Coxsackievirus B3 Induces Autophagic Response in Cardiac Myocytes in vivo
Xia Zhai1#, Bing Bai2#, Bohai Yu1, Tanying Wang1, Huapeng Wang3, Yao Wang3, Huiyan Li2, Lei Tong1, Yan Wang1, Fengmin Zhang1, Wenran Zhao3*, and Zhaohua Zhong1*
1Department of Microbiology, Harbin Medical University, 194 Xuefu Road, Harbin 150086, China; E-mail: zhonghzh@hrbmu.edu.cn2Department of Cardiology, The First Affiliated Hospital of Harbin Medical University, 23 Youzheng Street, Harbin 150001, China
3Department of Cell Biology, Harbin Medical University, 194 Xuefu Road, Harbin 150086, China; E-mail: zhaowr@ems.hrbmu.edu.cn; zhaowenran2002@aliyun.com
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received November 20, 2014; Revision received February 9, 2015
Viral myocarditis is a common disease that contributes to dilated cardiomyopathy or heart failure. Coxsackievirus B (CVB) is one of the major causative pathogens of viral myocarditis. Previous studies have shown that autophagy is exploited to promote CVB replication in cell lines. To study whether cardiac myocytes respond to CVB infection in a similar way, viral myocarditis was established by the inoculation of 3-week-old BALB/c mice with CVB3. Electron microscopic observation showed that autophagosome-like vesicles were induced in the cardiac myocytes of mice infected by CVB3 at 3, 5, and 7 days after viral infection. The lipidated microtubule-associated protein 1 light chain 3 (LC3), LC3-II, was also significantly increased in both myocardium and the cardiac myocytes extracted from the ventricles of mice infected with CVB3. The increased LC3-II coincided with high level of viral RNA and proteins in both myocardium and isolated cardiac myocytes. Moreover, viral protein synthesis was significantly decreased in primary cardiac myocytes by the treatment with 3-methyladenine, an inhibitor of autophagy. The expression and the phosphorylation of extracellular signal regulated kinase (ERK) were also increased in both myocardium and in the isolated cardiac myocytes of the virus-infected mice, while the interplay of ERK with autophagic response remains to be studied. This study demonstrated that cardiac myocytes respond to CVB3 infection by increased formation of autophagosomes in vivo, which might be exploited for viral replication.
KEY WORDS: coxsackievirus B, cardiac myocytes, myocardium, autophagy, autophagosomeDOI: 10.1134/S0006297915080052
Abbreviations: Atg, autophagy protein; CVB3, coxsackievirus B3; ERK, extracellular signal-regulated kinase; LC3, microtubule-associated protein 1 light chain 3; 3-MA, 3-methyladenine; pfu, plaque-forming unit; RT-qPCR, quantitative reverse transcription polymerase chain reaction.
Viral myocarditis is a common illness worldwide that can lead to severe
consequence or death in infants and young adults [1-3]. Enteroviruses, especially
coxsackievirus B (CVB), are the most common pathogens that contribute
to viral myocarditis [4, 5].
CVB is a group of non-enveloped, single-stranded RNA viruses of the
Enterovirus genus, Picornaviridae family. Although most
CVB-related cardiac illnesses are subclinical, severe viral myocarditis
can lead to heart failure or sudden cardiac death [6-8]. About 10% of symptomatic
patients eventually develop to dilated cardiomyopathy [9-11]. Previous studies have
shown that both of the virus-induced damage to the heart and the host
immune response play a role in the pathogenesis of CVB-related
myocarditis, while the precise mechanism of viral myocarditis remains
to be resolved [12-14].
Studies in recent years have shown that autophagy is involved the
process of CVB infection [15-20].
Macroautophagy, hereafter referred as autophagy, is a significant cellular catabolic process in which long-lived proteins and damaged organelles are degraded in lysosomes [21]. The principal role of autophagy is to supply nutrient and energy to prolong the survival of the cell under stress conditions such as starvation, cellular damage, and pathogen invasion [22, 23]. Autophagic response begins by the growth of a small, flat membrane sac (known as isolation membrane or phagophore) of unknown origin. This membrane sac elongates, curves, and finally fuses its ends to form a double-membrane vesicle termed autophagosome. The autophagosome then fuses with lysosome to form autophagolysosome, in which the sequestered materials are degraded by lysosomal enzymes [24, 25]. Increasing evidence has shown that autophagy plays a crucial role in viral infection. Some viruses can evade autophagy, while others, such as CVB, may exploit autophagic response to promote viral replication [17, 18].
Studies have shown that autophagy is utilized to promote CVB replication. CVB3 and CVB4 induce the formation of autophagosomes in vitro, and inhibited autophagy decreases virion production [17-19]. It is believed that CVB3 inhibits the fusion of autophagosomes with lysosomes in order to provide sufficient membrane structures upon which viral RNA replication complex assemble [26]. The interplay between CVB and autophagy has also been demonstrated in vivo. Like the findings in HeLa cells, CVB3 induces the formation of autophagosomes and inhibits the fusion of autophagosomes with lysosomes, thus leading to the generation of megaphagosomes [18]. Specific disruption of autophagy in pancreatic acinar cells markedly reduced CVB replication, especially at the early stage of viral infection. Disrupted autophagy also improved pancreatic function [17]. These data seem to suggest that autophagy might be a potential therapeutic target for CVB infection, since autophagy is utilized to promote both viral replication and cellular damage. However, it remains unexplored whether autophagy plays a similar role in cardiac myocytes, the primary target of CVB.
