, DNMTs, and miRNAs by natural compounds either alone or in combination with similar compounds having structural and functional similarity. However, further investigations of other natural compounds as effective therapeutic epigenetic agents need to fully explore the potential of these compounds in the treatment of cancer and other diseases as well as those based on structure–activity relationship.
REFERENCES
1.Baylin, S. B., Esteller, M., Rountree, M. R.,
Bachman, K. E., Schuebel, K., and Herman, J. G. (2001) Aberrant
patterns of DNA methylation, chromatin formation and gene expression in
cancer, Hum. Mol. Genet., 10, 687-692.
2.Yoo, C. B., and Jones, P. A. (2006) Epigenetic
therapy of cancer: past, present and future, Nat. Rev. Drug
Discov., 5, 37-50.
3.Bird, A. (2002) DNA methylation patterns and
epigenetic memory, Genes Dev., 16, 6-21.
4.Illingworth, R., Kerr, A., Desousa, D., Jorgensen,
H., Ellis, P., Stalker, J., Jackson, D., Clee, C., Plumb, R., Rogers,
J., Humphray, S., Cox, T., Langford, C., and Bird, A. (2008) A novel
CpG island set identifies tissue-specific methylation at developmental
gene loci, PLoS Biol., 6, e22.
5.Esteller, M. (2007) Cancer epigenomics: DNA
methylomes and histone-modification maps, Nat. Rev. Genet.,
8, 286-298.
6.Chen, T., Hevi, S., Gay, F., Tsujimoto, N., He, T.,
Zhang, B., Ueda, Y., and Li, E. (2007) Complete inactivation of DNMT1
leads to mitotic catastrophe in human cancer cells, Nat. Genet.,
39, 391-396.
7.Okano, M., Bell, D. W., Haber, D. A., and Li, E.
(1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de
novo methylation and mammalian development, Cell, 99,
247-257.
8.Szyf, M. (2005) DNA methylation and demethylation
as targets for anticancer therapy, Biochemistry (Moscow),
70, 533-549.
9.Stefanska, B., Salame, P., Bednarek, A., and
Fabianowska-Majewska, K. (2011) Comparative effects of retinoic acid,
vitamin D and resveratrol alone and in combination with adenosine
analogues on methylation and expression of phosphatase and tensin
homologue tumor suppressor gene in breast cancer cells, Br. J.
Nutr., 107, 781-790.
10.Tate, P. H., and Bird, A. P. (1993) Effects of
DNA methylation on DNA-binding proteins and gene expression, Curr.
Opin. Genet. Dev., 3, 226-231.
11.Hatziapostolou, M., and Iliopoulos, D. (2011)
Epigenetic aberrations during oncogenesis, Cell Mol. Life Sci.,
68, 1681-1702.
12.Kouzarides, T. (2007) Chromatin modifications and
their function, Cell, 128, 693-705.
13.Fullgrabe, J., Kavanagh, E., and Joseph, B.
(2011) Histone onco-modifications, Oncogene, 30,
3391-3403.
14.Yang, X. J., and Seto, E. (2007) HATs and HDACs:
from structure, function and regulation to novel strategies for therapy
and prevention, Oncogene, 26, 5310-5318.
15.Kondo, Y. (2009) Epigenetic cross-talk between
DNA methylation and histone modifications in human cancers, Yonsei
Med. J., 50, 455-463.
16.Spange, S., Wagner, T., Heinzel, T., and Kramer,
O. H. (2009) Acetylation of non-histone proteins modulates cellular
signaling at multiple levels, Int. J. Biochem. Cell Biol.,
41, 185-198.
17.Jenuwein, T., and Allis, C. D. (2001) Translating
the histone code, Science, 293, 1074-1080.
18.Bannister, A. J., and Kouzarides, T. (2011)
Regulation of chromatin by histone modifications, Cell Res.,
21, 381-395.
19.Xhemalce, B., Dawson, M. A., and Bannister, A. J.
(2011) Histone modifications, in Encyclopedia of Molecular
Cell Biology and Molecular Medicine, Epigenetic Regulation and
Epigenomics, 2nd Edn. (Meyers, R. A., ed.) Wiley-VCH, Weinheim,
Germany, pp. 1-45.
20.Cruickshank, M. N., Besant, P., and Ulgiati, D.
(2010) The impact of histone post-translational modifications on
developmental gene regulation, Amino Acids, 39,
1087-1105.
21.Shilatifard, A. (2006) Chromatin modifications by
methylation and ubiquitination. Implications in the regulation of gene
expression, Annu. Rev. Biochem., 75,
243-269.
22.Weake, V. M., and Workman, J. L. (2008) Histone
ubiquitination: triggering gene activity, Mol. Cell, 29,
653-663.
23.Johnson, E. S. (2004) Protein modification by
SUMO, Annu. Rev. Biochem., 73, 355-382.
24.Garcia-Dominguez, M., and Reyes, J. C. (2009)
SUMO association with repressor complexes, emerging routes for
transcriptional control, Biochim. Biophys. Acta, 1789,
451-459.
25.Cedar, H., and Bergman, Y. (2009) Linking DNA
methylation and histone modification: patterns and paradigms, Nat.
Rev. Genet., 10, 295-304.
