REVIEW: Amyloids: from Pathogenesis to Function
A. A. Nizhnikov1,2*, K. S. Antonets1,2, and S. G. Inge-Vechtomov1,2
1Department of Genetics and Biotechnology, St. Petersburg State University, 199034 St. Petersburg, Russia; E-mail: ant.nizhnikov@gmail.com2St. Petersburg Branch of Vavilov Institute of General Genetics, Russian Academy of Sciences, 199034 St. Petersburg, Russia
* To whom correspondence should be addressed.
Received March 3, 2015; Revision received March 30, 2015
The term “amyloids” refers to fibrillar protein aggregates with cross-β structure. They have been a subject of intense scrutiny since the middle of the previous century. First, this interest is due to association of amyloids with dozens of incurable human diseases called amyloidoses, which affect hundreds of millions of people. However, during the last decade the paradigm of amyloids as pathogens has changed due to an increase in understanding of their role as a specific variant of quaternary protein structure essential for the living cell. Thus, functional amyloids are found in all domains of the living world, and they fulfill a variety of roles ranging from biofilm formation in bacteria to long-term memory regulation in higher eukaryotes. Prions, which are proteins capable of existing under the same conditions in two or more conformations at least one of which having infective properties, also typically have amyloid features. There are weighty reasons to believe that the currently known amyloids are only a minority of their real number. This review provides a retrospective analysis of stages in the development of amyloid biology that during the last decade resulted, on one hand, in reinterpretation of the biological role of amyloids, and on the other hand, in the development of systems biology of amyloids, or amyloidomics.
KEY WORDS: amyloid, prion, protein, β-sheet, yeast, amyloidomics, amyloidosisDOI: 10.1134/S0006297915090047
AMBIGUITY OF THE CONCEPT “AMYLOID”
Although amyloids are under study in hundreds of laboratories throughout the world, up to now there is no conventional concept of “amyloid”. Mainly, this is because earlier researchers in this field considered as amyloid only extracellular fibrillar protein inclusions (most frequently pathological ones) that are formed in human and animal tissues [1, 2]. In some cases, this point of view is still used [3, 4].
Later, researchers ceased to regard the amyloid localization as of paramount importance, but on accumulation of data paid more attention to structural features of amyloid fibrils. From this standpoint, amyloids can be determined as non-branching protein fibrils consisting of monomers linked mainly due to hydrogen bonds between β-strands of intermolecular β-sheets arranged perpendicularly to the lateral axis of a fibril. This variant of fibril structure is usually named “cross-β” [5]. β-Sheets of an amyloid fibril can be arranged in parallel and in the register (similar amino acids of neighboring β-strands are one above the other and are bound with hydrogen bonds) [6-8]. Such arrangement of β-sheets is inherent in many amyloids, but not in all. Thus, among fibrils formed by a mutant variant of the human amyloid β-peptide (1-40 a.a.), there are fibrils with both parallel and antiparallel orientation of β-strands [9, 10], whereas some short amyloidogenic peptides form fibrils with antiparallel β-sheets [11, 12]. Fibrils of the infectious amyloid prion protein HET-s of the ascomycetes Podospora anserina seem to display a specific arrangement called β-solenoid [13]. Thus, scrupulous analysis of amyloid fibrils formed by different proteins reveals their structural heterogeneity, but it allows us to conclude that the cross-β structure is really a universal feature. It should be noted that the organization of amyloid fibrils is more complicated because they consist not of one but of several protofilaments laterally bound between themselves with hydrogen bonds. The protofilaments can be arranged not linearly but twisted helically [14, 15].
Another determination of amyloids is based not on their structure but on their unique physical features. First, true amyloid fibrils bind Congo Red stain, which has an affinity for β-sheets [16, 17] that results in apple-green birefringence in polarized light [18]. Second, protein aggregates analyzed for the presence of amyloid features are to have fibrillar structure and demonstrate characteristic cross-β pattern at two-dimensional X-ray diffraction [19]. Amyloid can also be determined as a fibrillar protein aggregate demonstrating cross-β pattern in X-ray diffraction analysis and birefringence in polarized light on staining with Congo Red, although this determination describes not the cause but the consequence: these two features of amyloid are due to their cross-β structure. There are also other specific features inherent in at least a sufficient number of known amyloids (some amyloids have not been analyzed for the presence of these features). The most important features are the resistance of amyloid aggregates to ionic detergents [20-22] and also their ability to bind fluorescent stains, thioflavin-T [23-25] and -S [26]. In total, the determination of amyloid as a fibrillar protein aggregate with the cross-β structure is the most adequate and reflecting the causal relationship.
There is no doubt that amyloids are important for medicine as lethal pathogens, some of which are also infectious. Moreover, some discoveries, especially those of the last decade, led to a complete reinterpretation of their biological role. Amyloids are now considered not only as lethal pathogens, but also as a variant of protein quaternary structure that is required in some cases for realizing key biological functions.
This review presents a retrospective analysis of the main stages and events in amyloid biology that within the last decade resulted, on one hand, in the understanding of their functional significance and, on the other hand, established a basis for appearance of systems biology of amyloids, or amyloidomics. The main discoveries described in this work, as well as their association with the number and functions of known amyloids, are shown in the figure. Data on the biological diversity of pathologic and functional amyloids and of prions are summarized, respectively, in Tables 1-3.
Scheme illustrating the key discoveries in amyloid biology, as well as association of these discoveries with the number and functions of known amyloids. a) Vertical arrows indicate trends in the development of amyloid biology, and horizontal arrows indicate the dates and key events; b) changes in the distribution of identified amyloids according to their functional and pathogenic role. Prions are shown as a separate group, and their number is given considering those with amyloid features not described; c) distribution of identified amyloids according to systematics. Groups of prokaryotes and eukaryotes are indicated, and human amyloids are shown separately
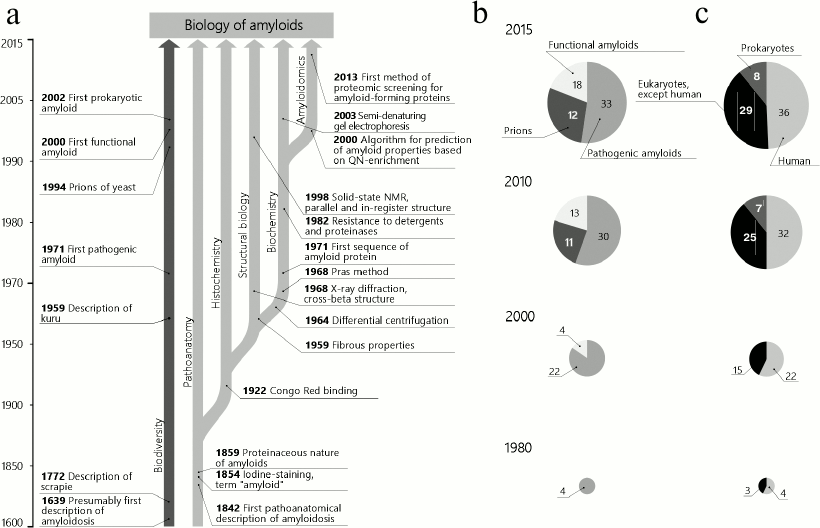
Table 1. Pathological amyloids
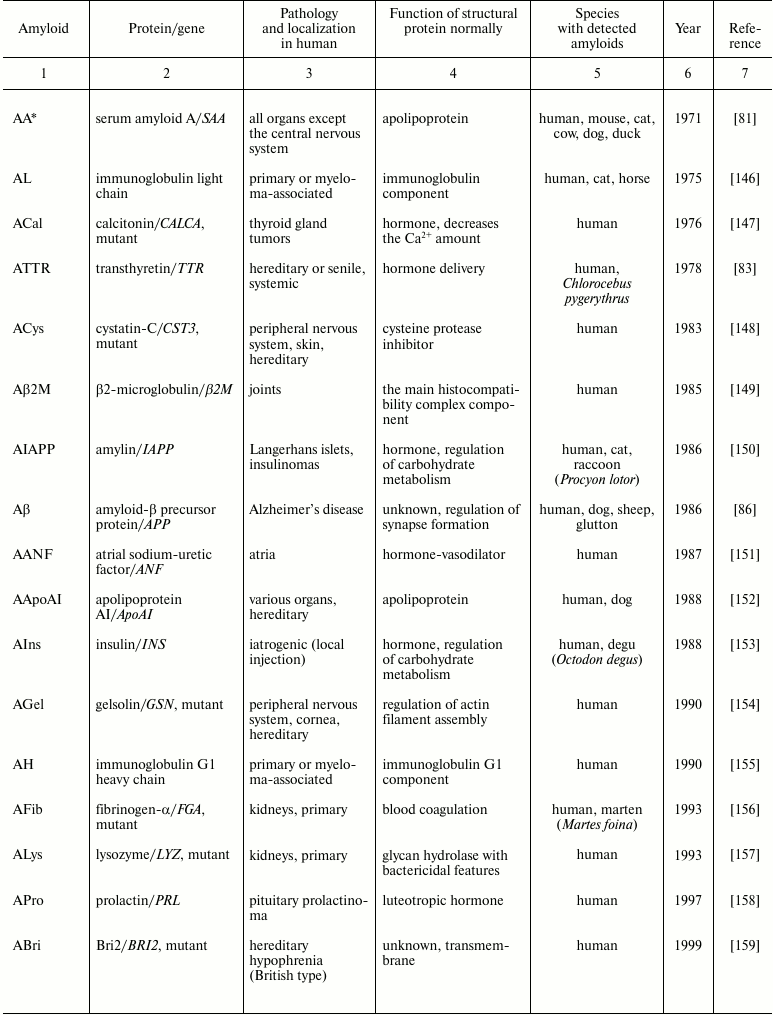
* The amyloid nomenclature is given in the correspondence with
recommendations of the International Society of Amyloidosis [3, 4].