In cardiac myocytes, autophagy is essential to maintain cellular structure and function [27]. Studies have shown that autophagy protein 5 (Atg5)-deficient mice developed significant cardiac dysfunction and ventricle dilation, while enhanced autophagy by the overexpression of Atg7 significantly improved cardiac function and survival of autophagy-deficient mice [28]. In contrast, basal autophagy might be dispensable in pancreatic acinar cells, since deficiency of Atg5 in these cells seems have little impact on pancreatic morphology and function [17]. These studies suggest that the biological importance of autophagy can vary in different types of cells. In addition, the activation of autophagy has been detected in diverse forms of cardiac conditions such as ischemia, ischemia–reperfusion, and genetic cardiomyopathy [23-27, 29].
Given the importance of autophagy in cardiac myocytes, the present study examined whether CVB3 could induce autophagic response in cardiac myocytes by using a mouse model of viral myocarditis. We also studied the role of autophagy in viral replication in cultured primary cardiac myocytes treated with 3-methyladenine (3-MA), an autophagy inhibitor. Overall, our data provide direct evidence that CVB3 infection induces autophagic response in cardiac myocytes in vivo, which might be exploited to facilitate viral replication.
MATERIALS AND METHODS
Antibodies and chemical reagents. Polyclonal antibody against enterovirus VP1 (M 7064; Dako, USA), antibody against LC3-I/II (L7543; Sigma, USA), anti-ERK1/2 and anti-phospho-ERK1/2 antibodies (9102L and 9101; Cell Signaling, USA), and antibody against β-actin (TA09; Zhongshan Golden Bridge, China) were used at dilution of 1 : 1000. Goat anti-mouse horseradish peroxidase-conjugated secondary antibody (2B-2305; Zhongshan Golden Bridge) was used at dilution of 1 : 5000. 3-MA was purchased from Sigma-Aldrich (USA).
Cell culture and virus propagation. HeLa cells were cultured in RPMI 1640 medium (Life Technologies, USA) supplemented with 5% fetal bovine serum (FBS) (Biological Industries, Israel) and penicillin-streptomycin. CVB3 Nancy strain was obtained from the Center for Endemic Disease Control of China. The virus was propagated in HeLa cells in RPMI 1640 medium supplemented with 5% FBS. Virus titer was determined by plaque assay as described previously [30].
Infection of mice. BALB/c mice were purchased from the Laboratory Animal Center of Harbin Medical University. The animal experiment was carried out in accordance with the Regulation for the Use of Laboratory Animals of Harbin Medical University. The experimental protocol used in this study was approved by the committee for the use of experimental animals of Harbin Medical University. Male BALB/c mice at the age of three weeks were inoculated intraperitoneally with 1.2·108 pfu of CVB3 in 0.3 ml of saline. The mice were sacrificed by cervical dislocation at day 0, 3, 5, and 7 post-infection (p.i.). The heart was fixed by retrograde perfusion through the thoracic aorta for 10 min with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4), at room temperature. Immediately after the perfusion, the heart was removed and the ventricle was isolated. The ventricle was subjected to histopathological observation, preparation of ultra-thin sections for electron microscopy, and extraction of RNA and proteins.
Primary cardiomyocyte isolation and infection. Primary cultures of ventricular cardiac myocytes from BALB/c mice were prepared as described previously [31, 32]. Purity of the preparation was monitored visually by phase-contrast microscopy. Cardiac myocytes isolated from the mice infected with CVB3 were subjected to extraction of protein and Western blot analysis. Isolated cardiac myocytes (105/ml) were cultured for 2 h. Cells were then treated with 40 µM 3-MA for 1 h and infected with CVB3 at 104 pfu/ml for 8 h. Proteins were extracted and subjected to Western blot analysis.
Quantitative reverse transcription polymerase chain reaction (RT-qPCR). CVB3 RNA was determined by RT-qPCR with a LightCycler 2.0 (Roche, Switzerland). SybrGreen master mix (TaKaRa, China) was used according to the manufacturer’s instructions. Briefly, total RNA was extracted from the cardiomyocytes cultured in 6-well plates by 500 µl TRIzol reagent (Invitrogen, USA) and resolved in 20 µl ddH2O. RNA (1 µl) and the antisense primer were used for cDNA synthesis using the PrimeScript RT reagent (TaKaRa, Japan). To perform real-time PCR, each 20 µl of reaction mixture contain 1× SybrGreen master mix, l µl of cDNA product, and 600 nM of forward and reverse primers. GAPDH mRNA was used as loading control. Each reaction was performed in triplicates. The cycle threshold (Ct) was used to measure the level of RNA by the 2–ΔΔCt method [33]. The primer sequences for CVB3 RNA are 5′-GCACACACCCTCAAACCAGA-3′ (sense) and 5′-ATGAAACACGGACACCCAAAG-3′ (antisense). The primers for GAPDH are 5′-AGGGCATCTTGGGCTACAC-3′ (sense) and 5′-CATACCAGGAAATGAGCTTGA-3′ (antisense).
Western blot. Mouse ventricles or primary cardiac myocytes were washed with PBS and lysed with RIPA buffer (Thermo, USA) containing protease inhibitor cocktail and 1% phenylmethylsulfonyl fluoride (PMSF; Beyotime, China) on ice for 15 min. Cell lysates were harvested and centrifuged at 12,000g for 10 min at 4°C. The protein concentration in the supernatant was determined by the Bradford assay with Protein Assay Kit (Bio-Rad, USA). Proteins were separated by 15% SDS-PAGE and then transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, USA). The membrane was blocked for 1 h with 5% skim milk dissolved in 1× TBS containing 0.3% Tween 20. The membrane was probed overnight with primary antibodies at 4°C. After three washes with TBS containing 0.3% Tween 20 and 5% skim milk, the membrane was incubated with anti-mouse horseradish peroxidase-conjugated secondary antibody (Dako) for 2 h at room temperature. Proteins in the cellular or tissue lysates were visualized with the enhanced chemiluminescence technique using SuperSignal West Pico chemiluminescent substrate (Thermo, USA). Actin was used as loading control.