26.Lehnertz, B., Ueda, Y., Derijck, A.,
Braunschweig, U., Perez-Burgos, L., Kubicek, S., Chen, T., Li, E.,
Jenuwein, T., and Peters, A. (2003) Suv39h-mediated histone H3 lysine 9
methylation directs DNA methylation to major satellite repeats at
pericentric heterochromatin, Curr. Biol., 13,
1192-1200.
27.Tachibana, M., Matsumura, Y., Fukuda, M., Kimura,
H., and Shinkai, Y. (2008) G9a/GLP complexes independently mediate H3K9
and DNA methylation to silence transcription, EMBO J.,
27, 2681-2690.
28.Zhao, Q., Rank, G., Tan, Y. T., Li, H., Moritz,
R. L., Simpson, R. J., Cerruti, L., Curtis, D. J., Patel, D. J., Allis,
C. D., Cunningham, J. M., and Jane, S. M. (2009) PRMT5-mediated
methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA
methylation in gene silencing, Nat. Struct. Mol. Biol.,
16, 304-311.
29.Esteve, P. O., Chin, H. G., Benner, J., Feehery,
G. R., Samaranayake, M., Horwitz, G. A., Jacobsen, S. E., and Pradhan,
S. (2009) Regulation of DNMT1 stability through SET7-mediated lysine
methylation in mammalian cells, Proc. Natl. Acad. Sci. USA,
106, 5076-5081.
30.Calin, G. A., and Croce, C. M. (2006) MicroRNA
signatures in human cancers, Nat. Rev. Cancer, 6,
857-866.
31.Winter, J., Jung, S., Keller, S., Gregory, R. I.,
and Diederichs, S. (2009) Many roads to maturity: microRNA biogenesis
pathways and their regulation, Nat. Cell Biol., 11,
228-234.
32.Guil, S., and Esteller, M. (2009) DNA methylomes,
histone codes and miRNAs: tying it all together, Int. J. Biochem.
Cell Biol., 41, 87-95.
33.Lujambio, A., Portela, A., Liz, J., Melo, S. A.,
Rossi, S., Spizzo, R., Croce, C. M., Calin, G. A., and Esteller, M.
(2010) CpG island hypermethylation-associated silencing of noncoding
RNAs transcribed from ultraconserved regions in human cancer,
Oncogene, 29, 6390-6401.
34.Cox, P. M., and Goding, C. R. (1991)
Transcription and cancer, Br. J. Cancer, 63, 651-662.
35.Taby, R., and Issa, J. P. J. (2010) Cancer
epigenetics, CA Cancer J. Clin., 60, 376-392.
36.Paluszczak, J., and Baer-Dubowska, W. (2005)
Epigenome and cancer: new possibilities of cancer prevention and
therapy? Postepy Biochem., 51, 244-250.
37.Reik, W., and Dean, W. (2001) DNA methylation and
mammalian epigenetics, Electrophoresis, 22,
2838-2843.
38.Upadhyay, A. K., and Cheng, X. (2011) Dynamics of
histone lysine methylation: structures of methyl writers and erasers,
Prog. Drug Res., 67, 107-124.
39.Fuks, F., Hurd, P. J., Wolf, D., Nan, X., Bird,
A. P., and Kouzarides, T. (2003) The methyl-CpG-binding protein MeCP2
links DNA methylation to histone methylation, J. Biol. Chem.,
278, 4035-4040.
40.Iorio, M. V., Piovan, C., and Croce, C. M. (2010)
Interplay between microRNAs and the epigenetic machinery: an intricate
network, Biochim. Biophys. Acta, 1799, 694-701.
41.Zhou, H., Hu, H., and Lai, M. (2010) Non-coding
RNAs and their epigenetic regulatory mechanisms, Biol. Cell,
102, 645-655.
42.Hanahan, D., and Weinberg, R. A. (2011) Hallmarks
of cancer: the next generation, Cell, 144, 646-674.
43.Sporn, M. B. (2011) Perspective: the big C
– for chemoprevention, Nature, 471, S10-11.
44.Deocaris, C. C., Widodo, N., Wadhwa, R., and
Kaul, S. C. (2008) Merger of Ayurveda and tissue culture-based
functional genomics: inspirations from systems biology, J. Transl.
Med., 6, 14.
45.Aggarwal, B. B., and Gehlot, P. (2009)
Inflammation and cancer: how friendly is the relationship for cancer
patients? Curr. Opin. Pharmacol., 9, 351-369.
46.Li, J. W. H., and Vederas, J. C. (2009) Drug
discovery and natural products: end of an era or an endless frontier?
Science, 325, 161-165.
47.Matouk, C. C., and Marsden, P. A. (2008)
Epigenetic regulation of vascular endothelial gene expression, Circ.
Res., 102, 873-887.
48.Reddy, L., Odhav, B., and Bhoola, K. D. (2003)
Natural products for cancer prevention: a global perspective,
Pharmacol. Therap., 99, 1-13.
49.Devasagayam, T. P., Tilak, J. C., Boloor, K. K.,
Sane, K. S., Ghaskadbi, S. S., and Lele, R. D. (2004) Free radicals and
antioxidants in human health: current status and future prospects,
J. Assoc. Physicians India, 52, 794-804.
50.Dashwood, R. H., and Ho, E. (2007) Dietary
histone deacetylase inhibitors: from cells to mice to man, Semin.
Cancer Biol., 17, 363-369.