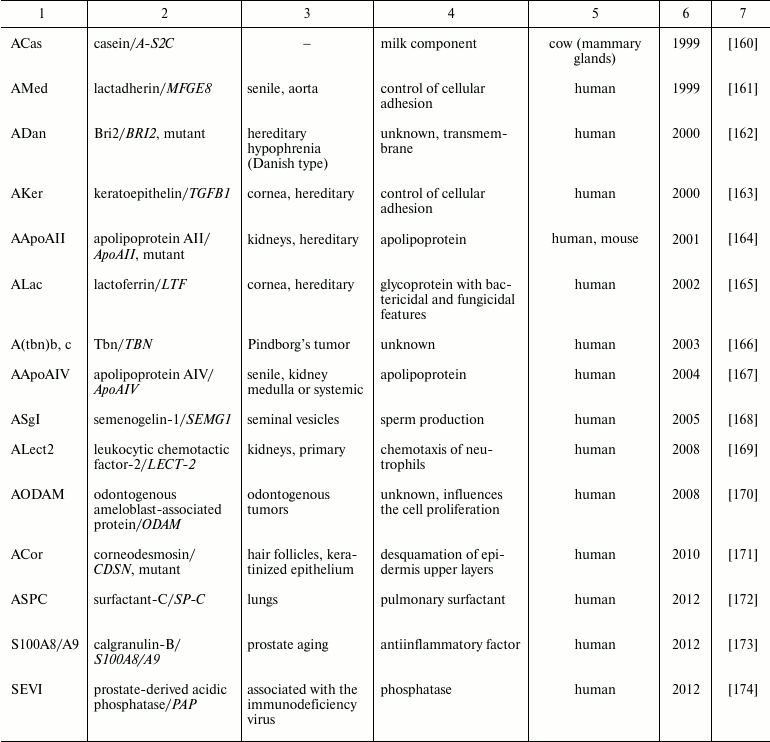
Table 2. Functional amyloids
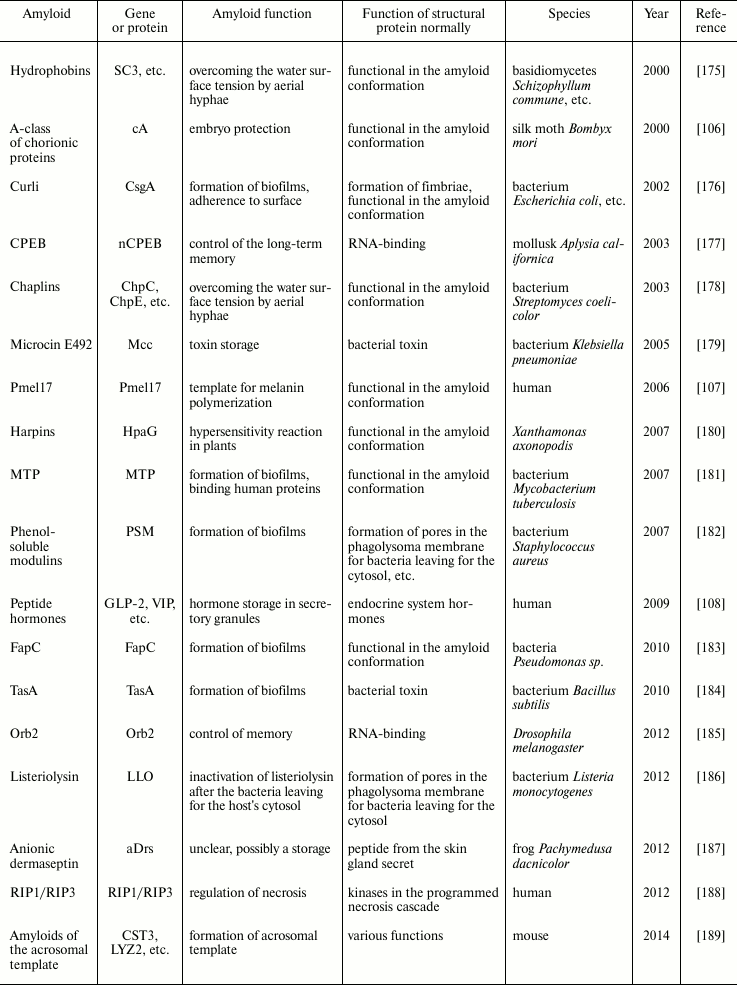
Table 3. Prions
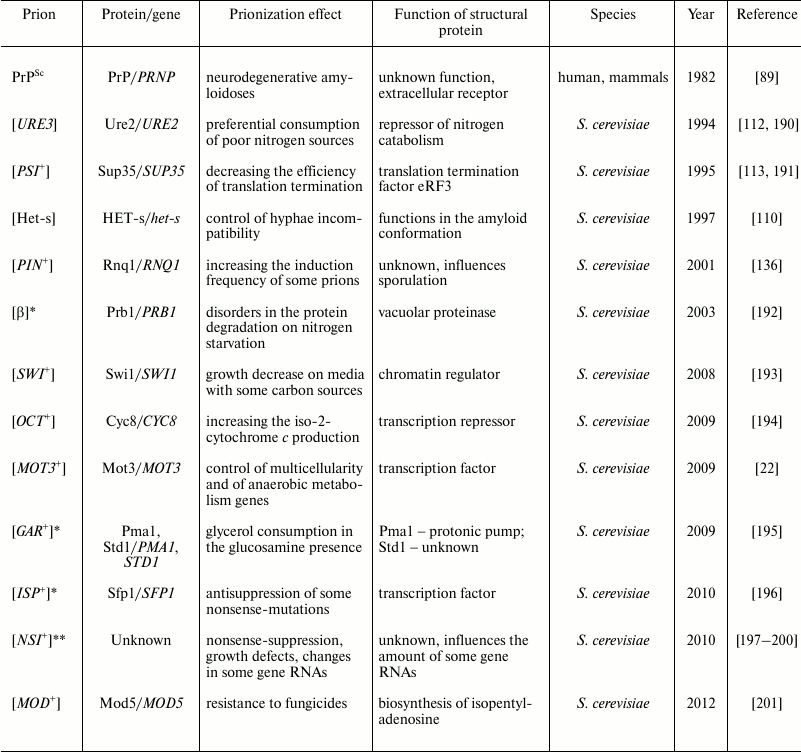
* The relation with amyloidogenesis is not established.
** Prion-like factor.
PATHOANATOMIC DESCRIPTION OF AMYLOIDOSES (FROM XIX CENTURY UNTIL
PRESENT)
Pathological changes in internal organs caused by some amyloidoses have been known for a very long time. Thus, according to Robert Kyle [27], an amyloidosis-affected organ seems to be first described in 1639 in the book Responsionum et Curationum Medicinalium written by the physician and dramatist Nicolao Fontano (his true name was Fonteyn) [28]. Fontano described the autopsy of a teenager whose spleen was strongly increased in volume and contained large white inclusions. Somewhat later, Thomas Bartholin reported the autopsy of a woman whose spleen was so changed that it was difficult to cut it with a knife, and the cutting was accompanied by a sound similar to that on sawing wood [27, 29]. Antoine Portal was probably the first who reported about liver amyloidosis (1789) [27], and in 1842 Carl Rokitansky found that patients with syphilis, tuberculosis, or mercuric poisoning could have their liver greatly enlarged because of infiltration with a gray substance (“waxy” liver) [30].
Manifestations of some amyloidoses, especially of neurodegenerative ones, are also known for a long time. One of the first reliably described amyloidoses is scrapie, a lethal infectious neurodegenerative amyloidosis of sheep caused by PrPSc, which is an amyloid isoform of the prion protein PrP. Schneider et al. analyzed the history of the description of this disease [31] and found that the first available work mentioning scrapie was published in 1772 [32]. In this work, it was said that scrapie was known in England for 40 years, i.e. from 1732. However, no bibliography study known to us mentioned a work of 1732. Nevertheless, Schneider thinks [31] that scrapie was known in Germany long before 1772, but not to biologists and physicians, but to “ökonomen”, i.e. to farmers and landlords, who made efforts to conceal from purchasers the presence of this disease in their flocks and also unsuccessfully attempted to find out its cause. In addition to Schneider, many authors reported that scrapie was described in the XVII [33] and even in the XV [34] centuries (but without concrete references). There is also the most intriguing hypothesis of R. Wickner: he supposed that scrapie was known in ancient China more than 2000 years ago [35]. Wickner proposed this hypothesis after analyzing a hieroglyph that meant “pruritus”, which seemed to consist of three parts (disease, sheep, and just pruritus) [36]. However, this interesting hypothesis was met negatively by Chinese researchers, who reported that all hieroglyphs had changed with time and their present meaning did not correlate with the ancient one. Moreover, the component treated by Wickner as “sheep” is really a phonetic one and does not have any sense [36]. Thus, it can be asserted that symptoms of prion amyloidoses in animals were reliably described in the third quarter of the XVIII century. Similar human diseases, first of all Kuru, were described only in the middle of the XX century [37].
The term “amyloid” (starch-like) is a derivative of Latin “amylum” and Greek “amylon” (starch); this term was initially introduced by M. Schleiden in 1838 for describing starch conglomerates normally present in plant cells [38]. R. Virchow in 1854 [39] found that inclusions of the “waxy” liver could be stained with iodine similarly to starch (the reaction was discovered in 1814 by Colin and de Claubry) [40]. Therefore, in his further works Virchow called these deposits amyloids, considering them starch derivatives [38]. But, already in 1859 Freidrich and Kekule showed that amyloids did not contain a substance chemically similar to starch or cellulose but were enriched with nitrogen and were protein-like [1, 38]. However, until the end of his life, Virchow considered amyloids to be polysaccharides [41]. Now it is known that amyloid inclusions isolated from mammalian tissues, in addition to the major fibrillar protein component, also contain proteoglycans [42] and glycosaminoglycans [43, 44], which is an explanation of their stainability with iodine.
Thus, pathoanatomical and histological studies on amyloids were not only the first and basic stage in the development of the biology of amyloids, but were also actively used later, already in the XX century, and are used up to now, resulting in detection of about 30 amyloids pathologic to human [3] (Table 1). Significant progress in this line of studies has been contributed by introduction of some approaches for detecting amyloids, biochemical methods of their isolation and purification, as well as studies on the structure of amyloid fibrils, which will be considered below.
DEVELOPMENT OF HISTOCHEMICAL METHODS FOR DETECTION OF AMYLOIDS
(FROM 1922 UNTIL PRESENT)
After the discovery that amyloid inclusions could be stained with iodine, investigations by light microscopy were developed very intensively. At that time, a test proving the amyloid nature of a studied protein inclusion was extremely required. In 1922, H. Bennhold [16] found that amyloid could bind the stain Congo Red. The exact mechanism of this binding is still unclear, but it results in apple-green birefringence in polarized light [17] that was first shown by Divry and Florkin in 1927 [18]. Bennhold’s method improved in 1962 by Putchler [45] allowed researchers to easily diagnose amyloidoses on histological sections. Now Congo Red binding is a main test for the amyloid nature of protein aggregates. The sensitivity of this method can be increased by analyzing the autofluorescence of Congo Red [46, 47].
Interesting modifications of the application of Congo Red were developed later, these providing the primary classification of amyloid inclusions due to differential staining. Thus, a preliminary autoclaving of tissue specimens for 30 min at 120°C results in the loss of the affinity of amyloid-A for Congo Red [48], and increasing the autoclaving duration to 120 min results in a similar effect in amyloids formed by the immunoglobulin light chain [48]; some other amyloids are sensitive to treatment with potassium permanganate [49] or with alkaline guanidine [50].
