Repair of Clustered Damage and DNA Polymerase Iota
E. A. Belousova1 and O. I. Lavrik1,2*
1Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia; fax: +7 (383) 333-3677; E-mail: lavrik@niboch.nsc.ru2Novosibirsk State University, 630090 Novosibirsk, Russia; fax: +7 (383) 363-4333
* To whom correspondence should be addressed.
Received November 27, 2014; Revision received January 30, 2015
Multiple DNA lesions occurring within one or two turns of the DNA helix known as clustered damage are a source of double-stranded DNA breaks, which represent a serious threat to the cells. Repair of clustered lesions is accomplished in several steps. If a clustered lesion contains oxidized bases, an individual DNA lesion is repaired by the base excision repair (BER) mechanism involving a specialized DNA polymerase after excising DNA damage. Here, we investigated DNA synthesis catalyzed by DNA polymerase iota using damaged DNA templates. Two types of DNA substrates were used as model DNAs: partial DNA duplexes containing breaks of different length, and DNA duplexes containing 5-formyluracil (5-foU) and uracil as a precursor of apurinic/apyrimidinic sites (AP) in opposite DNA strands. For the first time, we showed that DNA polymerase iota is able to catalyze DNA synthesis using partial DNA duplexes having breaks of different length as substrates. In addition, we found that DNA polymerase iota could catalyze DNA synthesis during repair of clustered damage via the BER system by using both undamaged and 5-foU-containing templates. We found that hPCNA (human proliferating cell nuclear antigen) increased efficacy of DNA synthesis catalyzed by DNA polymerase iota.
KEY WORDS: clustered lesions, DNA polymerase iota, translesion synthesis (TLS), base excision repair, oxidized basesDOI: 10.1134/S0006297915080064
Ionizing radiation, UV radiation, or oxidative stress result in generation of free radicals inside cells; the latter interact with heterocyclic bases of DNA and produce a large number of DNA lesions localized within one or two turns of a DNA helix known as clustered DNA damage [1]. Such clusters may be composed of several types of DNA lesions that undergo repair depending on their structure. In particular, in mammalian cells damage caused by alkylating agents or reactive oxygen species is repaired in mammalian cells using the base excision repair (BER) system [2]. The process starts from cleavage of N–C glycosidic bond between a damaged base and the sugar-phosphate backbone by a specific DNA glycosidase, followed by cleavage of the DNA strand due to endonuclease activity of AP-endonuclease [3]. Then the 5′-terminal sugar phosphate residue (dRp-fragment) is excised due to a lyase activity, followed by DNA polymerase gap filling; finally, the ends of a single-stranded DNA break are ligated by DNA ligase. It should be noted that during repair of clustered lesion via the BER mechanism, DNA polymerase is forced to synthesize DNA using a damaged template. In mammalian cells, DNA polymerase beta is the main enzyme that performs DNA synthesis during the BER process. It provides with low fidelity replication and has a dRp-lyase activity [4]. At the same time, this enzyme is able to catalyze DNA synthesis using damaged templates [5, 6]. Over the last few years, a number of novel DNA polymerases have been discovered in human cells, including DNA polymerase iota [7]. The function of this enzyme in cells is not yet clear. However, it is evident that the enzyme has DNA polymerase and dRp-lyase activities, and so it can be considered to play DNA polymerase function during the BER process [8]. DNA polymerase iota belongs to the family Y DNA polymerases that are involved in translesion synthesis [9]. Moreover, DNA polymerase iota exhibits rather low fidelity of DNA synthesis upon copying undamaged templates; however, this enzyme is able to create base pairs using non-canonic interactions, which substantially elevates fidelity of translesion synthesis. No doubt, this gives ground to assume that DNA polymerase iota might be involved in such a complex process as repair of DNA clustered lesions.