51.Arasaradnam, R. P., Commane, D. M., Bradburn, D.,
and Mathers, J. C. (2008) A review of dietary factors and its influence
on DNA methylation in colorectal carcinogenesis, Epigenetics,
3, 193-198.
52.Link, A., Balaguer, F., and Goel, A. (2010)
Cancer chemoprevention by dietary polyphenols: promising role for
epigenetics, Biochem. Pharmacol., 80, 1771-1792.
53.Aggarwal, B. B., Prasad, S., Reuter, S.,
Kannappan, R., Yadev, V. R., Park, B., Kim, J. H., Gupta, S. C.,
Phromnoi, K., Sundaram, C., Chaturvedi, M. M., and Sung, B. (2011)
Identification of novel anti-inflammatory agents from Ayurvedic
medicine for prevention of chronic diseases: “reverse
pharmacology” and “bedside to bench” approach,
Curr. Drug Targets, 12, 1593-1653.
54.Surh, Y. J. (2003) Cancer chemoprevention with
dietary phytochemicals, Nat. Rev. Cancer, 3, 768-780.
55.Landis-Piwowar, K. R., Milacic, V., and Dou, Q.
P. (2008) Relationship between the methylation status of dietary
flavonoids and their growth-inhibitory and apoptosis-inducing
activities in human cancer cells, J. Cell Biochem., 105,
514-523.
56.Paluszczak, J., Krajka-Kuzniak, V., and
Baer-Dubowska, W. (2010) The effect of dietary polyphenols on the
epigenetic regulation of gene expression in MCF7 breast cancer cells,
Toxicol. Lett., 192, 119-125.
57.Meeran, S. M., Ahmed, A., and Tollefsbol, T. O.
(2010) Epigenetic targets of bioactive dietary components for cancer
prevention and therapy, Clin. Epigenet., 1, 101-116.
58.Szarc vel Szic, K., Ndlovu, M. N., Haegeman, G.,
and Vanden Berghe, W. (2010) Nature or nurture: let food be your
epigenetic medicine in chronic inflammatory disorders, Biochem.
Pharmacol., 80, 1816-1832.
59.Cai, Y. Z., Mei, S., Jie, X., Luo, Q., and Corke,
H. (2006) Structure-radical scavenging activity relationships of
phenolic compounds from traditional Chinese medicinal plants, Life
Sci., 78, 2872-2888.
60.Xiao, X., Shi, D., Liu, L., Wang, J., Xie, X.,
Kang, T., and Deng, W. (2011) Quercetin suppresses cyclooxygenase-2
expression and angiogenesis through inactivation of p300 signaling,
PloS One, 6, e22934.
61.De Boer, V. C., de Goffau, M. C., Arts, I. C.,
Hollman, P. C., and Keijer, J. (2006) SIRT1 stimulation by polyphenols
is affected by their stability and metabolism, Mech. Ageing
Dev., 127, 618-627.
62.Yeung, F., Hoberg, J. E., Ramsey, C. S., Keller,
M. D., Jones, D. R., Frye, R. A., and Mayo, M. W. (2004) Modulation of
NFκB-dependent transcription and cell survival by the SIRT1
deacetylase, EMBO J., 23, 2369-2380.
63.Lee, W. J., Chen, Y. R., and Tseng, T. H. (2011)
Quercetin induces FasL-related apoptosis, in part, through promotion of
histone H3 acetylation in human leukemia HL-60 cells, Oncol.
Rep., 25, 583-591.
64.Ye, R., Goodarzi, A. A., Kurz, E. U., Saito, S.,
Higashimoto, Y., Lavin, M. F., Appella, E., Anderson, C. W., and
Lees-Miller, S. P. (2004) The isoflavonoids genistein and quercetin
activate different stress signaling pathways as shown by analysis of
site-specific phosphorylation of ATM, p53 and histone H2AX, DNA
Repair, 3, 235-244.
65.Tan, S., Wang, C., Lu, C., Zhao, B., Cui, Y.,
Shi, X., and Ma, X. (2009) Quercetin is able to demethylate the
p16INK4a gene promoter, Chemotherapy, 55, 6-10.
66.Abdulla, A., Zhao, X., and Yang, F. (2013)
Natural polyphenols inhibit lysine-specific demethylase-1 in
vitro, J. Biochem. Pharmacol. Res., 1, 56-63.
67.Lee, W. J., Shim, J. Y., and Zhu, B. T. (2005)
Mechanisms for the inhibition of DNA methyltransferases by tea
catechins and bioflavonoids, Mol. Pharmacol., 68,
1018-1030.
68.Grahaman, H. N. (1992) Green tea composition,
consumption and polyphenol chemistry, Prev. Med., 21,
334-350.
69.Huang, J., Plass, C., and Gerhauser, C. (2011)
Cancer chemoprevention by targeting the epigenome, Curr. Drug
Targets, 12, 1925-1956.
70.Yang, C., Lambert, J., and Sang, S. (2009)
Antioxidative and anti-carcinogenic activities of tea polyphenols,
Arch. Toxicol., 83, 11-21.
71.Li, Y., and Tollefsbol, T. O. (2010) Impact on
DNA methylation in cancer prevention and therapy by bioactive dietary
components, Curr. Med. Chem., 17, 2141-2151.
72.Singh, B. N., Shankar, S., and Srivastava, R. K.