Metachromatic stains, such as Methyl Violet and Crystal Violet [51], Toluidine Blue, etc. [2], were also proposed for staining amyloid inclusions. Thus, Crystal Violet stains red some amyloids, whereas the adjacent tissues remain blue. Unfortunately, these methods have low sensitivity and specificity, which prevented their wide application [52].
In addition to Congo Red, fluorescent stains thioflavin-T [23-25] and -S [26] became widely used. The binding of thioflavin-T with amyloid fibrils is accompanied by a red shift of the emission spectrum, and the binding of thioflavin-S is accompanied by an increase in its fluorescence [53]. Staining with thioflavin-S gives rather high background fluorescence, and therefore it is usually applied for staining amyloid inclusions on histological preparations but not for a widely used quantitative analysis of the in vitro kinetics of amyloid fibril formation. For such experiments Congo Red [54] or thioflavin-T are used [55]. Thioflavin-T is nearly a universal stain for amyloids, although with some amyloids it binds rather poorly [56].
Later, from the 1980s, methods for immunodetection of amyloid were actively developed. Antibodies were prepared to the majority of known pathologic amyloids, which resulted in more effective diagnosis of amyloidoses [57]. Along this line, there is an interesting attempt to create “conformational” antibodies [58] and oligonucleotide DNA-aptamers [59] that would be able to distinguish not a sequence of a definite protein, but all or the majority of proteins in amyloid conformation. These approaches were shown to be effective for detection of certain amyloids, but it is too soon to speak about their universality.
Thus, the spectrum of amyloid detection methods based on specific binding with definite compounds is very large, and this trend is under continuous active development. New effective stains appear [60, 61], as well as modes of their use for subsequent identification of amyloids. In particular, there is an approach for visualization of amyloid inclusion on a histological section by staining with Congo Rot or another stain, the cutting this inclusion with a microdissection microscope, and the identification of the molecular composition by mass spectrometry [62]. However, this method can only be used for identification of amyloids that form sufficiently large inclusions, thus limiting its application.
STRUCTURAL STUDIES ON AMYLOIDS (FROM 1959 UNTIL PRESENT)
Binding of the same stains by different amyloid inclusions suggested a structural similarity of the amyloids. The first data confirming this hypothesis were obtained in 1959 in a classic work of Cohen and Calkins, who used electron microscopy for studies on amyloids [63]. They investigated amyloid inclusions from histological preparations of various tissues and organs and showed that all they were formed by filaments (or fibrils) 75-100 Å in thickness and 1000-16,000 Å in length [63]. Then a model of a filament consisting of several protein strands, protofilaments [1, 64], was proposed, which was general for that time. These protofilaments had about 30 Å diameter and were linked to each other along the lateral axis, forming a tubular structure, a filament. Filaments can join as a pile and form a fibril. Thus, three levels of the amyloid organization can be determined: protofilament, filament, and fibril [65]. At present, the terminology is slightly confused because the modern concept of fibril often corresponds to the filament concept in the old works.
Introduction of in vitro approaches was the next step in studies on amyloid structures. Improvement of purification methods (see next section) allowed researchers to isolate from tissues amyloid fibrils and study their structure on the molecular level. First, it was important to demonstrate that amyloids retained the fibrillar structure upon their isolation from tissues and that their structure could be analyzed in vitro [66]. Then, X-ray diffraction was introduced, which allowed researchers to begin study on secondary structure (i.e. arrangement of the β-sheets) of protein molecules in amyloid fibrils [5, 67-69]. As a result, a common secondary cross-β structure was revealed in all studied amyloids, with scattering signals of about 4.7 Å (corresponded to distance between neighbor β-strands in a β-sheet) and of about 10 Å (corresponded to distance between β-sheets) [14]. Later, it became clear that studies on the fine structure of amyloids were rather difficult because they did not crystallize and were poorly soluble. This prevented application of X-ray crystallography and nuclear magnetic resonance approaches [7]. Nevertheless, studies by cryoelectron microscopy confirmed that protofilaments formed amyloid fibrils and showed that protofilaments: (i) were arranged not linearly, but twisted along the lateral axis, and (ii) fibrils could be formed by different numbers of protofilaments, and their binding to each other could vary significantly [70-73]. Solid-state nuclear magnetic resonance not requiring the studied object to be crystallized was another qualitative step in the study of amyloid structure on the atomic level. Using this method, detailed structural models were designed for some amyloids, and the arrangement of β-sheets was shown to be parallel and in register [11, 13, 74-76].
Amyloids are a rather heterogeneous group at the tertiary structure level: various amyloids were shown to have parallel and antiparallel arrangement of β-sheets [9, 10], as well as β-solenoid organization [13]. It seems that there is no common tertiary structure of amyloid, but a number of different structures can exist, some of which have been modeled recently in the work of Smaoui et al. [77]. It is also necessary to return to the yet unclear problem considered in the work of Inoue et al. [78], who compared by electron microscopy the organization of amyloid fibrils in situ with the data obtained earlier in vitro [78]. They concluded that formation of amyloid inclusions in situ occur with involvement of chondroitin sulfate and heparan sulfate, which are likely to change the structure of fibrils. Thus, structural data obtained in vitro seem to unable to completely characterize the picture of amyloid fibril organization in vivo. This problem can only be solved with development of new and better approaches to analyzing the structure of amyloids.
BIOCHEMICAL ANALYSIS OF AMYLOIDS (FROM 1964 UNTIL PRESENT)
Progress in biochemical purification and analysis of amyloids is closely associated with their ordered structure are responsible for their unique features. Amyloid fibrils are insoluble in salt solutions. This feature was used for isolation and purification of fibrils using differential centrifugation [79], which was the first approach for extracting amyloids. A method of aqueous extraction, or the Pras method [66], was proposed already in 1968. With this method, amyloids are initially separated from proteins soluble in salt solutions through precipitation by differential centrifugation and then separated from insoluble proteins by suspending with subsequent centrifugation in aqueous solution to retain amyloids in the supernatant fraction [66]. By this method, it was shown that amyloids could be formed by various proteins [80-84]. In 1971, the first sequences of amyloid-producing proteins were identified in the works of Glenner et al. [80] and Benditt et al. [81] as a fragment of the immunoglobulin light chain and amyloid-A, respectively, and just from that year the study on amyloids as specific proteins began.
The following very important stage in the biochemical characterization of amyloids was the detection of their insolubility in some protein-denaturing agents, such as ionic detergents (sodium dodecyl sulfate [85]), chaotropic agents, formic acid, guanidine chloride [86], and also of their resistance to proteinases [87, 88]. The resistance to sodium dodecyl sulfate of amyloid β-peptide causing Alzheimer’s disease was used for its isolation and identification [86]. A similar resistance of PrPSc amyloid to proteinase K allowed Bolton et al. to identify it as an agent of infectious neurodegenerative human and animal amyloidoses [89]. In fact, the variety of now existing methods for isolation and purification of amyloids can be reduced to two main modes, which are used either together or separately: differential centrifugation, and treatment with different protein-denaturing agents.
Among the relatively new approaches, semi-denaturing detergent agarose gel electrophoresis (SDD-AGE) should be noted [21]. This approach also includes the treatment of amyloids with ionic detergents, which were shown to dissociate amyloid fibrils to oligomers. These oligomers enter agarose gel and can be visualized, in particular with antibodies. An important advantage of this method is the possibility of evaluating the size of the amyloid oligomers [21, 90]. The SDD-AGE method with subsequent additional separation by two-dimensional gel electrophoresis allows researchers to analyze not only amyloids themselves, but also proteins involved in their aggregates [91].
AMYLOIDOMICS (FROM 2000 UNTIL PRESENT)
The accumulation of experimental data on various amyloid-forming proteins was favorable for development of systems approaches, both theoretical and experimental, for searching for new amyloids on the proteome level. The theoretical approaches capable of predicting amyloid features of proteins are based on searching for a resemblance between the amino acid sequence of a studied protein and those of known amyloid-forming proteins. An early study in this line was a work from Weissman’s laboratory in 2000 that noted the enrichment with asparagine (N) and glutamine (Q) of three known yeast infectious amyloids, prions. They compiled a list of 100 yeast proteins most enriched with these amino acids [92]. Later, another algorithm was proposed for detecting N/Q-rich domains by searching lowest probability sequences (LPS) [93]. This approach was optimized in our work using an original algorithm of sequence analysis based on ranking of probabilities (SARP) [94]. The list of 170 yeast proteins obtained using LPS includes all known N/Q-rich yeast prions, and this indicates its efficiency. Moreover, the N/Q-rich domains of many proteins from this list can produce amyloid-like aggregates in vivo on their overproduction [22]. Thus, detection in the proteome of proteins enriched with N and Q is promising for searching for new amyloids. By contrast, it is difficult to predict amyloids without N/Q-rich domains, but just such amyloids actually form the majority of amyloids now known. Specific features of amino acid sequences of amyloidogenic regions of such proteins are insufficiently studied, but some of their features are known, in particular, their richness with nonpolar and uncharged polar amino acids. A series of algorithms has been developed for predicting amyloid properties in proteins not enriched with N/Q-tracts [95-97]. Unfortunately, these algorithms are mainly based on indirect data (e.g. on peptide aggregation in vitro) and cannot be adequately used for analyzing amyloid features of full-size proteins. Experimental detection of new amyloids will contribute to improvement of these algorithms.
Until recently, there was no experimental method for detecting amyloidogenic proteins on the proteome level. However, in 2013-2014 two methods of this type were published: TAPI (Technique for Amyloid Purification and Identification) [98], and PSIA (Proteomic Screening and Identification of Amyloids) [99]. Both methods include the treatment with ionic detergents and differential centrifugation for purification amyloid fractions. The TAPI method additionally uses polyacrylamide gel electrophoresis for purification from monomeric proteins. The PSIA method uses a modified approach for amyloid isolation proposed in the work of Kushnirov et al. [100]. Proteins are separated by liquid chromatography with mass spectrometry in TAPI method [98] and by two-dimensional differential gel electrophoresis (2D-DIGE) in PSIA, which provide for highly accurate comparison of molecular compositions of two specimens [99]. It should be noted that proteins revealed by these two methods are only candidates for the role of amyloids, and their amyloid features need to be tested by additional experiments.