In this work, we investigated the ability of DNA polymerase iota to catalyze DNA synthesis during base excision repair including use of a damaged template. Two groups of DNA substrates were used as model structures. First group contained partial DNA duplexes having breaks of different length: 1, 2, and 5 nucleotide residues with 3′-hydroxyl and 5′-phosphate groups at the ends. Second group included double-stranded DNA duplexes with 5-formyluracil (5-foU) and uracil as a precursor of apurinic/apyrimidinic sites (AP) in opposite DNA strands shifted by one nucleotide residue. The AP site was created using uracil-DNA glycosidase, which specifically cleaves the glycosidic bond in a deoxyuridine residue within DNA. Then the repair of the DNA substrate was performed using BER proteins. A kinetic approach was used as the main method with determining Km, Vmax, and kcat. By comparing the kinetic parameters obtained after repair of a single (AP site) and a clustered (5-foU/AP site) DNA lesion occurring with/without additional protein factors, it would be possible to assess efficacy of repair for a clustered lesion of defined composition. Moreover, by knowing kinetic characteristics, this would give an idea about the specificity of a certain reaction relative to the type of lesion within a cluster as well as about the opportunity for an investigated enzyme and/or protein factor to participate in repair of a clustered lesion.
MATERIALS AND METHODS
The following materials and reagents were used in the study: bovine serum albumin, BSA (New England Biolabs, USA), T4-polynucleotide kinase (5000 U/ml) (Biosan, Russia), [γ-32P]ATP with specific activity 5000 Ci/mmol (Laboratory of Biotechnology, ICBFM SB RAS), dNTP (Promega, USA), unmodified and uracil-containing oligonucleotides (GenSet, Switzerland), and reagents for running electrophoresis and the main buffer components (Sigma, USA). Other extra pure and analytical grade reagents and buffer components used in the study were produced in Russia.
The formyl group of thymine was generated as described earlier [5].
Wild type hPCNA (human proliferating cell nuclear antigen) was purified and kindly provided by I. O. Petruseva (Laboratory of Bioorganic Chemistry of Enzymes, ICBFM SB RAS). Uracil-DNA glycosylase (UDG) from E. coli and human apurinic/apyrimidinic endonuclease-1 (hAPE1) were purified and kindly provided by S. N. Khodyreva (Laboratory of Bioorganic Chemistry of Enzymes, ICBFM SB RAS).
Recombinant human DNA polymerase iota was purified from E. coli strain RW644 transformed with plasmid DNA pCT14 as published earlier [10]. Plasmid DNA was kindly provided by Prof. R. Woodgate and Dr. E. G. Frank (National Institutes of Health, Rockville, USA). Recombinant human DNA polymerase iota contained C-terminal histidine tag, which facilitates protein purification from cellular extract by using metal-chelate chromatography on Ni-NTA-agarose. Further purification was conducted using hydroxyapatite and cation exchange resin. After dialysis of the target fractions, the yield of the target protein was 1.15 mg purified from 3 liters of E. coli culture medium.
Preparation of 5′-[γ-32P]-labeled oligonucleotides. Radioactive label was incorporated into the 5′-end of the oligonucleotide using T4 polynucleotide kinase as described [11]. The reaction mixture (10 µl) contained 0.5 µM primer, 5 MBq [γ-32P]ATP, and 5 U T4 polynucleotide kinase. The reaction was performed at 37°C for 30 min followed by adding ATP at final concentration 1 mM and then further incubated overnight at 4°C. Nucleotide material was extracted from the appropriate gel lane (that was visualized by autoradiography) and transferred onto DE-81 paper by electroelution in 50 mM Tris-borate buffer, pH 8.3. The product was eluted from the DE-81 paper by five portions (20 µl per portion) of hot 3 M LiClO4. The eluate was supplemented with 1.2 ml of cooled acetone (4°C) and kept at –40°C for 1 h. The resulting precipitate was collected by centrifugation, washed twice with 1 ml of cooled (4°C) acetone, air dried, and dissolved in water to the required concentration. The final radioactively labeled oligonucleotides were used to obtain double-stranded DNA substrates by mixing solutions containing several oligonucleotides at 1 : 1 molar ratio and incubating at 97°C for 5 min followed by gradual cooling to room temperature.
AP site was generated immediately before using DNA substrates by adding uracil-DNA glycosylase. The reaction mixture (in 10 µl water) containing 1 pmol double-stranded 5′-[32P]-labeled U-containing DNA substrate and 0.1 activity unit of UDG was incubated at 37°C for 30 min for further use. Complete uracil excision was confirmed by running gel electrophoresis (depicted in figures). All subsequent reactions were performed in the presence of 0.15 mM MgCl2 or MnCl2 and standard components of TDB buffer: 50 mM Tris-HCl, pH 8.0, 0.5 mM DTT, and 0.25 mg/ml BSA.