Electron microscopy. Ventricles from CVB3-infected mice were fixed in 2% glutaraldehyde for 24 h and post-fixed in 1% osmium tetroxide (Electron Microscopy Sciences, USA). The tissues were then subjected to preparation of ultrathin sections. Ultrathin sections were prepared using an ultramicrotome (Reichert-Jung, Germany) and stained with saturated aqueous uranyl acetate and lead citrate (both from Electron Microscopy Sciences) at room temperature. Sections were examined with a transmission electron microscope (H7650; Hitachi, Japan).
Statistical analysis. Data are presented as the mean ± standard deviation. Differences between the test and control groups were analyzed by Student’s t-test using Prism software. Significance was set at p < 0.05 (*) and p < 0.01 (**).
RESULTS AND DISCUSSION
Autophagosome-like vesicles are induced in myocardium infected by CVB3. To determine the autophagic response in cardiac myocytes, we evaluated the two criteria that represent the activation of autophagy. First, we determined the morphological alteration of the CVB3-infected myocardium by analyzing the double-membrane autophagosomes, the prominent character of autophagy. Second, we analyzed the lipidated form of the microtubule-associated protein 1 light chain 3 (LC3) in the myocardium and in the isolated cardiac myocytes from CVB3-infected mice.
We began this study by examining the formation of autophagosomes in myocardium after viral infection. To establish the mouse model of viral myocarditis, BALB/c mice at the age of three weeks were inoculated intraperitoneally with 4·108 pfu/ml of CVB3 in 0.3 ml of saline. The ventricles of the mice were collected at 0, 3, 5, and 7 days p.i. and subjected to histological observation. Focal myocyte necrosis with inflammatory infiltrate was observed at 3, 5, and 7 days p.i. (Fig. 1A). Since myocarditis is primarily characterized by inflammation of myocardium [11, 34, 35], we considered that CVB3 infection led to acute myocarditis in the mice.
Fig. 1. Autophagosome-like vesicles induced by CVB3 infection in cardiac myocytes of mice. BALB/c mice at the age of 3 weeks were infected with CVB3 (4·108 pfu/ml, 0.3 ml per mouse). The myocardial tissues were collected at 0, 3, 5, and 7 days p.i. and subjected to histopathological and electron microscopic observation. A) Light microscopy after HE staining shows that the infiltration of inflammatory cells in the myocardial tissues appeared at days 3, 5, and 7 p.i. Myocardial infarction was dispersed among cardiac muscle at the time points of 3, 5, and 7 days p.i. Severe damage in cardiac muscle occurred at day 7 p.i. Representative histological micrographs are presented (n = 5). Amplification: 100× (upper row); 200× (lower row). B) Electron microscopy shows that severely damaged myofibrils and disorganized mitochondria appeared in the cytoplasm of cardiac myocytes infected with CVB3. Small double-membrane-enclosed vesicles (arrowheads) in size ranging from 0.2 to 0.5 µm diameter were dispersed in the proximity of mitochondria (M) (c, d). a, b) The micrographs taken from the control mice. Amplification: 8000× (a, c); 15,000× (b, d). Representative electron micrographs obtained at day 3 p.i. are presented.
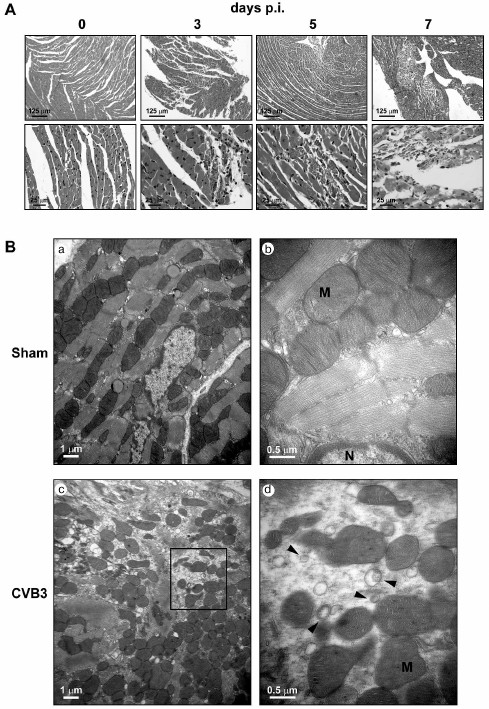
Ventricles collected from the mice at 0, 3, and 5 days p.i. were subjected to the preparation of ultrathin sections. Transmission electron microscopy (TEM) observation showed that myofibrils were well organized with orderly distributed mitochondria in between in the cardiac myocytes of the control mice (Fig. 1B). At day 3 p.i., organized myofibrils disappeared and irregularly shaped mitochondria were crowded in the cytoplasm of the necrotic cells (Fig. 1B). Clusters of autophagosome-like vesicles with double membranes 0.20-0.35 µm in size were observed (Fig. 1B). These double-membrane-enclosed vesicles were very likely autophagosomes since they contained cytosolic contents, small ribosome-like dots. Although there was no intact cardiac myocytes in the CVB3-infected region seen by TEM, it was unlikely that these autophagosome-like vesicles were from non-myocytes, since the myocyte is characterized by containing abundant mitochondria in the cytoplasm [36]. Unlike the effect of CVB3 on HeLa cells, the number of the double-membrane vesicles was much fewer than that in HeLa cells. In addition, the size of the autophagosome-like vesicles formed in the myocardium was smaller than that in pancreatic acinar cells after CVB3 infection, in which megaphagosomes were found [17, 18]. Nevertheless, our observation at least demonstrated that autophagosome-like vesicles were indeed induced in the myocardium during CVB3 infection.