It is obvious that systems biology of amyloids, or amyloidomics, is only at the beginning of its development. This discipline has been contributed to by both theoretical and experimental studies, but it is still unclear what specific features of the amino acid sequence of a protein determine its ability to form amyloids. Short amyloidogenic fragments capable of aggregating in vitro can be detected in many proteins using bioinformatics [95-97], but full-length proteins in the majority of cases are unable to aggregate in vivo. Therefore, it is now necessary to understand not what sequences are amyloidogenic, but what ones can prevent amyloidogenesis. Undoubtedly, obtaining a large massive of experimental data on proteins capable of forming amyloids in vivo has to be the main condition for solving this problem, and both TAPI and PSIA are likely to be useful.
FROM PATHOGENESIS TO FUNCTION: PROTEIN INHERITANCE
Studies on amyloids were started because of amyloid-caused pathologies, especially in human. This was favorable for the active development of amyloid biology because the known pathologic amyloids in most cases formed large inclusions convenient for histochemical characterization and for development of biochemical methods for isolation of amyloids as the earlier mentioned method of Pras [66]. Now more than 30 human amyloids have been shown to be associated with different pathologies (Table 1). Moreover, such diseases as Huntington’s disease [101], Parkinson’s disease [102], and tauopathies [103] are associated with an amyloid-like aggregation of proteins huntingtin (a mutant variant with an elongated polyglutamine tract), synuclein, and tau, respectively. It should be noted that all amyloid-associated diseases are still incurable, and the incidence of some of them is rather high. Thus, type II diabetes, or diabetes mellitus associated with formation of IAPP amyloids in the pancreas affects, by some evaluations, 400 million people (5% of the World’s population) [104]. The neurodegenerative Alzheimer’s disease associated with amyloid β-peptide aggregation affects one of three persons after 90 years of age [105]. Some tumors are also accompanied by amyloidogenesis of certain proteins (Table 1). The problem of amyloidoses can become even more urgent because many of them are age-related diseases and thus seriously increase in the human lifetime.
As a result, during a rather long-term story of studies on amyloids they were considered only as lethal pathogens. Nevertheless, this paradigm was destroyed just at the border of the XX and XXI centuries, in 2000, due to discovery that organisms normally form some amyloids responsible for some important functions. The first work in this line was a demonstration of amyloid properties of the protective proteins of the silk moth Bombyx mori oocyte chorion [106]. This report was followed by a number of works that showed that amyloids really performed very important functions in very different organisms, from bacteria to human (Table 2). In particular, in humans amyloids participate in the control of melanin polarization [107] and the storage of many hormones [108]. Amyloid-like features were found even in biofilms of archaea [109]. Thus, functional amyloids seem to be distributed in all three domains of life. It is now obvious that amyloids can be not only pathogens, but are a widely distributed variant of functional quaternary structure of proteins.
Studies on infectious proteins, prions, the majority of which are amyloids, were also started by investigations of pathologies. In 1982, Prusiner et al. identified the prion PrPSc [89], which causes in human and other mammals lethal infectious amyloidoses including Kuru and Creutzfeldt–Jakob disease. Later, prions were found not only in mammals, but also in ascomycete fungi. The prion HET-s of the ascomycete Podospora anserina was described already in 1997 [110], but its amyloid features were shown after 2000 [111]. This prion controls the incompatibility of hyphae and can be considered an infectious functional amyloid. For the yeast Saccharomyces cerevisiae, many prions have been described, which in the majority of cases are not pathogenic and in some cases even can be called functional (Table 3). More exactly, the “prion” concept is beyond the framework of the concept “amyloid”: not all prions form amyloids, and not all amyloids are prions.
Just the identification of some cytoplasmic hereditary factors as prions in the yeast S. cerevisiae [112, 113] promoted the creation of the concept of protein inheritance based on the idea of conformational or spatial templates [114, 115]. The template principle was first proposed in 1928 by N. K. Koltsov for explaining chromosome replication [116] and later was applied by F. Crick to transcription and translation as the central dogma of molecular biology [117, 118]. The template principle of Koltsov–Crick described the replication of the linear sequence of biological macromolecules – linear templates, or type I templates.
Conformational, or spatial, (type II) templates characterize the ability of some proteins to act as templates for the spatial stacking of homological or heterological polypeptide chains. However, this statement does not implicate the ability of the protein to replicate their primary structure and does not contradict the general ideas about the synthesis of polypeptide chains on ribosomes directed by mRNAs of the corresponding structural genes. The real existence of type II template processes (TP II) is based on the so-called “protein transformation” in the yeast S. cerevisiae. Thus, protein extracts of cells carrying the prion [PSI+] transfer the corresponding phenotypical trait (the ability for omnipotent nonsense-suppression) to the cells [psi–]. Moreover, a replication of the prion [PSI+] was shown in vitro with subsequent introduction of prion fibrils into cells through transformation [119-121]. As half-mockingly noted G. Fink, molecular biology history could be quite another if protein transformation had been discovered earlier than the DNA-mediated transformation [122].
Universal features of type I template processes (TP I) are their ambiguity (“errors” of replication) which are compensated by repair or correction [123, 124]. The balance between ambiguity and correction is expressed in the optimization of the accuracy of TP I during their evolution. All three TP I (replication, transcription, translation) have three stages (initiation, elongation, termination) that are affected by ambiguity, the level of which is optimized by the correction processes.
Are these characteristics of TP I also inherent in TP II? Stages of initiation and elongation have been described for TP II. Initiation, or nucleation, is a spatial rearrangement of a precursor protein of prion or amyloid without changes in its primary structure, and the subsequent joining to it of a molecule (molecules) of the same (or another) protein. During the joining, this protein is spatially transformed in the image of the primer protein molecule, which is in the amyloid conformation. Then the protofilament is elongated due to joining and amyloid conversion of new molecules of the corresponding protein. It is still unclear whether a specific termination stage of this process really exists. At least in the case of prions, the termination can be represented by cleavage of a fibril with specific chaperones – disaggregases, e.g. Hsp104, as shown for the yeast prion [PSI+] [125-127]. This results in production of oligomers – fragments or “seeds” of a prion, which serve for elongation of new fibrils in the daughter cells. Other chaperones are also involved in this process [128], but their role in prion “multiplication” in not completely clear.
The ambiguity of TP II is also manifested by presence of the so-called prion strains consisting of a protein with the same primary structure but different in physicochemical features and phenotypic characteristics [129-131]. Different pH-dependent conformational rearrangements of the amyloid β-peptide precursor protein can also exist [132, 133]. Whether such ambiguity is associated only with the initiation stage or also with the elongation stage of TP II is now under study on the prion [PSI+] model [134]. It is not yet established whether a correction mechanism exists during amyloidogenesis. However, the participation of chaperones makes correction probable.
It can be supposed that amyloids and/or prions are likely to form network (or networks), which can be called “amyloidome” as a part of proteome. These ideas are consequences of the works of I. L. Derkatch et al. who showed the interaction of prions [PSI+] and [PIN+] in S. cerevisiae [135, 136] and postulated the existence of such prion or amyloid networks in the cell. This is confirmed by data on the interaction of different mammalian amyloids, mainly under in vitro conditions, as well as on the influence of yeast prions on the frequency of their mutual induction [137]. It must also be taken into account that β-sheets potentially responsible for amyloid formation and aggregation are present in many proteins but remain disguised in their tertiary structure. β-Sheets are usually flanked with specific amino acid residues (gatekeepers) preventing their aggregation and participation in amyloidogenesis [138]. Nevertheless, the inner β-structures of proteins can be exposed sporadically (more often at mature age). As C. Dobson thinks, this can lead to appearance of nonspecific amyloidoses [139, 140], or we suppose that this can be a manner of involving into amyloid structure of “usual” proteins unable to undergo amyloidogenesis.
TP I and TP II are realized with mutual dependence. Thus, the TP I ambiguity differently influences the structure of proteins and, hence, TP II. Potential mobility of prionogenic regions was many times demonstrated experimentally: the fusion of the prionogenic region of protein Sup35 with a “usual” protein made it capable to form prion [141, 142]. Such translocation of prionogenic regions could be possible (however, not detected up to now) due to chromosome rearrangements providing evolution of amyloidome. This could also occur in the case of regions responsible for formation of noninfectious amyloids. It seems that infectious and noninfectious amyloids have more features in common than generally believed [143].
Some examples of the influence of TP II on TP I are known, in particular, in cases of prionization of translation and transcription factors (Table 3) that lead to changes in the expression of many genes. Nonrandom changes in the expression (transcription) of 314 genes are shown in Alzheimer’s disease [144], but this effect can be due not only to aggregation of the Aβ peptide, but also by other causes. The chromosome disjunction frequency in somatic cells is increased in Alzheimer’s disease [145]. Undoubtedly, examples of pleotropic manifestations of amyloidoses will increase.
Thus, it becomes more obvious that amyloid structures initially described as pathologic are widely distributed and can present functional variants of the quaternary structure of proteins. Moreover, the data obtained show the existence of prion and amyloid networks with structure and dynamics provided for by conformational templates. These networks and their biological role are now under study using new methods of theoretical and experimental amyloidomics.
The authors are grateful to Dr. A. P. Galkin (St. Petersburg State University and Institute of General Genetics, Russian Academy of Sciences) for his attentive reading the manuscript.
This work was supported by the grant of the President of the Russian Federation (MK-4854.2015.4), the Russian Foundation for Basic Research (project No. 14-04-31838), and the St. Petersburg Government. The authors acknowledge St. Petersburg State University for research grants (1.50.2543.2013 and 0.37.696.2013).
REFERENCES
1.Sipe, J. D., and Cohen, A. S. (2000) Review:
history of amyloid fibril, J. Struct. Biol., 130,
88-89.
2.Buxbaum, J. N., and Linke, R. P. (2000) A molecular
history of the amyloidoses, J. Mol. Biol., 421,
142-159.
3.Sipe, J. D., Benson, M. D., Buxbaum, J. N., Ikeda,
S., Merlini, G., Saraiva, M. J., and Westermark, P. (2012) Amyloid
fibril protein nomenclature: 2012 recommendations from the Nomenclature
Committee of the International Society of Amyloidosis, Amyloid,
19, 167-170.
4.Sipe, J. D., Benson, M. D., Buxbaum, J. N., Ikeda,
S., Merlini, G., Saraiva, M. J., and Westermark, P. (2014) Nomenclature
2014: amyloid fibril proteins and clinical classification of
amyloidosis, Amyloid, 21, 221-224.
5.Eanes, E. D., and Glenner, J. (1968) X-Ray
diffraction studies on amyloid filaments, Histochem. Cytochem.,
16, 673-677.
6.Wickner, R. B., Edskes, H. K., Bateman, D. A.,
Kelly, A. C., Gorkovskiy, A., Dayani, Y., and Zhou, A. (2013) Amyloids
and yeast prion biology, Biochemistry, 52, 1514-1527.