(2011) Green tea catechin, epigallocatechin-3-gallate (EGCG):
mechanisms, perspectives and clinical applications, Biochem.
Pharmacol., 82, 1807-1821.
73.Fang, M., Wang, Y., Ai, N., Hou, Z., Sun, Y., Lu,
H., Welsh, W., and Yang, C. (2003) Tea polyphenol
(−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and
reactivates methylation-silenced genes in cancer cell lines, Cancer
Res., 63, 7563-7570.
74.Pandey, M., Shukla, S., and Gupta, S. (2010)
Promoter demethylation and chromatin remodeling by green tea
polyphenols leads to re-expression of GSTP1 in human prostate cancer
cells, Int. J. Cancer, 126, 2520-2533.
75.Nandakumar, V., Vaid, M., and Katiyar, S. K.
(2011) (–)-Epigallocatechin-3-gallate reactivates silenced tumor
suppressor genes Cip1/p21 and p16INK4a by reducing DNA methylation and
increasing histones acetylation in human skin cancer cells,
Carcinogenesis, 32, 537-544.
76.Kim, S. O., and Kim, M. R. (2013)
(–)-Epigallocatechin 3-gallate inhibits invasion by inducing the
expression of Raf kinase inhibitor protein in AsPC-1 human pancreatic
adenocarcinoma cells through the modulation of histone deacetylase
activity, Int. J. Oncol., 42, 349-358.
77.Groh, I. A., Chen, C., Luske, C., Cartus, A. T.,
and Esselen, M. (2013) Plant polyphenols and oxidative metabolites of
the herbal alkenylbenzene methyleugenol suppress histone deacetylase
activity in human colon carcinoma cells, J. Nutr. Metab.,
2013, 821082.
78.Thakur, V. S., Gupta, K., and Gupta, S. (2012)
Green tea polyphenols increase p53 transcriptional activity and
acetylation by suppressing class I histone deacetylases, Int. J.
Oncol., 41, 353-361.
79.Lee, Y. H., Kwak, J., Choi, H. K., Choi, K. C.,
Kim, S., Lee, J., Jun, W., Park, H. J., and Yoon, H. G. (2012) EGCG
suppresses prostate cancer cell growth modulating acetylation of
androgen receptor by anti-histone acetyltransferase activity, Int.
J. Mol. Med., 30, 69-74.
80.Lee, J. M., and Yoon, H. G. (2009)
Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor,
inhibits EBV-induced B lymphocyte transformation via suppression of
RelA acetylation, Cancer Res., 69, 583-592.
81.Gao, Z., Xu, Z., Hung, M. S., Lin, Y. C., Wang,
T., Gong, M., Zhi, X., Jablon, D. M., and You, L. (2009) Promoter
demethylation of WIF-1 by epigallocatechin-3-gallate in lung cancer
cells, Anticancer Res., 29, 2025-2030.
82.Landis-Piwowar, K. R., Huo, C., Chen, D.,
Milacic, V., Shi, G., Chan, T. H., and Dou, Q. P. (2007) A novel
prodrug of the green tea polyphenol
(−)-epigallocatechin-3-gallate as a potential anticancer agent,
Cancer Res., 67, 4303-4310.
83.Krishnaswamy, K. (2008) Traditional Indian spices
and their health significance, Asia Pac. J. Clin. Nutr., 17
(Suppl. 1), 265-268.
84.Goel, A., and Aggarwal, B. B. (2010) Curcumin,
the golden spice from Indian saffron, is a chemosensitizer and
radiosensitizer for tumors and chemoprotector and radioprotector for
normal organs, Nutr. Cancer, 62, 919-930.
85.Kunnumakkara, A. B., Anand, P., and Aggarwal, B.
B. (2008) Curcumin inhibits proliferation, invasion, angiogenesis and
metastasis of different cancers through interaction with multiple cell
signaling proteins, Cancer Lett., 69, 199-225.
86.Dicato, M., and Diederich, M. (2013) Curcumin as
a regulator of epigenetic events, Mol. Nutr. Food Res.,
57, 1619-1629.
87.Kang, J., Chen, J., Shi, Y., Jia, J., and Zhang,
Y. (2005) Curcumin induced histone hypoacetylation: the role of
reactive oxygen species, Biochem. Pharmacol., 69,
1205-1213.
88.Marcu, M. G., Jung, Y. J., Lee, S., Chung, E. J.,
Lee, M. J., Trepel, J., and Neckers, L. (2006) Curcumin is an inhibitor
of p300 histone acetyltransferase, Med. Chem., 2,
169-174.
89.Reuter, S., Eifes, S., Dicato, M., Aggarwal, B.
B., and Diederich, M. (2008) Modulation of anti-apoptotic and survival
pathways by curcumin as a strategy to induce apoptosis in cancer cells,
Biochem. Pharmacol., 76, 1340-1351.
90.Gupta, S. C., Sundaram, C., Reuter, S., and
Aggarwal, B. B. (2010) Inhibiting NF-κB activation by small
molecules as a therapeutic strategy, Biochim. Biophys. Acta,
1799, 775-787.
91.Davis, C. D., and Ross, S. A. (2007) Dietary
components impact histone modifications and cancer risk, Nutr.
Rev., 65, 88-94.