DNA synthesis was catalyzed by DNA polymerase iota as follows. When necessary, 5′-[32P]-labeled double-stranded DNA substrates were first treated with UDG (see above). Then the reaction mixture was supplemented with TDB buffer, MgCl2 or MnCl2, and hAPE1 at final concentration 0.15 mM and 0.001 µM, respectively, followed by incubation at 37°C for 5 min to complete cleavage of the AP-site-containing DNA strand that was confirmed by gel electrophoresis (see footnotes in figures). DNA synthesis catalyzed by DNA polymerase iota was performed in reaction mixture (10 µl) containing 0.01 µM DNA polymerase iota, 0.01 µM 5′-[32P]-labeled DNA substrate, 50 µM dNTP {{anchor|OLELINK6}} {{anchor|OLELINK7}} or their mixture, 0.15 mM MgCl2 or MnCl2 in TDB buffer at 37°C for 20 min. Then the reaction was stopped by cooling the sample on ice. The reaction products were separated by running 20% PAGE under denaturing conditions as described in [12] and visualized by radioautography using a Molecular Imager FX (BioRad, USA). The data were collected and analyzed using Quantity One software (BioRad).
Michaelis constants and rates of DNA synthesis for dNTP on different DNA substrates were measured in two stages. At the first stage, kinetic dependences for incorporation of dNMP into relevant DNA substrates by DNA polymerase iota were analyzed. The reaction mixtures (80 µl) containing 0.01 µM 5′-[32P]-labeled DNA substrate, 0.01 µM DNA polymerase iota, 50 µM dNTP, and 0.15 mM MgCl2 or MnCl2 in TDB buffer were incubated at 37°C. Aliquots (10 µl) were collected at defined intervals (0, 1′, 3′, 5′, 10′, 15′, 20′, 40′). The reaction was stopped by cooling the samples on ice. Reaction products were separated as described above. The data were analyzed using OriginPro7.5 software (MicroCal Software, USA). At the second stage, Michaelis constants (Km) and rates of DNA synthesis catalyzed by DNA polymerase iota were measured by varying dNTP concentrations at estimated time periods based on the results obtained during the first stage of the experiments. dNTP concentrations ranged within 0.005-200 µM. Concentrations of other components used in the reaction were kept unchanged. Reaction products were analyzed as described above. The data were processed using a Michaelis–Menten kinetic model. All experiments were repeated at least three times.
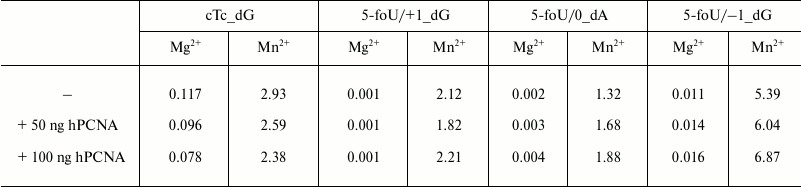
Effect of hPCNA on DNA synthesis catalyzed by DNA polymerase iota was analyzed as follows. The reaction mixtures (10 µl) containing 0.01 µM 5′-[32P]-labeled DNA substrate, 0.01 µM DNA polymerase iota, varying concentrations of dNTP, 0.15 mM MgCl2 or MnCl2, and hPCNA at final concentration 0, 50, or 100 ng in TDB buffer were incubated at 37°C for 20 min, and then reaction was stopped by cooling the samples on ice. Reaction products were analyzed as described above.
Binding of DNA polymerase iota to DNA substrates was evaluated using the gel-retardation method. To do this, the reaction mixtures (10 µl) containing 0.01 µM 5′-[32P]-labeled DNA substrate, 0.15 mM MgCl2 in TDB buffer, as well as 0.001-2 µM of DNA polymerase iota were incubated at 37°C for 5 min, supplemented with loading buffer (4% glycerin and 0.005% bromophenol blue), and then loaded on a gel at 10-12 V/cm. Products of the complex formation were separated in 8% polyacrylamide gel (AA/BisAA = 30 : 1) in 1× TBE, at 4°C and 10-12 V/cm, until the dye migrated half way. Then, the gel was dried and visualized by radioautography using Molecular Imager FX (BioRad). The data were collected and analyzed by using the Quantity One software. Dissociation constants Kd were evaluated using the MicroCal Origin 7.5 software with the hyperbolic equation y = A·x/(Kd + x), where the variables were presented as concentration of DNA polymerase (x) and of protein–nucleic acid complex (y); the latter was measured as a percentage of radioactive DNA–protein complex relative to total amount of radioactivity per lane; A – maximum concentration of protein–nucleic acid complex formed during reaction.