LC3-II is markedly increased in myocardium infected by CVB3. The assembly of autophagosomes involves the conversion of cytosolic LC3-I into lipidated form LC3-II, which is recruited to the membrane of autophagosomes [37]. Thus, the processing of LC3-I to LC3-II represents the appearance of autophagosomes or autophagic response. To determine the autophagic response in the myocardium, BALB/c mice at the age of three weeks were inoculated with CVB3. Myocardial tissues were collected at days 0, 3, 5, and 7 p.i. Viral RNA was determined by RT-qPCR. LC3-I/II and VP1 were determined by Western blotting (Fig. 2).
As shown in Fig. 2a, both LC3-I and LC3-II were undetectable in the myocardium of uninfected tissues, while the levels of both proteins were increased at days 3, 5, and 7 p.i., suggesting the change of in vivo gene expression in response to CVB3 infection. The ratio of LC3-II to LC3-I was dramatically increased at day 3 p.i. (Fig. 2b), corresponding to the accumulation of autophagosome-like vesicles (Fig. 1B, c and d). The increased level of LC3-II coincided with markedly increased synthesis of viral RNA (Fig. 2d) and proteins (Fig. 2c). The ratio of LC3-II to LC3-I then declined at days 5 and 7 p.i., when viral RNA and proteins also decreased. These data can suggest that both the virus production and the synthesis of cellular proteins are affected by the necrosis of the cardiac myocytes and their clearance by phagocytes. This implication is supported by our previous study in which the levels of viral RNA and proteins were gradually decreased in the myocardium of suckling mice infected with CVB3 [38].
Fig. 2. Increased LC3-II level in the tissue of myocardium infected with CVB3. BALB/c mice were infected with CVB3. Ventricles of the mice were collected at different time intervals p.i. and subjected to Western blot analysis (a) and RT-qPCR (d). b, c) Densitometric measurements of LC3 and VP1 in the Western blot normalized to β-actin. Experiments were repeated three times. ERK1/2 and p-ERK1/2 are dephospho- and phospho-forms of ERK protein kinase of types 1 and 2.
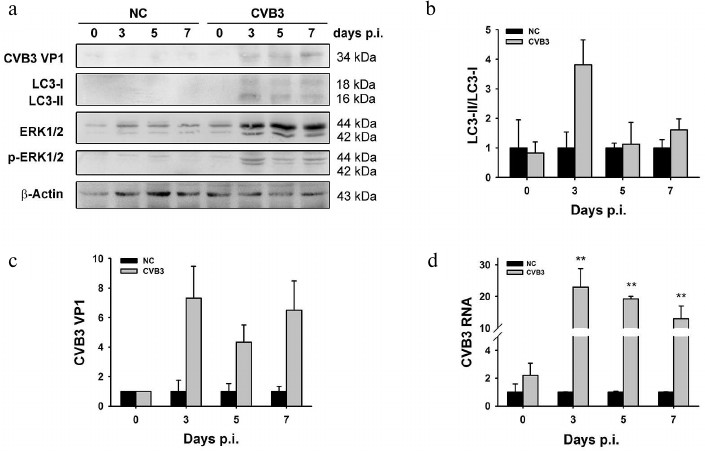
The autophagic response also coincided with the activation of extracellular signal regulated kinase (ERK) (Figs. 2a and 3b), while the association between autophagic response and ERK was not studied here. Although the intensity of autophagic response may vary in different context, our study indicates that the increased LC3-II is associated with CVB3 replication in myocardium.
In vivo study has shown that p62, the adaptor protein that is degraded together with autophagosomes, was accumulated in CVB3-infected pancreatic acinar cells, indicating that CVB3 replication blocks autophagic flux [39, 40]. In contrast, a study by Wong et al. showed that the level of p62 remained unchanged in HeLa cells during CVB infection [20]. Similarly, change in p62 level was not found in the present study (data not shown). Although cellular p62 level is widely considered as one of the markers of autophagic flux [41], it remains to be clarified whether p62 is exclusively degraded by autophagosomes. A study by Bardag-Gorce et al. found that p62 was increased when the proteasome was inhibited, indicating that p62 may also be degraded by proteasomes [42]. Moreover, p62 has been found to play a critical role in the delivery of K63-polyubiquitinated proteins for degradation through the ubiquitin–proteasome system (UPS) [43]. Since CVB3 infection promotes the UPS [44, 45], the upregulated UPS might explain the unchanged p62 in the present study. Nonetheless, it remains to be clarified if the increased level of LC3-II was due to increased formation of autophagosomes or inhibited processing of the autophagosomes by lysosomal enzymes.
Previous studies have indicated that ERK activation is required for the replication of enteroviruses 71 (EV71) [46-48]. In addition, studies have shown that ERK signaling regulates the expression of autophagy and lysosomal genes or interacts with LC3 [49, 50]. Although the induction of autophagic response was accompanied by increased expression and the phosphorylation of ERK during CVB3 infection (Figs. 2a and 3b), here we cannot conclude that ERK activation is required for the autophagic response or that it is only a concomitant event.