92.Chen, Y., Shu, W., Chen, W., Wu, Q., Liu, H., and
Cui, G. (2007) Curcumin, both histone deacetylase and p300/CBP-specific
inhibitor, represses the activity of nuclear factor κB and Notch
1 in Raji cells, Basic Clin. Pharmacol. Toxicol., 101,
427-433.
93.Meja, K. K., Rajendrasozhan, S., Adenuga, D.,
Biswas, S. K., Sundar, I. K., Spooner, G., Marwick, J. A., Chakravarty,
P., Fletcher, D., Whittaker, P., Megson, I. L., Kirkham, P. A., and
Rahman, I. (2008) Curcumin restores corticosteroid function in
monocytes exposed to oxidants by maintaining HDAC2, Am. J. Respir.
Cell Mol. Biol., 39, 312-323.
94.Lee, S. J., Krauthauser, C., Maduskuie, V.,
Fawcett, P. T., Olson, J. M., and Rajasekaran, S. A. (2011)
Curcumin-induced HDAC inhibition and attenuation of medulloblastoma
growth in vitro and in vivo, BMC Cancer,
11, 144.
95.Fu, S., and Kurzrock, R. (2010) Development of
curcumin as an epigenetic agent, Cancer, 116,
4670-4676.
96.Kuck, D., Singh, N., Lyko, F., and Medina-Franco,
J. L. (2010) Novel and selective DNA methyltransferase inhibitors:
docking-based virtual screening and experimental evaluation, Bioorg.
Med. Chem., 18, 822-829.
97.Mirza, S., Sharma, G., Parshad, R., Gupta, S. D.,
Pandya, P., and Ralhan, R. (2013) Expression of DNA methyltransferases
in breast cancer patients and to analyze the effect of natural
compounds on DNA methyltransferases and associated proteins, J.
Breast Cancer, 16, 23-31.
98.Yu, J., Peng, Y., Wu, L. C., Xie, Z., Deng, Y.,
Hughes, T., He, S., Mo, X., Chiu, M., Wang, Q. E., He, X., Liu, S.,
Grever, M. R., Chan, K. K., and Liu, Z. (2013) Curcumin down-regulates
DNA methyltransferase 1 and plays an anti-leukemic role in acute
myeloid leukemia, PLoS One, 8, e55934.
99.Jha, A. K., Nikbakht, M., Parashar, G.,
Shrivastava, A., Capalash, N., and Kaur, J. (2010) Reversal of
hypermethylation and reactivation of the RARbeta2 gene by natural
compounds in cervical cancer cell lines, Folia Biol. (Praha),
56, 195-200.
100.Liu, Y. L., Yang, H. P., Gong, L., Tang, C. L.,
and Wang, H. J. (2011) Hypomethylation effects of curcumin,
demethoxycurcumin and bis-demethoxycurcumin on WIF-1 promoter in
non-small-cell lung cancer cell lines, Mol. Med. Report,
4, 675-679.
101.Khor, T. O., Huang, Y., Wu, T. Y., Shu, L.,
Lee, J., and Kong, A. N. (2011) Pharmacodynamics of curcumin as DNA
hypomethylation agent in restoring the expression of Nrf2 via promoter
CpGs demethylation, Biochem. Pharmacol., 82,
1073-1078.
102.Shu, L., Khor, T. O., Lee, J. H., Boyanapalli,
S. S., Huang, Y., Wu, T. Y., Saw, C. L., Cheung, K. L., and Kong, A. N.
(2011) Epigenetic CpG demethylation of the promoter and reactivation of
the expression of Neurog1 by curcumin in prostate LNCaP cells, AAPS
J., 13, 606-614.
103.Link, A., Balaguer, F., Shen, Y., Lozano, J.
J., Leung, H. C., Boland, C. R., and Goel, A. (2013) Curcumin modulates
DNA methylation in colorectal cancer cells, PLoS One, 8,
e57709.
104.Gerhauser, C. (2013) Cancer chemoprevention and
nutriepigenetics: state of the art and future challenges, Top. Curr.
Chem., 329, 73-132.
105.Hua, W. F., Fu, Y. S., Liao, Y. J., Xia, W. J.,
Chen, Y. C., Zeng, Y. X., Kung, H. F., and Xie, D. (2010) Curcumin
induces down-regulation of EZH2 expression through the MAPK pathway in
MDAMB-435 human breast cancer cells, Eur. J. Pharmacol.,
637, 16-21.
106.Valinluck, V., and Sowers, L. C. (2007)
Inflammation-mediated cytosine damage: a mechanistic link between
inflammation and the epigenetic alterations in human cancers, Cancer
Res., 67, 5583-5586.
107.Parasramka, M. A., Ho, E., Williams, D. E., and
Dashwood, R. H. (2012) MicroRNAs, diet, and cancer: new mechanistic
insights on the epigenetic actions of phytochemicals, Mol.
Carcinog., 51, 213-230.
108.Saini, S., Majid, S., and Dahiya, R. (2010)
Diet, microRNAs and prostate cancer, Pharm. Res., 27,
1014-1026.
109.Gupta, N. K., and Dixit, V. K. (2011)
Bioavailability enhancement of curcumin by complexation with
phosphatidylcholine, J. Pharm. Sci., 100, 1987-1995.
110.Valls, J., Millan, S., Marti, M. P., Borras,
E., and Arola, L. (2009) Advanced separation methods of food
anthocyanins, isoflavones and flavanols, J. Chromatogr. A,
1216, 7143-7172.