RESULTS AND DISCUSSION
Heterocyclic bases in nucleic acids undergo chemical modifications under the influence of different exogenous and endogenous factors. Occurrence of free radicals in cells results in appearance of a large amount of damages in genomic DNA localized within one or two turns of a DNA helix, i.e. clustered lesions [1]. Such clusters can contain two or more different types of DNA lesions, and mechanism of their repair depends on their nature. AP sites and 5-foU are the most common lesions of genomic DNA [13, 14]. Both types of such lesions represent a significant threat to cells, as replicative DNA polymerases are able to incorporate any of the four dNMP opposite to 5-foU, whereas an AP site has no coding bases at all. Also, a cluster simultaneously containing a 5-foU and an AP site as individual lesions represents an additional threat to cells. Generally, such lesions are repaired via the BER system [15, 16].
Repair of the clustered DNA lesions is a complicated process that results in sequential, stepwise restoring of DNA integrity. According to modern understanding, if an AP site is present within a double-stranded clustered lesion, it would be repaired first [17]. Such strategy helps to avoid occurrence of double-stranded breaks, which is the most toxic type of DNA lesions. If a clustered lesion contains an oxidative moiety, the individual lesions would be repaired via the BER mechanism, which includes the gap filling following the DNA damage excise. In this case, DNA polymerase should use the damaged template, i.e. to perform a translesion synthesis. In mammalian cells, this role is designated to the family Y proteins, to which DNA polymerase iota relates [18]. At present, the role of DNA polymerase iota in higher eukaryotes is unclear. It is assumed that this protein may play a role in the TLS process during replication of damaged DNA and potentially participates in maturation of immunoglobulin genes [19, 20]. Moreover, DNA polymerase iota possesses biochemical properties necessary for its implication in the BER process as it has both DNA polymerase and dRp-lyase activities [8].
By taking into consideration these facts, here we primarily investigated the capacity of DNA polymerase iota to catalyze DNA synthesis using DNA substrates containing breaks of different length. To model this, we selected partial DNA duplexes having 1, 2, or 5-nucleotide residue breaks, which contain thymidine at +1 position of the template strand relative to the 3′-end of the primer (Table 1).
Table 1. DNA substrates used in the
study

Notes: Primer length is length of the priming oligonucleotide after
treatment with APE1; X is thymine or 5-formyluracil;
Uf is 5-formyluracil; O is AP
site; asterisk (*) is 32P-radioacive label.
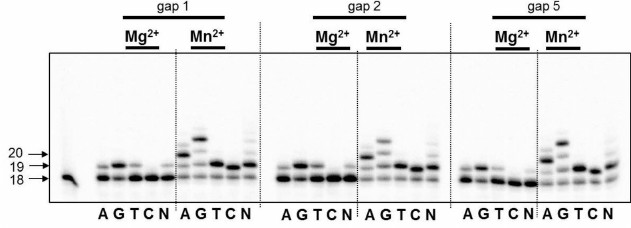
DNA polymerase iota as a nucleotide exchange enzyme is characterized by stringently requiring Mn2+ ions as a cofactor in in vitro experiments [21]. Moreover, Woodgate and Frank demonstrated that: (i) high concentration of Mg2+ (>0.5 mM) or Mn2+ (>0.2 mM) ions significantly decreases the enzyme activity; (ii) the capacity of DNA polymerase iota to bypass different lesions on the template strand varies in the presence of Mg2+ or Mn2+ ions [21]. In our study, 0.15 mM Mg2+ or Mn2+ was used for reactions of DNA synthesis. We found for the first time that DNA polymerase iota is able to catalyze incorporation of dNMP into partial DNA duplexes containing breaks of different length (Fig. 1). Despite the incorporation of a correct dAMP complementary to T-base of the template strand, the specificity of DNA polymerase iota was low corresponding to its biochemical properties [22]. Irrespective of the length of the break, DNA polymerase iota is able to catalyze DNA synthesis in the presence of any of four dNMP.
Fig. 1. Specificity of dNMP incorporation catalyzed by DNA polymerase iota in the presence of Mg2+ or Mn2+ ions using DNA substrates having 1, 2, or 5-nucleotide residue breaks. Left, arrows depict position of 5′-[32P]-labeled oligonucleotides: 18 – length of the initial oligonucleotide; 19, 20 – length of the products of DNA synthesis.