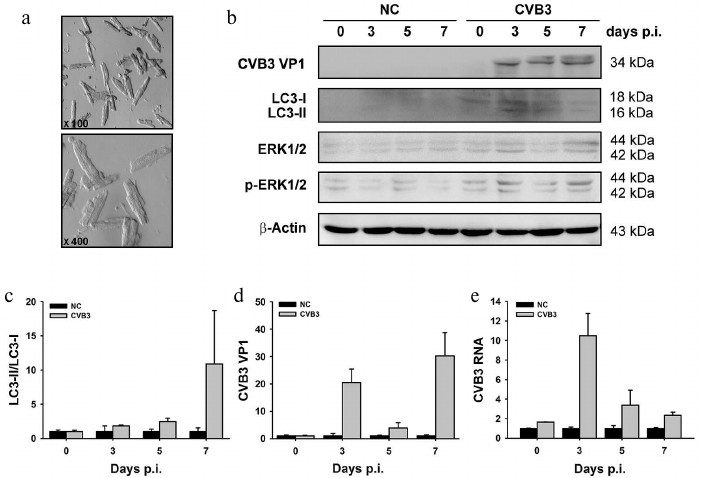
LC3-II is increased in cardiac myocytes isolated from CVB3-infected myocardium. Heart is composed of multiple cell types. The permanent cellular constituents of the heart include cardiac myocytes, fibroblasts, endothelial cells, and vascular smooth muscle cells [51, 52]. During stressful conditions such as inflammation, a population of lymphocytes, macrophages, and master cells are recruited to the heart [4, 53, 54]. Although morphological observation by electron microscopy showed the presence of the autophagosome-like vesicles (Fig. 1B), there is no direct evidence that demonstrates autophagic response occurred in the cardiac myocytes. Therefore, we isolated cardiac myocytes from the mouse ventricles after CVB infection and determined the change in LC3. As shown in Fig. 3c, significantly increased LC3-II was obtained at day 7 p.i. Like the change observed in the myocardium, the increased level of LC3-II coincided with increased synthesis of viral protein VP1 and the activation of ERK (p-ERK) (Fig. 3, b and d). Although the fold changes of LC3-II/LC3-I, viral RNA, and viral protein obtained from the isolated cardiac myocytes did not match exactly with that from the tissue of myocardium (compare with Fig. 2), these data at least demonstrate that autophagic response was indeed induced in the cardiac myocytes during CVB3 infection. Our results indicate that as the key target of CVB3, cardiac myocytes also respond to viral infection by increasing the accumulation of autophagosomes, which may provide the intracellular membrane required for viral RNA replication [55].
Fig. 3. Changes of LC3-II/LC3-I ratio (c) and levels of viral RNA (e) and protein VP1 (d) in cardiomyocytes isolated from myocardium of mice infected with CVB3. a) Electron photographs of isolated rod-shaped cardiac myocytes. LC3-II was increased in CVB3-infected cardiac myocytes. b, e) Viral RNA and proteins were extracted from the isolated cardiac myocytes and subjected to Western blot analysis (b) and RT-qPCR (e). c, d) Densitometric measurements of LC3 and VP1 in the Western blot normalized to β-actin. The experiments were repeated three times. NC, myocytes from control animals; CVB3, myocytes from animals infected with the virus.
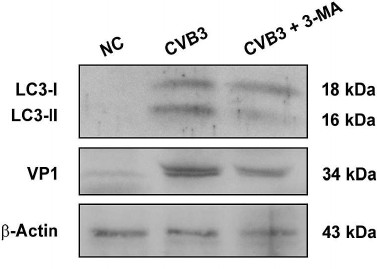
CVB3 replication is partially downregulated by an autophagy inhibitor in cardiac myocytes. It has been reported that autophagy stimulated CVB3 replication both in vitro and in vivo [17-20]. To show whether such interplay between autophagy and CVB3 replication also exists in cardiac myocytes, cardiac myocytes isolated from the ventricles of healthy mice were infected with CVB3 and then treated with 3-MA, an autophagy inhibitor. LC3-I, LC3-II, and VP1 were determined by Western blotting. As shown in Fig. 4, LC3-II was significantly increased after CVB3 infection, while it was markedly decreased by the treatment with 3-MA. Correspondingly, the synthesis of viral proteins was also significantly reduced in the cardiac myocytes by the inhibition of autophagy, indicating that autophagic response might be exploited by CVB3 to facilitate viral replication. Our results are consistent with a report that the disruption of autophagic gene Atg5 in pancreatic acinar cells dramatically reduced CVB3 replication and improved pancreatic function [17]. It was postulated that CVB3 replication is inhibited due to the lack of intracellular membrane. However, from the data presented here, we can only conclude that autophagy benefits CVB3 replication, while the precise mechanism remains to be studied further.
Fig. 4. Blocking the formation of autophagosomes inhibited CVB3 replication. Cardiac myocytes isolated from mice were infected with CVB3 with or without pretreatment with 3-MA. Proteins were extracted and subjected to Western blot analysis. The experiment was repeated three times.
A study has shown that EV71 also induces autophagy, and viral production decreased in the presence of 3-MA while it increased with treatment by autophagy inducer [56]. It seems that the induction of autophagy and the accumulation of autophagosomes might be a common mechanism used by enteroviruses to promote viral replication. Therefore, autophagy might be a potential target for anti-CVB treatment. While mice infected with EV71 showed alleviated symptom and decreased viral load by treatment with 3-MA [57], it remains to be elucidated if the autophagy inhibitor would have similar effect on CVB replication in vivo.
In summary, the present study demonstrated that cardiac myocytes respond to CVB3 infection by increased formation of autophagosomes in vivo, which might be exploited by viral replication. These findings indicate that targeting the autophagic pathway is a potential therapeutic strategy for CVB-induced myocarditis.