111.Barnes, S. (1995) Effect of genistein on in
vitro and in vivo models of cancer, J. Nutr., 125
(Suppl. 3), 777S-783S.
112.Basak, S., Pookot, D., Noonan, E. J., and
Dahiya, R. (2008) Genistein downregulates androgen receptor by
modulating HDAC6-Hsp90 chaperone function, Mol. Cancer Ther.,
7, 3195-3202.
113.Vardi, A., Bosviel, R., Rabiau, N., Adjakly,
M., Satih, S., Dechelotte, P., Boiteux, J. P., Fontana, L., Bignon, Y.
J., Guy, L., and Bernard-Gallon, D. J. (2010) Soy phytoestrogens modify
DNA methylation of GSTP1, RASSF1A, EPH2 and BRCA1 promoter in prostate
cancer cells, In vivo, 24, 393-400.
114.Lattrich, C., Lubig, J., Springwald, A.,
Goerse, R., Ortmann, O., and Treeck, O. (2011) Additive effects of
trastuzumab and genistein on human breast cancer cells, Anticancer
Drugs, 22, 253-261.
115.Li, W., Frame, L. T., Hirsch, S., and Cobos, E.
(2010) Genistein and hematological malignancies, Cancer Lett.,
296, 1-8.
116.Banerjee, S., Li, Y., Wang, Z., and Sarkar, F.
H. (2008) Multi-targeted therapy of cancer by genistein, Cancer
Lett., 269, 226-242.
117.Zhang, Z., Wang, C. Z., Du, G. J., Qi, L. W.,
Calway, T., He, T. C., Du, W., and Yuan, C. S. (2013) Genistein induces
G2/M cell cycle arrest and apoptosis via ATM/p53-dependent pathway in
human colon cancer cells, Int. J. Oncol., 43,
289-296.
118.Majid, S., Kikuno, N., Nelles, J., Noonan, E.,
Tanaka, Y., Kawamoto, K., Hirata, H., Li, L. C., Zhao, H., Okino, S.
T., Place, R. F., Pookot, D., and Dahiya, R. (2008) Genistein induces
the p21WAF1/CIP1 and p16INK4a tumor suppressor genes in prostate cancer
cells by epigenetic mechanisms involving active chromatin modification,
Cancer Res., 68, 2736-2744.
119.Li, Y., Chen, H., Hardy, T. M., and Tollefsbol,
T. O. (2013) Epigenetic regulation of multiple tumor-related genes
leads to suppression of breast tumorigenesis by dietary genistein,
PloS One, 8, e54369.
120.Dagdemir, A., Durif, J., Ngollo, M., Bignon, Y.
J., and Bernard-Gallon, D. (2013) Histone lysine trimethylation or
acetylation can be modulated by phytoestrogen, estrogen or anti-HDAC in
breast cancer cell lines, Epigenomics, 5, 51-63.
121.Fang, M., Chen, D., and Yang, C. S. (2007)
Dietary polyphenols may affect DNA methylation, J. Nutr., 137
(Suppl. 1), 223S-228S.
122.Majid, S., Dar, A. A., Ahmad, A. E., Hirata,
H., Kawakami, K., Shahryari, V., Saini, S., Tanaka, Y., Dahiya, A. V.,
Khatri, G., and Dahiya, R. (2009) BTG3 tumor suppressor gene promoter
demethylation, histone modification and cell cycle arrest by genistein
in renal cancer, Carcinogenesis, 30, 662-670.
123.Kikuno, N., Shiina, H., Urakami, S., Kawamoto,
K., Hirata, H., Tanaka, Y., Majid, S., Igawa, M., and Dahiya, R. (2008)
Genistein mediated histone acetylation and demethylation activates
tumor suppressor genes in prostate cancer cells, Int. J. Cancer,
123, 552-560.
124.Li, Y., Vandenboom, T. G., Kong, D., Wang, Z.,
Ali, S., Philip, P. A., and Sarkar, F. H. (2009) Upregulation of
miR-200 and let-7 by natural agents leads to the reversal of
epithelial-to-mesenchymal transition in gemcitabine-resistant
pancreatic cancer cells, Cancer Res., 69, 6704-6712.
125.Parker, L. P., Taylor, D. D., Kesterson, J.,
Metzinger, D. S., and Gercel-Taylor, C. (2009) Modulation of microRNA
associated with ovarian cancer cells by genistein, Eur. J. Gynecol.
Oncol., 30, 616-621.
126.Kim, B. R., Jeon, Y. K., and Nam, M. J. (2011)
A mechanism of apigenin-induced apoptosis is potentially related to
anti-angiogenesis and anti-migration in human hepatocellular carcinoma
cells, Food Chem. Toxicol., 47, 1626-1632.
127.Pandey, M., Kaur, P., Shukla, S., Abbas, A.,
Fu, P., and Gupta, S. (2012) Plant flavone apigenin inhibits HDAC and
remodels chromatin to induce growth arrest and apoptosis in human
prostate cancer cells: in vitro and in vivo study,
Mol. Carcinog., 51, 952-962.
128.Calderon-Montano, J. M., Burgos-Moron, E.,
Perez-Guerrero, C., and Lopez-Lazaro, M. (2011) A review on the dietary
flavonoid kaempferol, Mini Rev. Med. Chem., 11,
298-344.