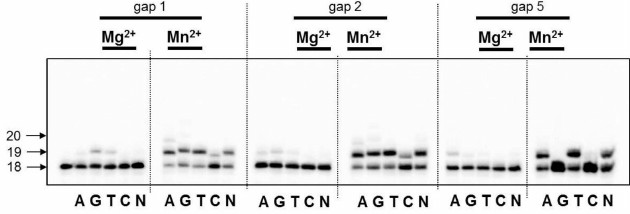
5-Formyluracil (5-foU) is the main oxidation product of the methyl group in thymine residues under exposure to ionizing and UVA radiation. The presence of 5-foU in the DNA does not lead to replication block on both strands, however, results in transition or transversion mutations [13]. When 5-foU is located within a clustered lesion, it increases the requirements for the relevant reparative DNA polymerase. DNA polymerase iota could play a role of a specific DNA polymerase. This explains why in the next step we examined the capacity of DNA polymerase iota to perform DNA synthesis across 5-foU within partial DNA duplexes having breaks of different length (Fig. 2). It turned out that in this case, DNA polymerase iota also does not exhibit strict specificity and catalyzes the incorporation of all the dNMP. Altogether, translesion synthesis was less effective compared to DNA synthesis performed using native templates.
Fig. 2. Specificity of dNMP incorporation catalyzed by DNA polymerase iota in the presence of Mg2+ or Mn2+ ions using DNA substrates containing 5-foU on one strand and 1, 2, or 5-nucleotide residue breaks on the other strand. Left, arrows depict position of 5′-[32P]-labeled oligonucleotides: 18 – length of the initial oligonucleotide; 19, 20 – length of the products of DNA synthesis.
It is known that fidelity and efficacy of DNA synthesis catalyzed by different DNA polymerases is markedly increased in the presence of additional protein factors. Among them is a human proliferating cell nuclear antigen (hPCNA) that acts not only as a processivity factor, but also is responsible for coordination of DNA polymerases during translesion synthesis [23, 24]. By using its structural PIP-motif, hPCNA interacts with DNA polymerase iota and stimulates the processivity of DNA synthesis depending on the structure of DNA template used [23]. Here, the efficacy of incorporation of a correct dAMP into DNA in the presence or absence of hPCNA was evaluated by measuring kcat/Km ratio, where Km is the Michaelis–Menten constant determined experimentally together with Vmax, the maximum rate of DNA synthesis; kcat is the rate constant for the catalytic stage of DNA synthesis calculated as Vmax/E0, where E0 is the concentration of DNA polymerase iota in the reaction mixture (Table 2). As a result, it was found that hPCNA decreases the efficacy of DNA synthesis using undamaged templates; however, it increases efficacy of DNA synthesis when DNA polymerase iota bypasses 5-foU. It should be noted that a cumulative effect of hPCNA expressed as its positive impact on efficacy of translesion synthesis by DNA polymerase iota and simultaneous negative impact on efficacy of DNA synthesis with native templates was higher for the gap1 structure that directly mimics a BER intermediate.
Table 2. Efficacy of DNA synthesis in the
presence of a correct dATP catalyzed by DNA polymerase iota using
partial DNA duplexes having breaks of different length,
kcat/Km, µM–1·sec–1
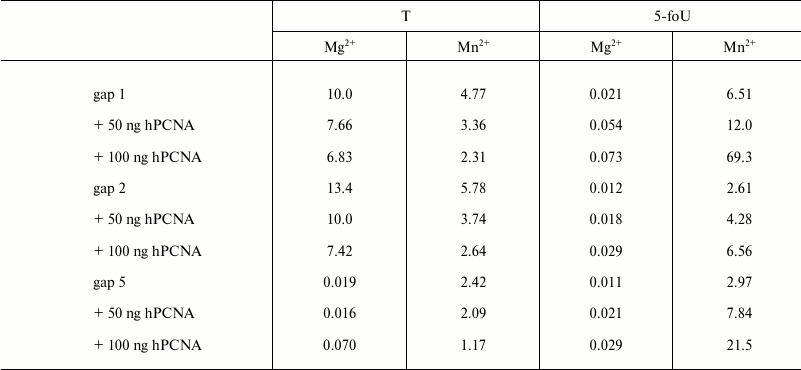
Note: The results are presented as the average value of three
independent experiments, SD ≤ 10%.