7.Tycko, R., and Wickner, R. B. (2013) Amyloid
structure: conformational diversity and consequences, Acc. Chem.
Res., 46, 1487-1496.
8.Toyama, B. H., and Weissman, J. S. (2011) Amyloid
structure: conformational diversity and consequences, in Annual
Review of Biochemistry, Vol. 80 (Kornberg, R. D., Raetz, C. R. H.,
Rothman, J. E., and Thorner, J. W., eds.), pp. 557-585.
9.Qiang, W., Yau, W. M., Luo, Y. Q., Mattson, M. P.,
and Tycko, R. (2012) Antiparallel β-sheet architecture in
Iowa-mutant β-amyloid fibrils, Proc. Natl. Acad. Sci. USA,
109, 4443-4448.
10.Tycko, R., Sciarretta, K. L., Orgel, J., and
Meredith, S. C. (2009) Evidence for novel β-sheet structures in
Iowa-mutant β-amyloid fibrils, Biochemistry, 48,
6072-6084.
11.Lansbury, P. T., Costa, P. R., Griffiths, J. M.,
Simon, E. J., Auger, M., Halverson, K. J., Kocisko, D. A., Hendsch, Z.
S., Ashburn, T. T., Spencer, R. G. S., Tidor, B., and Griffin, R. G.
(1995) Structural model for the β-amyloid fibril based on
interstrand alignment of an antiparallel-sheet comprising a C-terminal
peptide, Nat. Struct. Biol., 2, 990-998.
12.Nielsen, J. T., Bjerring, M., Jeppesen, M. D.,
Pedersen, R. O., Pedersen, J. M., Hein, K. L., Vosegaard, T.,
Skrydstrup, T., Otzen, D. E., and Nielsen, N. C. (2009) Unique
identification of supramolecular structures in amyloid fibrils by solid
state NMR spectroscopy, Angew. Chem. Int. Ed. Engl.,
48, 2118-2121.
13.Van Melckebeke, H., Wasmer, C., Lange, A., Eiso,
A. B., Loquet, A., Bockmann, A., and Meier, B. H. (2010)
Atomic-resolution three-dimensional structure of Het-s(218-289) amyloid
fibrils by solid state NMR spectroscopy, J. Am. Chem. Soc.,
132, 13765-13775.
14.Sunde, M., Serpell, L. C., Bartlam, M., Fraser,
P. E., Pepys, M. B., and Blake, C. C. (1997) Common core structure of
amyloid fibrils by synchrotron X-ray diffraction, J. Mol. Biol.,
273, 729-739.
15.Fitzpatrick, A. W., Debelouchina, G. T., Bayro,
M. J., Clare, D. K., Caporini, M. A., Bajaj, V. S., Jaroniec, C. P.,
Wang, L., Ladizhansky, V., Muller, S. A., MacPhee, C. E., Waudby, C.
A., Mott, H. R., De Simone, A., Knowles, T. P., Saibil, H. R.,
Vendruscolo, M., Orlova, E. V., Griffin, R. G., and Dobson, C. M.
(2013) Atomic structure and hierarchical assembly of a cross-β
amyloid fibril, Proc. Natl. Acad. Sci. USA, 110,
5468-5473.
16.Bennhold, H. (1922) Specific staining of amyloid
by Congo Red, Muench. Medizin. Wochensch., 69, 1537-1538
(German).
17.Steensma, D. P. (2001) Congo Red, Arch. Path.
Lab. Med., 125, 250-252.
18.Divry, P., and Florkin, M. (1927) Sur les
prcprietes optiques de l’amyloide, Compt. rend. Soc.
Biol., 97, 1808-1810 (French).
19.Eanes, E. D., and Glenner, G. G. (1968) X-Ray
diffraction studies on amyloid filaments, J. Histochem.
Cytochem., 16, 673-677.
20.Meyer, R. K., McKinley, M. P., Bowman, K. A.,
Braunfeld, M. B., Barry, R. A., and Prusiner, S. B. (1986) Separation
and properties of cellular and scrapie prion proteins, Proc. Natl.
Acad. Sci. USA, 83, 2310-2314.
21.Kryndushkin, D. S., Alexandrov, I. M.,
Ter-Avanesyan, M. D., and Kushnirov, V. V. (2003) Yeast
[PSI+] prion aggregates are formed by small Sup35
polymers fragmented by Hsp104, J. Biol. Chem., 278,
49636-49643.
22.Alberti, S., Halfmann, R., King, O., Kapila, A.,
and Lindquist, S. (2009) A systematic survey identifies prions and
illuminates sequence features of prionogenic proteins, Cell,
137, 146-158.
23.Vassar, P. S., and Culling, C. F. (1959)
Fluorescent stains, with special reference to amyloid and connective
tissues, Arch. Pathol., 68, 487-498.
24.Hobbs, J. R., and Morgan, A. D. (1963)
Fluorescence microscopy with thioflavin-T in the diagnosis of amyloid,
J. Pathol. Bacteriol., 86, 437-442.
25.Naiki, H., Higuchi, K., Hosokawa, M., and Takeda,
T. (1989) Fluorometric determination of amyloid fibrils in vitro
using the fluorescent dye thioflavin T1, Anal. Biochem.,
177, 244-249.
26.Schwartz, P. (1967) New patho-anatomic
observations on amyloidosis in the aged. Fluorescence microscopic
observations, in Proc. Symp. on Amyloidosis (Mandema, E.,
Ruinen, L., Scholten, J. H., and Cohen, A. S., eds.) Excerpta Medica,
Amsterdam, pp. 400-417.
27.Kyle, R. (2001) Amyloidosis: a convoluted story,
Br. J. Haematol., 114, 529-538.
28.Fonteyn, N. (1639) Responsionum et Curationum
Medicinalium, Amstelodami, Amsterdam.
29.Bartholin, T. (1641) Historiarum Anatomicarum
Rariorum, Apud Johannem Henrici, Amsterdam.
30.Rokitansky, C. (1842) Handbuch der Speciellen
Pathologischen Anatomica, Braumuller und Seidel, Vienna.
31.Schneider, K., Fangerau, H., Michaelsen, B., and
Raab, W. H.-M. (2008) The early history of the transmissible spongiform
encephalopathies exemplified by scrapie, Brain Res. Bull.,
77, 343-355.
32.Comber, T. (1772) Real Improvements in
Agriculture, London.
33.Gajdusek, D. C. (1967) Slow-virus infections of
the nervous system, New Engl. J. Med., 276, 392-400.
34.Brown, D. R. (ed.) (2005) Neurodegeneration
and Prion Disease, Springer, New York.
35.Wickner, R. B. (2005) Scrapie in ancient China?
Science, 309, 874.
36.Li, P., and Xing, H. (2006) Disease but no sheep,
Science, 311, 1867.
37.Zigas, V., and Gajdusek, D. C. (1959) Kuru:
clinical, pathological and epidemiological study of a recently
discovered acute progressive degenerative disease of the central
nervous system reaching epidemic proportions among natives of the
Eastern Highlands of New Guinea, PNG Med. J., 3,
1-31.
38.Kelly, J. J., Jr. (1987) Amyloidosis, in
Polyneuropathies Associated with Plasma Cell Dyscrasias (Kelly,
J. J., Jr., Kyle, R. A., and Latov, N., eds.), Topics in the
Neurosciences, Vol. 5, pp. 105-127.
39.Virchow, R. (1854) Ueber eine im Gehirn und
Ruckenmark des Menschen aufgefunde Substanz mit der chemishen Reaction
der Cellulose, Virchows Arch. Path. Anat., 6,
135-138.
40.Colin, J. J., and de Claubry, H. F. (1914) Sur
les combinaisons de l’iode avec les substances vegetales et
animals, Ann. Chimie, 90, 87-100.
41.Tanskanen, M. (2013) “Amyloid”
– Historical Aspects, InTech, Rijeka.
42.Niewold, T. A., Flores Landeira, J. M., van den
Heuvel, L. P., Ultee, A., Tooten, P. C., and Veerkamp, J. H. (1991)
Characterization of proteoglycans and glycosaminoglycans in bovine
renal AA-type amyloidosis, Virchows Arch. B Cell. Pathol. Incl. Mol.
Pathol., 60, 321-328.
43.Snow, A. D., Sekiguchi, R., Nochlin, D., Fraser,
P., Kimata, K., Mizutani, A., Arai, M., Schreier, W. A., and Morgan, D.
G. (1994) An important role of heparan sulfate proteoglycan (Perlecan)
in a model system for the deposition and persistence of fibrillar A
beta-amyloid in rat brain, Neuron, 12, 219-234.
44.Snow, A. D., Willmer, J., and Kisilevsky, R.
(1987) Sulfated glycosaminoglycans: a common constituent of all
amyloids? Lab. Invest., 56, 120-123.
45.Putchler, H., Sweat, F., and Levine, M. (1962) On
the binding of Congo Red by amyloid, J. Histochem. Cytochem.,
10, 355-364.
46.Putchler, H., and Sweat, F. (1965) Congo Red as a
stain for fluorescence microscopy of amyloid, J. Histochem.
Cytochem., 13, 693.
47.Linke, K. (2000) Highly sensitive diagnosis of
amyloid and various amyloid syndromes using Congo Red fluorescence,
Virchows Arch., 436, 439-448.
48.Kitamoto, T., Tateishi, J., Hikita, K., Nagara,
H., and Takeshita, I. (1985) A new method to classify amyloid fibril
proteins, Acta Neuropathol., 67, 272-278.
49.Wright, J. R., Calkins, E., and Humphrey, R. L.
(1977) Potassium permanganate reaction in amyloidosis, Lab.
Invest., 36, 274-281.
50.Tashima, T., Kitamoto, T., and Tateishi, J.
(1986) Histochemical classification of systemic amyloid fibril
proteins: alkaline guanidine method, Arch. Pathol. Lab.,
110, 885-888.
51.Carnes, W. H., and Forker, B. R. (1956)
Metachromasia of amyloid; a spectrophotometric study with particular
reference to the dye–chromotrope bond, Lab. Invest.,
5, 21-43.
52.Elghetany, M. T., and Saleem, A. (1988) Methods
for staining amyloid in tissues: a review, Stain Technol.,
63, 201-212.
53.LeVine, H., 3rd. (1999) Quantification of
beta-sheet amyloid fibril structures with thioflavin T, Methods
Enzymol., 309, 274-284.
54.Klunk, W. E., Pettegrew, J. W., and Abraham, D.
J. (1989) Quantitative evaluation of Congo Red binding to amyloid-like
proteins with a beta-pleated sheet conformation, J. Histochem.
Cytochem., 37, 1273-1281.