129.Berger, A., Venturelli, S., Kallnischkies, M.,
Bocker, A., Busch, C., Weiland, T., Noor, S., Leischner, C., Weiss, T.
S., Lauer, U. M., Bischoff, S. C., and Bitzer, M. (2013) Kaempferol, a
new nutrition-derived pan-inhibitor of human histone deacetylases,
J. Nutr. Biochem., 24, 977-985.
130.Marfe, G., Tafani, M., Indelicato, M.,
Sinibaldi-Salimei, P., Reali, V., Pucci, B., Fini, M., and Russo, M. A.
(2009) Kaempferol induces apoptosis in two different cell lines via Akt
inactivation, Bax and SIRT3 activation, and mitochondrial dysfunction,
J. Cell Biochem., 106, 643-650.
131.Lin, Y., Shi, R., Wang, X., and Shen,
H. M. (2008) Luteolin, a flavonoid with potential for cancer
prevention and therapy, Curr. Cancer Drug Targets, 8,
634-646.
132.Attoub, S., Hassan, A. H., Vanhoecke, B.,
Iratni, R., Takahashi, T., Gaben, A. M., Bracke, M., Awad, S., John,
A., Kamalboor, H. A., Al Sultan, M. A., Arafat, K., Gespach, C., and
Petroianu, G. (2011) Inhibition of cell survival, invasion, tumor
growth and histone deacetylase activity by the dietary flavonoid
luteolin in human epithelioid cancer cells, Eur. J. Pharmacol.,
651, 18-25.
133.Markaverich, B. M., and Vijjeswarapu, M. (2012)
Multiple sites of type II site ligand (luteolin and BMHPC) regulation
of gene expression in PC-3 cells, Int. J. Biomed. Sci.,
8, 219-232.
134.Da Silva Brandao, E. H., Oliveira, L. D.,
Landucci, L. F., Yumi Koga-Ito, C., and Jorge, A. O. C. (2007)
Antimicrobial activity of coffee-based solutions and their effects on
streptococcus mutants adherence, Braz. J. Oral. Sci., 6,
1274-1277.
135.Yashin, A., Yashin, Y., Wang, J. Y., and
Nemzer, B. (2013) Antioxidant and antiradical activity of coffee,
Antioxidants, 2, 230-245.
136.Lee, W. J., and Zhu, B. T. (2006) Inhibition of
DNA methylation by caffeic acid and chlorogenic acid, two common
catechol-containing coffee polyphenols, Carcinogenesis,
27, 269-277.
137.Van Breemen, R. B., and Pajkovic, N. (2008)
Multi-targeted therapy of cancer by lycopene, Cancer Lett.,
269, 339-351.
138.King-Batoon, A., Leszczynska, J. M., and Klein,
C. B. (2087) Modulation of gene methylation by genistein or lycopene in
breast cancer cells, Environ. Mol. Mutagen, 49,
36-45.
139.Okuda, T., Yoshida, T., and Hatano, T. (1989)
Ellagitannins as active constituents of medicinal plants, Planta.
Med., 55, 117-122.
140.Wen, X. Y., Wu, S. Y., Li, Z. Q., Zhang, J. J.,
Wang, G. F., Jiang, Z. H., and Wu, S. G. (2009) Ellagitannin (BJA3121),
an antiproliferative natural polyphenol compound, can regulate the
expression of miRNAs in HepG2 cancer cells, Phytother. Res.,
23, 778-784.
141.Pappa, G., Lichtenberg, M., Iori, R.,
Barillari, J., Bartsch, H., and Gerhauser, C. (2006) Comparison of
growth inhibition profiles and mechanisms of apoptosis induction in
human colon cancer cell lines by isothiocyanates and indoles from
Brassicaceae, Mutat. Res., 599, 76-87.
142.Izzotti, A., Calin, G. A., Arrigo, P., Steele,
V. E., Croce, C. M., and De Flora, S. (2009) Downregulation of microRNA
expression in the lungs of rats exposed to cigarette smoke, FASEB
J., 23, 806-812.
143.Lamprecht, S. A., and Lipkin, M. (2003)
Chemoprevention of colon cancer by calcium, vitamin D and folate:
molecular mechanisms, Nat. Rev. Cancer, 3, 601-614.
144.Kim, Y. I., Baik, H. W., Fawaz, K., Knox, T.,
Lee, Y. M., Norton, R., Libby, E., and Mason, J. B. (2001) Effects of
folate supplementation on two provisional molecular markers of colon
cancer: a prospective, randomized trial, Am. J. Gastroenterol.,
96, 184-195.
145.Ghoshal, K., Li, X., Datta, J., Bai, S.,
Pogribny, I., Pogribny, M., Huang, Y., Young, D., and Jacob, S. T.
(2006) A folate- and methyl-deficient diet alters the expression of DNA
methyltransferases and methyl CpG binding proteins involved in
epigenetic gene silencing in livers ofF344 rats, J. Nutr.,
136, 1522-1527.
146.Chagas, C. E., Bassoli, B. K., de Souza, C. A.,
Deminice, R., Junior, A. A., Paiva, S. A., Dagli, M. L., Ong, T. P.,
and Moreno, F. S. (2011) Folic acid supplementation during early
hepatocarcinogenesis: cellular and molecular effects, Int. J.
Cancer, 129, 2073-2082.