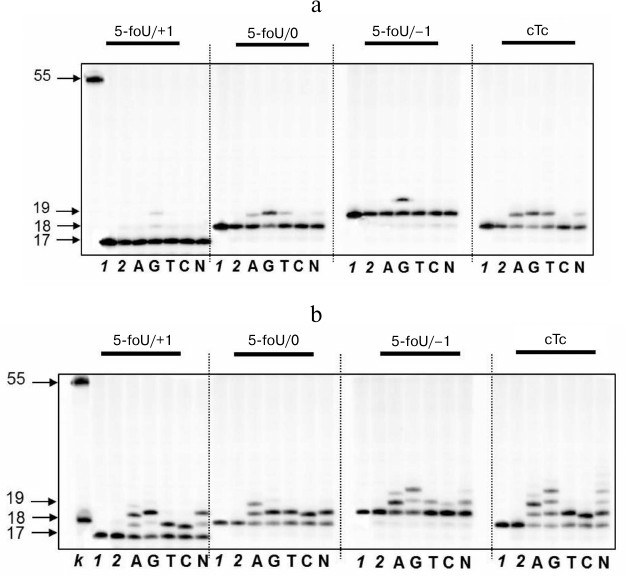
As mentioned above, repair of clustered lesions occurs sequentially and consists of several steps. If a clustered lesion consists of 5-foU and an AP site, repair would follow the BER mechanism via cleavage of the AP site with APE1 followed by translesion synthesis of the template strand, which is catalyzed by a specific DNA polymerase. In this case, the capacity of DNA polymerase iota to participate in this process was evaluated (Fig. 3). DNA duplexes that differed in reciprocal position of 5-formyluracil and AP site were used as DNA substrates (Table 1).
Fig. 3. Specificity of dNMP incorporation catalyzed by DNA polymerase iota in the presence of Mg2+ (a) or Mn2+ ions (b) using DNA substrates containing 5-foU on one strand and 1, 2, or 5-nucleotide residue breaks on the other strand. Left, arrows depict position of 5-[32P]-labeled oligonucleotides: 17, 18, 19 – length of the priming oligonucleotide after treatment with APE1; 55 – length of the initial oligonucleotide; lanes: k) 18 and 55 nt marker oligonucleotides; 1) control of reaction mixture treated with UDG; 2) control of reaction mixture treated with UDG and APE1.
The efficacy of DNA synthesis catalyzed by DNA polymerase is determined by several factors including the efficacy of binding between enzyme and substrate, which is characterized by a dissociation constant, Kd; the lower this value, the tighter are bonds in the protein–DNA complex. Binding of DNA polymerase iota to DNA duplexes was measured by the gel-retardation method (Fig. 4 and Table 3). As a control, we used DNA substrates that did not contain 5-foU. An AP site was generated using uracil-DNA glycosylase activity. Then, the AP site-containing strand within a DNA duplex was cleaved using APE1. Final DNA containing a single-stranded break with 3′-OH- and 5′-dRp-groups on one strand and 5-foU on the other strand were used as substrate to bind to DNA polymerase iota.
Fig. 4. Binding of DNA polymerase iota to different DNA substrates. Lanes: 1) control of reaction mixture treated with UDG; 2) control of reaction mixture treated with UDG and APE1; 3-10) varying concentrations of DNA polymerase iota.
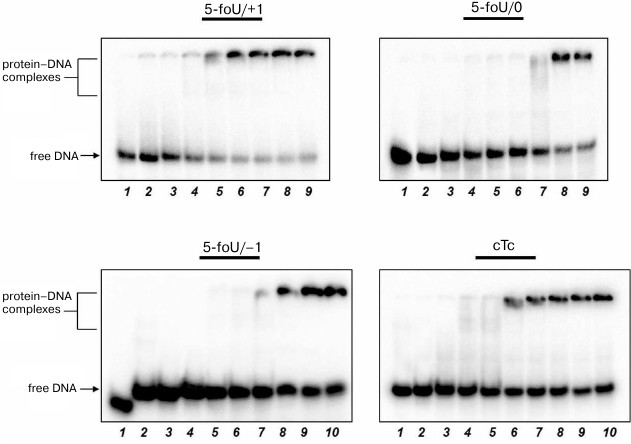
Table 3. Dissociation constants for
complexes between DNA polymerase iota and different DNA substrates,
Kd, µM

Note: The results are presented as the average value of three
independent experiments, SD ≤ 10%.