This work was supported by the grants of the National Natural Science Foundation of China to Zhaohua Zhong (Grant No. 81271825), Wenran Zhao (Grant No. 31270198), Tianying Wang (Grant No. 31300144) and Lei Tong (Grant No. 81101234). We thank Heilongjiang Provincial Key Laboratory of Pathogens and Immunity. We thank the technical support from Northern Translational Medicine Research Center of Harbin Medical University.
REFERENCES
1.Andreoletti, L., Leveque, N., Boulagnon, C.,
Brasselet, C., and Fornes, P. (2009) Viral causes of human myocarditis,
Arch. Cardiovasc. Dis., 102, 559-568.
2.Yajima, T. (2011) Viral myocarditis: potential
defense mechanisms within the cardiomyocyte against virus infection,
Future Microbiol., 6, 551-566.
3.Yajima, T., and Knowlton, K. U. (2009) Viral
myocarditis: from the perspective of the virus, Circulation,
119, 2615-2624.
4.Kawai, C. (1999) From myocarditis to
cardiomyopathy: mechanisms of inflammation and cell death: learning
from the past for the future, Circulation, 99,
1091-1100.
5.Bowles, N. E., Richardson, P. J., Olsen, E. G., and
Archard, L. C. (1986) Detection of coxsackie-B-virus-specific RNA
sequences in myocardial biopsy samples from patients with myocarditis
and dilated cardiomyopathy, Lancet, 1, 1120-1123.
6.Gaaloul, I., Riabi, S., Harrath, R., Evans, M.,
Salem, N. H., Mlayeh, S., Huber, S., and Aouni, M. (2012) Sudden
unexpected death related to enterovirus myocarditis: histopathology,
immunohistochemistry and molecular pathology diagnosis at post-mortem,
BMC Infect. Dis., 12, 212.
7.Caughey, R. W., Humphrey, J. M., and Thomas, P. E.
(2014) High-degree atrioventricular block in a child with acute
myocarditis, Ochsner J., 14, 244-247.
8.Steinke, K., Sachse, F., Ettischer, N.,
Strutz-Seebohm, N., Henrion, U., Rohrbeck, M., Klosowski, R., Wolters,
D., Brunner, S., Franz, W. M., Pott, L., Munoz, C., Kandolf, R.,
Schulze-Bahr, E., Lang, F., Klingel, K., and Seebohm, G. (2013)
Coxsackievirus B3 modulates cardiac ion channels, FASEB J.,
27, 4108-4121.
9.Spotnitz, M. D., and Lesch, M. (2006) Idiopathic
dilated cardiomyopathy as a late complication of healed viral
(coxsackie B virus) myocarditis: historical analysis, review of the
literature, and a postulated unifying hypothesis, Progr. Cardiovasc.
Dis., 49, 42-57.
10.Esfandiarei, M., and McManus, B. M. (2008)
Molecular biology and pathogenesis of viral myocarditis, Annu. Rev.
Pathol., 3, 127-155.
11.Doan, D., Rungta, S., Vikraman, N., and Rosman,
H. (2010) Fulminant coxsackie B myocarditis mimicking acute coronary
artery occlusion, Texas Heart Inst. J., 37, 500-501.
12.Kearney, M. T., Cotton, J. M., Richardson, P. J.,
and Shah, A. M. (2001) Viral myocarditis and dilated cardiomyopathy:
mechanisms, manifestations, and management, Postgrad. Med. J.,
77, 4-10.
13.Weithauser, A., Bobbert, P., Antoniak, S., Bohm,
A., Rauch, B. H., Klingel, K., Savvatis, K., Kroemer, H. K., Tschope,
C., Stroux, A., Zeichhardt, H., Poller, W., Mackman, N., Schultheiss,
H. P., and Rauch, U. (2013) Protease-activated receptor-2 regulates the
innate immune response to viral infection in a coxsackievirus
B3-induced myocarditis, J. Am. Coll. Cardiol., 62,
1737-1745.
14.Kong, Q., Wu, W., Yang, F., Liu, Y., Xue, Y.,
Gao, M., Lai, W., Pan, X., Yan, Y., Pang, Y., and Deng, Y. (2012)
Increased expressions of IL-22 and Th22 cells in the coxsackievirus
B3-induced mice acute viral myocarditis, Virol. J., 9,
232.
15.Robinson, S. M., Tsueng, G., Sin, J., Mangale,
V., Rahawi, S., McIntyre, L. L., Williams, W., Kha, N., Cruz, C.,
Hancock, B. M., Nguyen, D. P., Sayen, M. R., Hilton, B. J., Doran, K.
S., Segall, A. M., Wolkowicz, R., Cornell, C. T., Whitton, J. L.,
Gottlieb, R. A., and Feuer, R. (2014) Coxsackievirus B exits the host
cell in shed microvesicles displaying autophagosomal markers, PLoS
Pathog., 10, e1004045.
16.Li, M., Wang, X., Yu, Y., Yu, Y., Xie, Y., Zou,
Y., Ge, J., Peng, T., and Chen, R. (2014) Coxsackievirus B3-induced
calpain activation facilitates the progeny virus replication via a
likely mechanism related with both autophagy enhancement and apoptosis
inhibition in the early phase of infection: an in vitro study in
H9c2 cells, Virus Res., 179, 177-186.
17.Alirezaei, M., Flynn, C. T., Wood, M. R., and
Whitton, J. L. (2012) Pancreatic acinar cell-specific autophagy
disruption reduces coxsackievirus replication and pathogenesis in
vivo, Cell Host Microbe, 11, 298-305.