55.LeVine, H., 3rd. (1993) Thioflavin-T interaction
with synthetic Alzheimer’s disease beta-amyloid peptides:
detection of amyloid aggregation in solution, Protein Sci.,
2, 404-410.
56.Cloe, A. L., Orgel, J. P. R. O., Sachleben, J.
R., Tycko, R., and Meredith, S. C. (2011) The Japanese mutant Aβ
(ΔE22-Aβ1-39) forms fibrils instantaneously, with
low-thioflavin T fluorescence: seeding of wild-type Aβ1-40 into
atypical fibrils by ΔE22-Aβ1-39, Biochemistry,
50, 2026-2039.
57.Linke, R. P. (2102) On typing amyloidosis using
immunohistochemistry. Detailed illustrations, review and a note on mass
spectrometry, Progr. Histochem. Cytochem., 47,
61-132.
58.Greiner, E. R., Kelly, J. W., and
Palhano, F. L. (2014) Immunoprecipitation of amyloid fibrils by the use
of an antibody that recognizes a generic epitope common to amyloid
fibrils, PLoS One, 9, e105433.
59.Mitkevich, O. V., Kochneva-Pervukhova,
N. V., Surina, E. R., Benevolensky, S. V., Kushnirov, V.
V., and Ter-Avanesyan, M. D. (2012) DNA aptamers detecting generic
amyloid epitopes, Prion, 6, 400-406.
60.Chang, W. M., Dakanali, M., Capule, C. C.,
Sigurdson, C. J., Yang, J., and Theodorakis, E. A. (2011) ANCA: a
family of fluorescent probes that bind and stain amyloid plaques in
human tissue, ACS Chem. Neurosci., 2, 249-255.
61.Schmued, L., Raymick, J., Tolleson, W., Sarkar,
S., Zhang, Y. H., and Bell-Cohn, A. (2012) Introducing Amylo-Glo, a
novel fluorescent amyloid specific histochemical tracer especially
suited for multiple labeling and large scale quantification studies,
J. Neurosci. Methods, 209, 120-126.
62.Sethi, S., Vrana, J. A., Theis, J. D., Leung, N.,
Sethi, A., Nasr, S. H., Fervenza, F. C., Cornell, L. D., Fidler, M. E.,
and Dogan, A. (2012) Laser microdissection and mass spectrometry-based
proteomics aids the diagnosis and typing of renal amyloidosis,
Kidney Int., 82, 226-234.
63.Cohen, A. S., and Calkins, E. (1959) Electron
microscopic observation on a fibrous component in amyloid of diverse
origins, Nature, 183, 1202-1203.
64.Cohen, A. S., Shirahama, T., and Skinner, M.
(1982) Electron microscopy of amyloid, in Electron Microscopy of
Proteins (Harris, J. R., ed.) Academic Press, New York.
65.Shirahama, T., and Cohen, A. S. (1967)
High-resolution electron microscopic analysis of the amyloid fibril,
J. Cell Biol., 33, 679-708.
66.Pras, M., Schubert, M., Zucker-Franklin, D.,
Rimon, A., and Franklin, E. C. (1968) The characterization of soluble
amyloid prepared in water, J. Clin. Invest., 47,
924-933.
67.Bonar, L., Cohen, A. S., and Skinner, M. M.
(1969) Characterization of the amyloid fibril as a cross-beta protein,
Proc. Soc. Exp. Biol. Med., 131, 1373-1375.
68.Glenner, G. G., Eanes, E. D., Bladen, H. A.,
Linke, R. P., and Termine, J. D. (1974) Beta-pleated sheet fibrils. A
comparison of native amyloid with synthetic protein fibrils, J.
Histochem. Cytochem., 22, 1141-1158.
69.Sunde, M., and Blake, C. C. F. (1998) From the
globular to the fibrous state: protein structure and structural
conversion in amyloid formation, Q. Rev. Biophys., 31,
1-39.
70.Jimenez, J. L., Nettleton, E. J., and Saibil, H.
R. (2002) The protofilament structure of insulin amyloid fibrils,
PNAS, 99, 9196-9201.
71.Goldsbury, C., Goldie, K., Pellaud, J., Seelig,
J., Frey, P., Muller, S. A., Kistler, J., Cooper, G. J., and Aebi, U.
(2000) Amyloid fibril formation from full-length and fragments of
amylin, J. Struct. Biol., 130, 352-362.
72.Goldsbury, C. S., Wirtz, S., Muller, S. A.,
Sunderji, S., Wicki, P., Aebi, U., and Frey, P. (2000) Studies on the
in vitro assembly of Aβ1-40: implications for the search
for Aβ fibril formation inhibitors, J. Struct. Biol.,
130, 217-231.
73.Mizuno, N., Baxa, U., and Steven, A. C. (2011)
Structural dependence of HET-s amyloid fibril infectivity assessed by
cryoelectron microscopy, PNAS, 108, 3252-3257.
74.Benzinger, T. L. S., Gregory, D. M., Burkoth, T.
S., Miller-Auer, H., Lynn, D. G., Botto, R. E., and Meredith, S. C.
(1998) Propagating structure of Alzheimer’s β-amyloid(10-35)
is parallel β-sheet with residues in exact register, PNAS,
95, 13407-13412.
75.Petkova, A. T., Yau, W. M., and Tycko, R. (2006)
Experimental constraints on quaternary structure in Alzheimer’s
β-amyloid fibrils, Biochemistry, 45, 498-512.
76.Paravastu, A. K., Leapman, R. D., Yau, W. M., and
Tycko, R. (2008) Molecular structural basis for polymorphism in
Alzheimer’s β-amyloid fibrils, PNAS, 105,
18349-18354.
77.Smaoui, M. R., Poitevin, F., Delarue, M., Koehl,
P., Orland, H., and Waldispuhl, J. (2013) Computational assembly of
polymorphic amyloid fibrils reveals stable aggregates, Biophys.
J., 104, 683-693.
78.Inoue, S., Kuroiwa, M., Saraiva, M. J.,
Guimaraes, A., and Kisilevsky, R. (1998) Ultrastructure of familial
amyloid polyneuropathy amyloid fibrils: examination with
high-resolution electron microscopy, J. Struct. Biol.,
124, 1-12.
79.Cohen, A. S., and Calkins, E. (1964) The
isolation of amyloid fibrils and a study of the effect of collagenase
and hyaluronidase, J. Cell. Biol., 21, 481-486.
80.Glenner, G. G., Terry, W., Harada, M., Isersky,
C., and Page, D. (1971) Amyloid fibrils proteins: proof of homology
with immunoglobulin light chains by sequence analysis, Science,
172, 1150-1153.
81.Benditt, E. P., Eriksen, N., Hermodson, M. A.,
and Ericsson, L. H. (1971) The major proteins of human and monkey
amyloid substance: common properties including unusual N-terminal amino
acid sequences, FEBS Lett., 19, 169-173.
82.Benditt, E. P. (1976) The structure of amyloid
protein AA and evidence for a transmissible factor in the origin of
amyloidosis, in Amyloidosis (Wegelius, O., and Pasternack, A.,
eds.) Academic Press, London, pp. 323-337.
83.Costa, P. P., Figueira, A. S., and Bravo, F. R.
(1978) Amyloid fibril protein related to prealbumin in familial
amyloidotic polyneuropathy, PNAS, 75, 4499-4503.
84.Skinner, M., and Cohen, A. S. (1981) The
prealbumin nature of the amyloid protein in familial amyloid
polyneuropathy (FAP), Biochem. Biophys. Res. Commun., 99,
1326-1332.
85.Selkoe, D. J., Ihara, Y., and Salazar, F. J.
(1982) Alzheimer’s disease: insolubility of partially purified
paired helical filaments in sodium dodecyl sulfate and urea,
Science, 215, 1243-1245.
86.Selkoe, D. J., Abraham, C. R., Podlisny, M. B.,
and Duffy, L. K. (1986) Isolation of low-molecular-weight proteins from
amyloid plaque fibers in Alzheimer’s disease, J.
Neurochem., 46, 1820-1834.
87.Selkoe, D. J., lhara, Y., Abraham, C., Rasool, C.
G., and McCluskey, A. H. (1983) Biochemical and immunocytochemical
studies of Alzheimer paired helical filaments, in Banbury: Report
15: Biological Aspects of Alzheimer’s Disease (Katzman, R.,
ed.) Cold Spring Harbor, New York, pp. 125-134.
88.Yen, S.-H., and Kress, Y. (1983) The effect of
chemical reagents or proteases on the ultrastructure of paired helical
filaments, in Banbury: Report 15: Biological Aspects of
Alzheimer’s Disease (Katzman, R., ed.) Cold Spring Harbor,
New York, pp. 155-165.
89.Bolton, D. C., McKinley, M. P., and Prusiner, S.
B. (1982) Identification of a protein that purifies with the scrapie
prion, Science, 218, 1309-1311.
90.Bagriantsev, S. N., Kushnirov, V. V., and
Liebman, S. W. (2006) Analysis of amyloid aggregates using agarose gel
electrophoresis, Methods Enzymol., 412, 33-48.
91.Nevzglyadova, O. V., Artemov, A. V., Mittenberg,
A. G., Solovyov, K. V., Kostyleva, E. I., Mikhailova, E. V.,
Kuznetsova, I. M., Turoverov, K. K., and Soidla, T. R. (2009)
Prion-associated proteins in yeast: comparative analysis of isogenic
[PSI(+)] and [psi(–)] strains, Yeast,
26, 611-631.
92.Michelitsch, M. D., and Weissman, J. S. (2000) A
census of glutamine/asparagine-rich regions: implications for their
conserved function and the prediction of novel prions, PNAS,
97, 11910-11915.
93.Harrison, P. M., and Gerstein, M. (2003) A method
to assess compositional bias in biological sequences and its
application to prion-like glutamine/asparagine-rich domains in
eukaryotic proteomes, Genome Biol., 4, R40.
94.Antonets, K. S., and Nizhnikov, A. A. (2013) A
novel algorithm to assess compositional biases in protein sequences,
Evol. Bioinform., 9, 263-273.
95.Conchillo-Sole, O., de Groot, N. S., Aviles, F.
X., Vendrell, J., Daura, X., and Ventura, S. (2007) AGGRESCAN: a server
for the prediction and evaluation of “hot spots” of
aggregation in polypeptides, BMC Bioinform., 8, 65.
96.Maurer-Stroh, S., Debulpaep, M., Kuemmerer, N.,
Lopez de la Paz, M., Martins, I. C., Reumers, J., Morris, K. L.,
Copland, A., Serpell, L., Serrano, L., Schymkowitz, J. W., and
Rousseau, F. (2010) Exploring the sequence determinants of amyloid
structure using position-specific scoring templates, Nat.