It turned out that both the presence of 5-foU and its position in template strand relative to the 3′-hydroxyl group had no impact on affinity of DNA polymerase iota to DNA substrate. Thus, efficacy of DNA synthesis is determined directly by kcat/Km ratio (Table 4). Likewise, an impact of hPCNA on the efficacy of DNA synthesis in the presence of the correct dNTP during BER process was also evaluated by analyzing kcat/Km ratio (Table 4).
Table 4. Efficacy of DNA synthesis in the
presence of a correct dATP catalyzed by DNA polymerase iota by using
DNA substrates of BER system,
kcat/Km, µM–1·sec–1
Note: The results are presented as the average value of three
independent experiments, SD ≤ 10%.
The data presented here demonstrate that DNA polymerase iota is characterized by a quite low efficacy of DNA synthesis under incorporation of the correct dNMP directly opposite to 5-foU (5-foU/0), or if 5-foU is shifted in the 5′-direction relative to the 3′-end of the primer (5-foU/+1). However, when a lesion is shifted in 3′-end (5-foU/–1), it results in sharply increased magnitude of the kcat/Km ratio. hPCNA was found to increase the efficacy of DNA synthesis irrespective of the 5-foU position in the template strand.
Thus, here we demonstrated for the first time that DNA polymerase iota is able to catalyze DNA synthesis using as substrates partial DNA duplexes with breaks of different length. Also we examined the capacity of DNA polymerase iota to participate in the BER process during repair of an AP site existing both as an individual and clustered lesion together with 5-foU. For the first time, we demonstrated that DNA polymerase iota catalyzes the incorporation of the correct dNMP during repair of a clustered lesion via the BER system using 5-foU-containing templates. Furthermore, we showed that hPCNA increases the efficacy of DNA synthesis catalyzed by DNA polymerase iota. In addition, it was found that this enzyme exhibited the maximal efficacy during DNA synthesis using DNA structures wherein 5-foU was shifted into the primer region, i.e. creating a problem 3′-end for certain DNA polymerases. On one hand, these data indirectly evidence that DNA polymerase iota can participate in the TLS process during replication of damaged DNA; on the other hand, it opens new opportunities for this protein as a participant in repair of clustered DNA lesions.
This study was supported by the Russian Scientific Foundation (Grant No. 14-24-00038) and the Russian Foundation for Basic Research (Grant No. 14-04-00268).
REFERENCES
1.Eccles, L. J., O’Neill, P., and Lomax, M. E.
(2011) Delayed repair of radiation induced clustered DNA damage: friend
or foe? Mutat. Res., 711, 134-141.
2.Parikh, S. S., Mol, C. D., and Tainer, J. A. (1997)
Base excision repair enzyme family portrait: integrating the structure
and chemistry of an entire DNA repair pathway, Structure,
5, 1543-1550.
3.Barzilay, G., and Hickson, I. D. (1995) Structure
and function of apurinic/apyrimidinic endonucleases, Bioessays,
17, 713-719.
4.Srivastava, D. K., Berg, B. J., Prasad, R., Molina,
J. T., Beard, W. A., Tomkinson, A. E., and Wilson, S. H. (1998)
Mammalian abasic site base excision repair. Identification of the
reaction sequence and rate-determining steps, J. Biol. Chem.,
273, 21203-21209.
5.Belousova, E. A., Vasil’eva, I. A., Moor, N.
A., Zatsepin, T. S., Oretskaya, T. S., and Lavrik, O. I. (2013)
Clustered DNA lesions containing 5-formyluracil and AP site: repair via
the BER system, PLoS One, 8, e68576.
6.Belousova, E. A., and Lavrik, O. I. (2010) DNA
polymerases beta and lambda, and their roles in the DNA replication and
repair, Mol. Biol. (Moscow), 44, 947-965.
7.Burgers, P. M., Koonin, E. V., Bruford, E., Blanco,
L., Burtis, K. C., Christman, M. F., Copeland, W. C., Friedberg, E. C.,
Hanaoka, F., Hinkle, D. C., Lawrence, C. W., Nakanishi, M., Ohmori, H.,
Prakash, L., Prakash, S., Reynaud, C. A., Sugino, A., Todo, T., Wang,
Z., Weill, J. C., and Woodgate R. (2001) Eukaryotic DNA polymerases:
proposal for a revised nomenclature, J. Biol. Chem., 276,
43487-43490.