18.Kemball, C. C., Alirezaei, M., Flynn, C. T.,
Wood, M. R., Harkins, S., Kiosses, W. B., and Whitton, J. L. (2010)
Coxsackievirus infection induces autophagy-like vesicles and
megaphagosomes in pancreatic acinar cells in vivo, J.
Virol., 84, 12110-12124.
19.Yoon, S. Y., Ha, Y. E., Choi, J. E., Ahn, J.,
Lee, H., Kweon, H. S., Lee, J. Y., and Kim, D. H. (2008) Coxsackievirus
B4 uses autophagy for replication after calpain activation in rat
primary neurons, J. Virol., 82, 11976-11978.
20.Wong, J., Zhang, J., Si, X., Gao, G., Mao, I.,
McManus, B. M., and Luo, H. (2008) Autophagosome supports
coxsackievirus B3 replication in host cells, J. Virol.,
82, 9143-9153.
21.Kubli, D. A., and Gustafsson, A. B. (2014)
Cardiomyocyte health: adapting to metabolic changes through autophagy,
Trends Endocrinol. Metab., 25, 156-164.
22.Mei, Y., Thompson, M. D., Cohen, R. A., and Tong,
X. (2015) Autophagy and oxidative stress in cardiovascular diseases,
Biochim. Biophys. Acta, 1852, 243-251.
23.Jimenez, R. E., Kubli, D. A., and Gustafsson, A.
B. (2014) Autophagy and mitophagy in the myocardium: therapeutic
potential and concerns, Brit. J. Pharmacol., 171,
1907-1916.
24.White, E., Karp, C., Strohecker, A. M., Guo, Y.,
and Mathew, R. (2010) Role of autophagy in suppression of inflammation
and cancer, Curr. Opin. Cell Biol., 22, 212-217.
25.Bao, X. H., Naomoto, Y., Hao, H. F., Watanabe,
N., Sakurama, K., Noma, K., Motoki, T., Tomono, Y., Fukazawa, T.,
Shirakawa, Y., Yamatsuji, T., Matsuoka, J., and Takaoka, M. (2010)
Autophagy: can it become a potential therapeutic target? Int. J.
Mol. Med., 25, 493-503.
26.Luo, H., and McManus, B. M. (2012) Is autophagy
an avenue to modulate coxsackievirus replication and pathogenesis?
Future Microbiol., 7, 921-924.
27.Ghavami, S., Gupta, S., Ambrose, E., Hnatowich,
M., Freed, D. H., and Dixon, I. M. (2014) Autophagy and heart disease:
implications for cardiac ischemia-reperfusion damage, Curr. Mol.
Med., 14, 616-629.
28.Bhuiyan, M. S., Pattison, J. S., Osinska, H.,
James, J., Gulick, J., McLendon, P. M., Hill, J. A., Sadoshima, J., and
Robbins, J. (2013) Enhanced autophagy ameliorates cardiac
proteinopathy, J. Clin. Invest., 123, 5284-5297.
29.Kobayashi, S., and Liang, Q. (2015) Autophagy and
mitophagy in diabetic cardiomyopathy, Biochim. Biophys. Acta,
1852, 252-261.
30.Zhong, Z., Li, X., Zhao, W., Tong, L., Liu, J.,
Wu, S., Lin, L., Zhang, Z., Tian, Y., and Zhang, F. (2008) Mutations at
nucleotides 573 and 579 within 5′-untranslated region augment the
virulence of coxsackievirus B1, Virus Res., 135,
255-259.
31.Li, D., Wu, J., Bai, Y., Zhao, X., and Liu, L.
(2014) Isolation and culture of adult mouse cardiomyocytes for cell
signaling and in vitro cardiac hypertrophy, J. Vis. Exp.;
doi: 10.3791/51357, E-pub ahead of print.
32.Sreejit, P., Kumar, S., and Verma, R. S. (2008)
An improved protocol for primary culture of cardiomyocyte from neonatal
mice, In vitro Cell. Devel. Biol. Animal, 44, 45-50.
33.Slack, J. L., Bi, W., Livak, K. J., Beaubier, N.,
Yu, M., Clark, M., Kim, S. H., Gallagher, R. E., and Willman, C. L.
(2001) Preclinical validation of a novel, highly sensitive assay to
detect PML-RARalpha mRNA using real-time reverse-transcription
polymerase chain reaction, J. Mol. Diagn., 3,
141-149.
34.Leslie, K., Blay, R., Haisch, C., Lodge, A.,
Weller, A., and Huber, S. (1989) Clinical and experimental aspects of
viral myocarditis, Clin. Microbiol. Rev., 2, 191-203.
35.Reetoo, K. N., Osman, S. A., Illavia, S. J.,
Cameron-Wilson, C. L., Banatvala, J. E., and Muir, P. (2000)
Quantitative analysis of viral RNA kinetics in coxsackievirus
B3-induced murine myocarditis: biphasic pattern of clearance following
acute infection, with persistence of residual viral RNA throughout and
beyond the inflammatory phase of disease, J. Gen. Virol.,
81, 2755-2762.
36.Nakamura, H., Yamamoto, T., Yamamura, T., Nakao,
F., Umemoto, S., Shintaku, T., Yamaguchi, K., Liu, P., and Matsuzaki,
M. (1999) Repetitive coxsackievirus infection induces cardiac
dilatation in post-myocarditic mice, Japan. Circulation J.,
63, 794-802.
37.Sukseree, S., Rossiter, H., Mildner, M., Pammer,
J., Buchberger, M., Gruber, F., Watanapokasin, R., Tschachler, E., and
Eckhart, L. (2013) Targeted deletion of Atg5 reveals differential roles
of autophagy in keratin K5-expressing epithelia, Biochem. Biophys.