Methods, 7, 237-242.
97.Tsolis, A. C., Papandreou, N. C., Iconomidou, V.
A., and Hamodrakas, S. J. (2013) A consensus method for the prediction
of “aggregation-prone” peptides in globular proteins,
PLoS One, 8, e54175.
98.Kryndushkin, D., Pripuzova, N., Burnett, B. G.,
and Shewmaker, F. (2013) Non-targeted identification of prions and
amyloid-forming proteins from yeast and mammalian cells, J. Biol
Chem., 288, 27100-27111.
99.Nizhnikov, A. A., Alexandrov, A. I., Ryzhova, T.
A., Mitkevich, O. V., Dergalev, A. A., Ter-Avanesyan, M. D., and
Galkin, A. P. (2014) Proteomic screening for amyloid proteins, PLoS
One, 9, e116003.
100.Kushnirov, V. V., Alexandrov, I. M., Mitkevich,
O. V., Shkundina, I. S., and Ter-Avanesyan, M. D. (2006) Purification
and analysis of prion and amyloid aggregates, Methods,
39, 50-55.
101.DiFiglia, M., Sapp, E., Chase, K. O., Davies,
S. W., Bates, G. P., Vonsattel, J. P., and Aronin, N. (1997)
Aggregation of huntingtin in neuronal intranuclear inclusions and
dystrophic neurites in brain, Science, 277,
1990-1993.
102.Ueda, K., Fukushima, H., Masliah, E., Xia, Y.,
Iwai, A., Yoshimoto, M., Otero, D. A., Kondo, J., Ihara, Y., and
Saitoh, T. (1993) Molecular cloning of cDNA encoding an unrecognized
component of amyloid in Alzheimer disease, PNAS, 90,
11282-11286.
103.Grundke-Iqbal, I., Iqbal, K., Quinlan, M.,
Tung, Y. C., Zaidi, M. S., and Wisniewski, H. M. (1986)
Microtubule-associated protein tau. A component of Alzheimer paired
helical filaments, J. Biol. Chem., 261, 6084-6089.
104.Meetoo, D., McGovern, P., and Safadi, R. (2007)
An epidemiological overview of diabetes across the world, Br. J.
Nurs., 16, 1002-1007.
105.Brookmeyer, R., Evans, D. A., Hebert, L.,
Langa, K. M., Heeringa, S. G., Plassman, B. L., and Kukull, W. A.
(2011) National estimates of the prevalence of Alzheimer’s
disease in the United States, Alzheimers Dement., 7,
61-73.
106.Iconomidou, V. A., Vriend, G., and Hamodrakas,
S. J. (2000) Amyloids protect the silkmoth oocyte and embryo, FEBS
Lett., 479, 141-145.
107.Fowler, D. M., Koulov, A. V., Alory-Jost, C.,
Marks, M. S., Balch, W. E., and Kelly, J. W. (2006) Functional amyloid
formation within mammalian tissue, PLoS Biol., 4, e6.
108.Maji, S. K., Perrin, M. H., Sawaya, M. R.,
Jessberger, S., Vadodaria, K., Rissman, R. A., Singru, P. S., Nilsson,
K. P., Simon, R., Schubert, D., Eisenberg, D., Rivier, J., Sawchenko,
P., Vale, W., and Riek, R. (2009) Functional amyloids as natural
storage of peptide hormones in pituitary secretory granules,
Science, 325, 328-332.
109.Chimileski, S., Franklin, M. J., and Papke, R.
T. (2014) Biofilms formed by the archaeon Haloferax volcanii
exhibit cellular differentiation and social motility, and facilitate
horizontal gene transfer, BMC Biol., 12, 65.
110.Coustou, V., Deleu, C., Saupe, S., and
Begueret, J. (1997) The protein product of the het-s
heterokaryon incompatibility gene of the fungus Podospora
anserina behaves as a prion analog, PNAS, 94,
9773-9778.
111.Maddelein, M. L., Dos Reis, S., Duvezin-Caubet,
S., Coulary-Salin, B., and Saupe, S. J. (2002) Amyloid aggregates of
the HET-s prion protein are infectious, PNAS, 99,
7402-7407.
112.Wickner, R. B. (1994) [URE3] as an
altered Ure2 protein: evidence for a prion analog in Saccharomyces
cerevisiae, Science, 264, 566-569.
113.Wickner, R., Masison, D. C., and Edskes, H. K.
(1995) [PSI] and [URE3] as yeast prions, Yeast,
11, 1671-1685.
114.Inge-Vechtomov, S. G. (2000) Yeast prions and
the Central dogma of molecular biology, Vestnik Ross. Akad.
Nauk, 70, 195-202.
115.Inge-Vechtomov, S. G. (2003) Template principle
in biology (the past, present, future?) Ekol. Genet., 1,
6-15.
116.Koltsov, N. K. (1936) Hereditary molecules, in
Organization of the Cell [in Russian], Gos. Izdat. Biol. Med.
Lit., pp. 585-620.
117.Crick, F. H. C. (1958) On protein synthesis,
Symp. Soc. Exptl. Biol., 12, 138-163.
118.Crick, F. H. C. (1970) Central dogma of
molecular biology, Nature, 227, 561-563.
119.Paushkin, S. V., Kushnirov, V. V., Smirnov, V.
N., and Ter-Avanesyan, M. D. (1997) In vitro propagation of the
prion-like state of yeast Sup35 protein, Science, 277,
381-383.
120.Sparrer, H. E., Santoso, A., Szoka, F. C., and
Weissman, J. S. (2000) Evidence for the prion hypothesis: induction of
the yeast [PSI+] factor by in vitro-converted
Sup35 protein, Science, 289, 595-599.
121.Tanaka, M., Chien, P., Naber, N., Cooke, R.,
and Weissman, J. S. (2004) Conformational variations in an infectious
protein determine prion strain differences, Nature, 428,
323-328.
122.Fink, G. (2005) A transformation principle,
Cell, 120, 153-154.
123.Inge-Vechtomov, S. G. (2013) The template
principle: paradigm of modern genetics, Russ. J. Genet.,
49, 4-9.
124.Inge-Vechtomov, S. G. (2015) From chromosome
theory to the template principle, Russ. J. Genet., 51,
323-333.
125.Chernoff, Y. O., Lindquist, S. L., Ono, B.,
Inge-Vechtomov, S. G., and Liebman, S. W. (1995) Role of the chaperone
protein Hsp104 in propagation of the yeast prion-like factor
[PSI+], Science, 268, 880-884.
126.Paushkin, S. V., Kushnirov, V. V., Smirnov, V.
N., and Ter-Avanesyan, M. D. (1996) Propagation of the yeast prion-like
[psi+] determinant is mediated by oligomerization of the
SUP35-encoded polypeptide chain release factor, EMBO J.,
15, 3127-3134.
127.Kushnirov, V. V., and Ter-Avanesyan, M. D.
(1998) Structure and replication of yeast prions, Cell,
94, 13-16.
128.Liebman, S. W., and Chernoff, Y. O. (2012)
Prions in yeast, Genetics, 191, 1041-1072.
129.Krishnan, R., and Lindquist, S. (2005)
Structural insights into yeast prion illuminate nucleation and strain
diversity, Nature, 435, 765-772.
130.Tanaka, M., Collins, S. R., Tayama, B. H., and
Weissman, J. S. (2006) The physical basis of how prion conformation
determine strain phenotypes, Nature, 442, 585-589.
131.Foo, C. K., Ohhashi, Y., Kelly, M. J. S.,
Tanaka, M., and Weissman, J. S. (2011) Radically different amyloid
conformation dictate the seeding specificity of a chimeric Sup35 prion,
J. Mol. Biol., 408, 1-8.
132.Raychaudhuri, R., Lomakin, A., Bernstein, S.,
Zheng, X., Condron, M. M., Benedek, G. B., Bowers, M., and Teplow, D.
B. (2014) Gly25-Ser26 amyloid β-protein structural isomorphs
produce distinct Aβ42 conformational dynamics and assembly
characteristics, J. Mol. Biol., 426, 2422-2441.
133.Hoefgen, S., Dahms, S., Oertwig, K., and Than,
M. (2015) The amyloid precursor protein shows a pH-dependent
conformational switch in its E1 domain, J. Mol. Biol.,
427, 433-442.
134.Satput-Krishnan, P., and Serio, T. R. (2005)
Prion protein remodeling confers an immediate phenotypic switch,
Nature, 437, 262-265.
135.Derkatch, I. L., Bradley, M. E., Masse, S.,
Zadorsky, S. P., Polozkov, G. V., Inge-Vechtomov, S. G., and Liebman,
S. W. (2000) Dependence and independence of [PSI+]
and [PIN+]: a two prion system in yeast? EMBO
J., 19, 1942-1952.
136.Derkatch, I. L., Bradley, M. E., Hong, J. Y.,
and Liebman, S. W. (2001) Prions affect the appearance of other prions.
The story of [PIN+], Cell, 106,
171-182.
137.Sarell, C. J., Stockley, P. G., and Radford, S.
E. (2013) Assessing the causes and consequences of co-polymerization in
amyloid formation, Prion, 7, 359-368.
138.De Baets, G., Van Dare, J., Rousseau, F., and
Schymkowitz, J. (2014) A genome-wide sequence-structure analysis
suggests aggregation gate-keepers constitution evolutionary constrained
functional class, J. Mol. Biol., 426, 2405-2412.
139.Dobson, C. M. (2013) The amyloid phenomenon and
its significance, in Amyloid Fibrils and Prefibrillar Aggregates:
Molecular and Biological Properties (Otzen, D., ed.) Wiley-VCH.
140.Schnabel, J. (2010) Protein folding: the dark
side of proteins, Nature, 464, 828-829.
141.Borkhsenius, A. S., Sasnauskas, K., Gedvilaite,
A., and Inge-Vechtomov, S. G. (2002) Chimeric yeast prions with
unstable inheritance, Genetika, 38, 300-305.
142.Pogoda, A. A., Alenin, V. V., Borkhsenius, A.
S., Zadorsky, S. P., Manukhov, V. V., and Inge-Vechtomov, S. G. (2010)
[PIN+]-dependent induction of protease-resistant
amyloids by Ade2p proteins fused with prionizing NM domain of Sup35
protein of yeast Saccharomyces cerevisiae, Dokl. Biochem.
Biophys., 433, 183-186.
143.Rick, R. (2006) Infectious Alzheimer disease,
Nature, 444, 429-430.
144.Ricciarelly, R., d’Abramo, C., Massone,
S., Marinardi, U. M., Pronzato, M. A., and Tabaton, M. (2004)
Microarray analysis in Alzheimer’s disease and normal aging,
IUBMB Life, 56, 349-354.