8.Vidal, A. E., and Woodgate, R. (2009) Insights into
the cellular role of enigmatic DNA polymerase, DNA Repair,
8, 420-423.
9.Ohmori, H., Friedberg, E. C., Fuchs, R. P.,
Goodman, M. F., Hanaoka, F., Hinkle, D., Kunkel, T. A., Lawrence, C.
W., Livneh, Z., Nohmi, T., Prakash, L., Prakash, S., Todo, T., Walker,
G. C., Wang, Z., and Woodgate, R. (2001) The Y-family of DNA
polymerases, Mol. Cell, 8, 7-8.
10.Frank, E. G., McDonald, J. P., Karata, K.,
Huston, D., and Woodgate, R. (2012) A strategy for the expression of
recombinant proteins traditionally hard to purify, Analyt.
Biochem., 429, 132-139.
11.Yamshchikov, V. F. (1990) in Methods of
Molecular Genetics and Gene Engineering (Salganik, R. I., ed.) [in
Russian], Nauka, Novosibirsk, p. 28.
12.Yamshchikov, V. F. (1990) in Methods of
Molecular Genetics and Gene Engineering (Salganik, R. I., ed.) [in
Russian], Nauka, Novosibirsk, pp. 145-154.
13.Bjelland, S., Anensen, H., Knævelsrud, I.,
and Seeberg, E. (2001) Cellular effects of 5-formyluracil in DNA,
Mutat. Res., 486, 147-154.
14.Lindahl, T., and Nyberg, B. (1972) Rate of
depurination of native deoxyribonucleic acid, Biochemistry,
11, 3610-3618.
15.Barnes, D. E., Lindahl, T., and Sedgwick, B.
(1993) DNA repair, Curr. Opin. Cell. Biol., 5,
424-433.
16.Matsubara, M., Masaoka, A., Tanaka, T., Miyano,
T., Kato, N., Terato, H., Ohyama, Y., Iwai, S., and Ide, H. (2003)
Mammalian 5-formyluracil-DNA glycosylase. 1. Identification and
characterization of a novel activity that releases 5-formyluracil from
DNA, Biochemistry, 42, 4993-5002.
17.Georgakilas, A. G., O’Neill, P., and
Stewart, R. D. (2013) Induction and repair of clustered DNA lesions:
what do we know so far? Radiation Res., 180, 100-109.
18.Zhang, Y., Yuan, F., Wu, X., and Wang, Z. (2000)
Preferential incorporation of G opposite template T by the low-fidelity
human DNA polymerase iota, Mol. Cell. Biol., 20,
7099-7108.
19.Stallons, L. J., and McGregor, W. G. (2010)
Translesion synthesis polymerases in the prevention and promotion of
carcinogenesis, J. Nucleic Acids, 2010, pii: 643857.
20.Faili, A., Aoufouchi, S., Flatter, E., Gueranger,
Q., Reynaud, C. A., and Weill, J. C. (2013) Induction of somatic
hypermutation in immunoglobulin genes is dependent on DNA polymerase
iota, Nature, 419, 944-947.
21.Frank, E. G., and Woodgate, R. (2007) Increased
catalytic activity and altered fidelity of human DNA polymerase iota in
the presence of manganese, J. Biol. Chem., 282,
24689-24696.
22.Makarova, A. V., and Kulbachinskiy, A. V. (2012)
Structure of human DNA polymerase iota and the mechanism of DNA
synthesis, Biochemistry (Moscow), 77, 547-561.
23.Vidal, A. E., Kannouche, P., Podust, V. N., Yang,
W., Lehmann, A. R., and Woodgate, R. (2004) Proliferating cell nuclear
antigen-dependent coordination of the biological functions of human DNA
polymerase iota, J. Biol. Chem., 279, 48360-48368.
24.Bienko, M., Green, C. M., Crosetto, N., Rudolf,
F., Zapart, G., Coull, B., Kannouche, P., Wider, G., Peter, M.,
Lehmann, A. R., Hofmann, K., and Dikic, I. (2005) Ubiquitin-binding
domains in Y-family polymerases regulate translesion synthesis,
Science, 310, 1821-1824.