Res. Commun., 430, 689-694.
38.Tong, L., Lin, L., Wu, S., Guo, Z., Wang, T.,
Qin, Y., Wang, R., Zhong, X., Wu, X., Wang, Y., Luan, T., Wang, Q., Li,
Y., Chen, X., Zhang, F., Zhao, W., and Zhong, Z. (2013) MiR-10a*
upregulates coxsackievirus B3 biosynthesis by targeting the 3D-coding
sequence, Nucleic Acids Res., 41, 3760-3771.
39.Moscat, J., and Diaz-Meco, M. T. (2011) Feedback
on fat: p62-mTORC1-autophagy connections, Cell, 147,
724-727.
40.Deretic, V. (2010) Autophagy in infection,
Curr. Opin. Cell Biol., 22, 252-262.
41.Zhang, X. J., Chen, S., Huang, K. X., and Le, W.
D. (2013) Why should autophagic flux be assessed? Acta Pharmacol.
Sin., 34, 595-599.
42.Bardag-Gorce, F., Francis, T., Nan, L., Li, J.,
He Lue, Y., French, B. A., and French, S. W. (2005) Modifications in
P62 occur due to proteasome inhibition in alcoholic liver disease,
Life Sci., 77, 2594-2602.
43.Seibenhener, M. L., Babu, J. R., Geetha, T.,
Wong, H. C., Krishna, N. R., and Wooten, M. W. (2004) Sequestosome
1/p62 is a polyubiquitin chain binding protein involved in ubiquitin
proteasome degradation, Mol. Cell. Biol., 24,
8055-8068.
44.Si, X., Gao, G., Wong, J., Wang, Y., Zhang, J.,
and Luo, H. (2008) Ubiquitination is required for effective replication
of coxsackievirus B3, PloS One, 3, e2585.
45.Si, X., McManus, B. M., Zhang, J., Yuan, J.,
Cheung, C., Esfandiarei, M., Suarez, A., Morgan, A., and Luo, H. (2005)
Pyrrolidine dithiocarbamate reduces coxsackievirus B3 replication
through inhibition of the ubiquitin-proteasome pathway, J.
Virol., 79, 8014-8023.
46.Shi, W., Hou, X., Li, X., Peng, H., Shi, M.,
Jiang, Q., Liu, X., Ji, Y., Yao, Y., He, C., and Lei, X. (2013)
Differential gene expressions of the MAPK signaling pathway in
enterovirus 71-infected rhabdomyosarcoma cells, Brazil. J. Infect.
Dis., 17, 410-417.
47.Wang, B., Zhang, H., Zhu, M., Luo, Z., and Peng,
Y. (2012) MEK1-ERKs signal cascade is required for the replication of
enterovirus 71 (EV71), Antiviral Res., 93, 110-117.
48.Wong, W. R., Chen, Y. Y., Yang, S. M., Chen, Y.
L., and Horng, J. T. (2005) Phosphorylation of PI3K/Akt and MAPK/ERK in
an early entry step of enterovirus 71, Life Sci., 78,
82-90.
49.Colecchia, D., Strambi, A., Sanzone, S.,
Iavarone, C., Rossi, M., Dall’Armi, C., Piccioni, F., Verrotti di
Pianella, A., and Chiariello, M. (2012) MAPK15/ERK8 stimulates
autophagy by interacting with LC3 and GABARAP proteins,
Autophagy, 8, 1724-1740.
50.Settembre, C., Di Malta, C., Polito, V. A.,
Garcia Arencibia, M., Vetrini, F., Erdin, S., Erdin, S. U., Huynh, T.,
Medina, D., Colella, P., Sardiello, M., Rubinsztein, D. C., and
Ballabio, A. (2011) TFEB links autophagy to lysosomal biogenesis,
Science, 332, 1429-1433.
51.Ghavami, S., Cunnington, R. H., Yeganeh, B.,
Davies, J. J., Rattan, S. G., Bathe, K., Kavosh, M., Los, M. J., Freed,
D. H., Klonisch, T., Pierce, G. N., Halayko, A. J., and Dixon, I. M.
(2012) Autophagy regulates trans fatty acid-mediated apoptosis in
primary cardiac myofibroblasts, Biochim. Biophys. Acta,
1823, 2274-2286.
52.Simon, H. U. (2012) Autophagy in myocardial
differentiation and cardiac development, Circulation Res.,
110, 524-525.
53.Howard, C. M., and Baudino, T. A. (2014) Dynamic
cell–cell and cell–ECM interactions in the heart, J.
Mol. Cell. Cardiol., 70, 19-26.
54.Fujiu, K., and Nagai, R. (2013) Contributions of
cardiomyocyte-cardiac fibroblast-immune cell interactions in heart
failure development, Basic Res. Cardiol., 108, 357.
55.Gregoire, I. P., Rabourdin-Combe, C., and Faure,
M. (2012) Autophagy and RNA virus interactomes reveal IRGM as a common
target, Autophagy, 8, 1136-1137.
56.Huang, S. C., Chang, C. L., Wang, P. S., Tsai,
Y., and Liu, H. S. (2009) Enterovirus 71-induced autophagy detected
in vitro and in vivo promotes viral replication, J.
Med. Virol., 81, 1241-1252.
57.Lee, Y. R., Wang, P. S., Wang, J. R., and Liu, H.
S. (2014) Enterovirus 71-induced autophagy increases viral replication
and pathogenesis in a suckling mouse model, J. Biomed. Sci.,
21, 80.