147.Marsit, C. J., Eddy, K., and Kelsey, K. T.
(2006) MicroRNA responses to cellular stress, Cancer Res.,
66, 10843-10848.
148.Combs, G. F., Jr., and Gray, W. P. (1998)
Chemopreventive agents: selenium, Pharmacol. Ther., 79,
179-192.
149.Davis, C. D., Uthus, E. O., and Finley, J. W.
(2000) Dietary selenium and arsenic affect DNA methylation in vitro
in Caco-2 cells and in vivo in rat liver and colon, J.
Nutr., 130, 2903-2909.
150.Xiang, N., Zhao, R., Song, G., and Zhong, W.
(2008) Selenite reactivates silenced genes by modifying DNA methylation
and histones in prostate cancer cells, Carcinogenesis,
29, 2175-2181.
151.Brtko, J. (2007) Retinoids, rexinoids and their
cognate nuclear receptors: character and their role in chemoprevention
of selected malignant diseases, Biomed. Pap. Med. Fac. Univ.
Palacky. Olomouc. Czech. Repub., 151, 187-194.
152.Arany, I., Whitehead, W. E., Ember, I. A., and
Tyring, S. K. (2003) Dose-dependent
activation of p21waf1 transcription by all-trans-acid in cervical squamous carcinoma cells,
Anticancer Res., 23,
495-497.
153.Wu, Q., Zhang, M., Liu, S., Chen, Y., and
Su, W. (2002) Retinoic acid
receptor beta is required for anti-activator protein-1 activity by
retinoic acid in gastric cancer cells, Chin. Med. J., 115,
810-814.
154.Stefanska, B., Rudnicka, K., Bednarek, A., and
Fabianowska-Majewska, K. (2010) Hypomethylation and induction of retinoic acid
receptor beta 2 by concurrent action of adenosine analogues and natural
compounds in breast cancer cells, Eur. J. Pharmacol., 638,
47-53.
155.Das, S., Foley, N., Bryan, K., Watters, K. M.,
Bray, I., Murphy, D. M., Buckley, P. G., and Stallings, R. L.
(2010) MicroRNA mediates DNA
demethylation events triggered by retinoic acid during neuroblastoma
cell differentiation, Cancer
Res., 70,
7874-7881.
156.Garzon, R., Pichiorri, F., Palumbo, T.,
Visentini, M., Aqeilan, R., Cimmino, A., Wang, H., Sun, H., Volimia,
S., Alder, H., Calin, G. A., Liu, C. G., Andreeff, M., and Croce, C. M.
(2007) MicroRNA gene expression
during retinoic acid-induced differentiation of human acute
promyelocytic leukemia, Oncogene, 26,
4148-4157.
157.Alrawaiq, N. S., and Abdullah, A. (2014) An
evaluation of sulforaphane as a potential agent for disease prevention,
Res. J. Pharm. Biol. Chem. Sci., 5, 1335-1349.
158.Myzak, M. C., Hardin, K., Wang, R., Dashwood,
R. H., and Ho, E. (2006) Sulforaphane inhibits histone deacetylase
activity in BPH-1, LnCaP and PC-3 prostate epithelial cells,
Carcinogenesis, 27, 811-819.
159.Meeran, S. M., Patel, S. N., and Tollefsbol, T.
O. (2010) Sulforaphane causes epigenetic repression of hTERT expression
in human breast cancer cell lines, PLoS One, 5,
e11457.
160.Xiao, D., Lew, K. L., Zeng, Y., Xiao, H.,
Marynowski, S. W., Dhir, R., and Singh, S. V. (2006) Phenethyl
isothiocyanate-induced apoptosis in PC-3 human prostate cancer cells is
mediated by reactive oxygen species-dependent disruption of the
mitochondrial membrane potential, Carcinogenesis, 27,
2223-2234.
161.Wang, L. G., Liu, X. M., Fang, Y., Dai, W.,
Chiao, F. B., Puccio, G. M., Feng, J., Liu, D., and Chiao, J. M. (2008)
De-repression of the p21 promoter in prostate cancer cells by an
isothiocyanate via inhibition of HDACs and c-Myc, Int. J.
Oncol., 33, 375-380.
162.Izzotti, A., Calin, G. A., Steele, V. E.,
Cartiglia, C., Longobardi, M., Croce, C. M., and De Flora, S. (2010)
Chemoprevention of cigarette smoke-induced alterations of microRNA
expression in rat lungs, Cancer Prev. Res. (Phila Pa), 3,
62-72.
163.Gao, C., Jiang, X., Wang, H., Zhao, Z., and
Wang, W. (2013) Drug metabolism and pharmacokinetics of organosulfur
compounds from garlic, J. Drug Metab. Toxicol., 4, 5; http://dx.doi.org/10.4172/2157-7609.1000159.
164.Nian, H., Delage, B., Pinto, J. T., and
Dashwood, R. H. (2008) Allyl mercaptan, a garlic-derived organosulfur
compound, inhibits histone deacetylase and enhances Sp3 binding on the
P21WAF1 promoter, Carcinogenesis, 29, 1816-1824.
165.Druesne-Pecollo, N., Chaumontet, C., and
Latino-Martel, P. (2008) Diallyl disulfide increases histone
acetylation in colon cells in vitro and in vivo, Nutr.
Rev., 66 (Suppl. 1), S39-41.