145.Granic, A., Padmanabham, J., Norden, M., and
Potter, H. (2010) Alzheimer Aβ peptide induces chromosome
mis-segregation and aneuploidy, including trisomy 21: requirement for
tau and APP, Mol. Biol. Cell, 21, 511-520.
146.Westermark, P. (1975) Amyloid of medullary
carcinoma of the thyroid: partial characterization, Upsala J. Med.
Sci., 80, 88-92.
147.Sletten, K., Westermark, P., and Natvig, J. B.
(1976) Characterization of amyloid fibril proteins from medullary
carcinoma of the thyroid, J. Exp. Med., 143, 993-998.
148.Cohen, D. H., Feiner, H., Jensson, O., and
Frangione, B. (1983) Amyloid fibril in hereditary cerebral hemorrhage
with amyloidosis (HCHWA) is related to the gastroentero-pancreatic
neuroendocrine protein, gamma trace, J. Exp. Med., 158,
623-628.
149.Gejyo, F., Yamada, T., Odani, S., Nakagawa, Y.,
Arakawa, M., Kunitomo, T., Kataoka, H., Suzuki, M., Hirasawa, Y.,
Shirahama, T., Cohen, A. S., and Schmidt, K. (1985) A new form of
amyloid protein associated with chronic hemodialysis was identified as
2-microglobulin, Biochem. Biophys. Res. Commun., 129,
701-706.
150.Westermark, P., Wernstedt, C., Wilander, E.,
and Sletten, K. (1986) A novel peptide in the calcitonin gene related
peptide family as an amyloid fibril protein in the endocrine pancreas,
Biochem. Biophys. Res. Commun., 140, 827-831.
151.Johansson, B., Wernstedt, C., and Westermark,
P. (1987) Atrial natriuretic peptide deposited as atrial amyloid
fibrils, Biochem. Biophys. Res. Commun., 148,
1087-1092.
152.Nichols, W. C., Dwulet, F. E., Liepnieks, J.,
and Benson, M. D. (1988) Variant apolipoprotein AI as a major
constituent of a human hereditary amyloid, Biochem. Biophys. Res.
Commun., 156, 762-768.
153.Dische, F. E., Wernstedt, C., Westermark, G.
T., Westermark, P., Pepys, M. B., Rennie, J. A., Gilbey, S. G., and
Watkins, P. J. (1988) Insulin as an amyloid-fibril protein at sites of
repeated insulin injections in a diabetic patient, Diabetologia,
31, 158-161.
154.Maury, C. P., and Baumann, M. (1990) Isolation
and characterization of cardiac amyloid in familial amyloid
polyneuropathy type IV (Finnish): relation of the amyloid protein to
variant gelsolin, Biochim. Biophys. Acta, 1096,
84-86.
155.Eulitz, M., Weiss, D. T., and Solomon, A.
(1990) Immunoglobulin heavy-chain-associated amyloidosis, PNAS,
87, 6542-6546.
156.Benson, M. D., Liepnieks, J., Uemichi, T.,
Wheeler, G., and Correa, R. (1993) Hereditary renal amyloidosis
associated with a mutant fibrinogen alpha-chain, Nature Genet.,
3, 252-256.
157.Pepys, M. B., Hawkins, P. N., Booth, D. R.,
Vigushin, D. M., Tennent, G. A., Soutar, A. K., Totty, N., Nguyen, O.,
Blake, C. C., Terry, C. J., Feest, T. G., Zalin, T. G., and Hsuan, J.
J. (1993) Human lysozyme gene mutations cause hereditary systemic
amyloidosis, Nature, 362, 553-557.
158.Westermark, P., Eriksson, L., Engstrom, U.,
Enestrom, S., and Sletten, K. (1997) Prolactin-derived amyloid in the
aging pituitary gland, Am. J. Pathol., 150, 67-73.
159.Vidal, R., Frangione, B., Rostagno, A., Mead,
S., Revesz, T., Plant, G., and Ghiso, J. (1999) A stop-codon mutation
in the BRI gene associated with familial British dementia,
Nature, 399, 776-781.
160.Niewold, T. A., Murphy, C. L., Hulskamp-Koch,
C. A., Tooten, P. C., and Gruys, E. (1999) Casein-related amyloid,
characterization of a new and unique amyloid protein isolated from
bovine corpora amylacea, Amyloid, 6, 244-249.
161.Haggqvist, B., Naslund, J., Sletten, K.,
Westermark, G., Mucchiano, G., Tjernberg, L., Nordstedt, C., Engstrom,
U., and Westermark, P. (1999) Medin: an integral fragment of aortic
smooth muscle cell-produced lactadherin forms the most common human
amyloid, PNAS, 96, 8669-8674.
162.Vidal, R., Revesz, T., Rostagno, A., Kim, E.,
Holton, J. L., Bek, T., Bojsen-Møller, M., Braendgaard, H.,
Plant, G., Ghiso, J., and Frangione, B. (2000) A decamer duplication in
the 3′ region of the BRI gene originates an amyloid
peptide that is associated with dementia in a Danish kindred,
PNAS, 97, 4920-4925.
163.Korvatska, E., Henry, H., Mashima, Y., Yamada,
M., Bachmann, C., Munier, F. L., and Schorderet, D. F. (2000) Amyloid
and non-amyloid forms of 5q31-linked corneal dystrophy resulting from
kerato-epithelin mutations at Arg124 are associated with abnormal
turnover of the protein, J. Biol. Chem., 275,
11465-11469.
164.Benson, M. D., Liepnieks, J. J., Yazaki, M.,
Yamashita, T., Hamidi Asl, K., Guenther, B., and Kluve-Beckerman, B.
(2001) A new human hereditary amyloidosis: the result of a stop-codon
mutation in the apolipoprotein AII gene, Genomics, 72,
272-277.
165.Ando, Y., Nakamura, M., Kai, H., Katsuragi, S.,
Terazaki, H., Nozawa, T., Okuda, T., Misumi, S., Matsunaga, N., Hata,
K., Tajiri, T., Shoji, S., Yamashita, T., Haraoka, K., Obayashi, K.,
Matsumoto, K., Ando, M., and Uchino, M. (2002) A novel localized
amyloidosis associated with lactoferrin in the cornea, Lab.
Invest., 82, 757-766.
166.Solomon, A., Murphy, C. L., Weaver, K., Weiss,
D. T., Hrncic, R., Eulitz, M., Donnell, R. L., Sletten, K., Westermark,
G., and Westermark, P. (2003) Calcifying epithelial odontogenic
(Pindborg) tumor-associated amyloid consists of a novel human protein,
J. Lab. Clin. Med., 142, 348-355.
167.Bergstrom, J., Murphy, C. L., Weiss, D. T.,
Solomon, A., Sletten, K., Hellman, U., and Westermark, P. (2004) Two
different types of amyloid deposits – apolipoprotein A-IV and
transthyretin – in a patient with systemic amyloidosis, Lab.
Invest., 84, 981-988.
168.Linke, R. P., Joswig, R., Murphy, C. L., Wang,
S., Zhou, H., Gross, U., Rocken, C., Westermark, P., Weiss, D. T., and
Solomon, J. (2005) Senile seminal vesicle amyloid is derived from
semenogelin I, Lab. Clin. Med., 145, 187-193.
169.Benson, M. D., James, S., Scott, K., Liepnieks,
J. J., and Kluve-Beckerman, B. (2008) Leukocyte chemotactic factor 2: a
novel renal amyloid protein, Kidney Int., 74,
218-222.
170.Murphy, C. L., Kestler, D. P., Foster, J. S.,
Wang, S., Macy, S. D., Kennel, S. J., Carlson, E. R., Hudson, J.,
Weiss, D. T., and Solomon, A. (2008) Odontogenic ameloblast-associated
protein nature of the amyloid found in calcifying epithelial
odontogenic tumors and unerupted tooth follicles, Amyloid,
15, 89-95.
171.Caubet, C., Bousset, L., Clemmensen, O.,
Sourigues, Y., Bygum, A., Chavanas, S., Coudane, F., Hsu, C. Y., Betz,
R. C., Melki, R., Simon, M., and Serre, G. (2010) A new amyloidosis
caused by fibrillar aggregates of mutated corneodesmosin, FASEB
J., 24, 3416-3426.
172.Willander, H., Askarieh, G., Landreh, M.,
Westermark, P., Nordling, K., Keranen, H., Hermansson, E., Hamvas, A.,
Nogee, L. M., Bergman, T., Saenz, A., Casals, C., Aqvistg, J.,
Jornvall, H., Berglund, H., Presto, J., Knight, S. D., and Johansson,
J. (2012) High-resolution structure of a BRICHOS domain and its
implications for anti-amyloid chaperone activity on lung surfactant
protein C, PNAS, 109, 2325-2329.
173.Gharibyan, A. L., Raveh, D., and
Morozova-Roche, L. A. M. (2012) S100A8/A9 amyloidosis in the ageing
prostate: relating ex vivo and in vitro studies, Mol.
Biol., 849, 387-401.
174.Munch, J., Rucker, E., Standker, L., Adermann,
K., Goffinet, C., Schindler, M., Wildum, S., Chinnadurai, R., Rajan,
D., Specht, A., Gimenez-Gallego, G., Sanchez, P. C., Fowler, D. M.,
Koulov, A., Kelly, J. W., Mothes, W., Grivel, J. C., Margolis, L.,
Keppler, O. T., Forssmann, W. G., and Kirchhoff, F. (2007)
Semen-derived amyloid fibrils drastically enhance HIV infection,
Cell, 131, 1059-1071.
175.De Vocht, M. L., Reviakine, I., Wosten, H. A.,
Brisson, A., Wessels, J. G., and Robillard, G. T. (2000) Structural and
functional role of the disulfide bridges in the hydrophobin SC3, J.
Biol. Chem., 275, 28428-28432.
176.Chapman, M. R., Robinson, L. S., Pinkner, J.
S., Roth, R., Heuser, J., Hammar, M., Normark, S., and Hultgren, S. J.
(2002) Role of Escherichia coli curli operons in directing
amyloid fiber formation, Science, 295, 851-855.
177.Si, K., Giustetto, M., Etkin, A., Hsu, R.,
Janisiewicz, A. M., Miniaci, M. C., Kim, J. H., Zhu, H., and Kandel, E.
R. (2003) A neuronal isoform of CPEB regulates local protein synthesis
and stabilizes synapse-specific long-term facilitation in
Aplysia, Cell, 115, 893-904.
178.Claessen, D., Rink, R., de Jong, W., Siebring,
J., de Vreugd, P., Boersma, F. G. H., Dijkhuizen, L., and Wosten, H. A.
(2003) A novel class of secreted hydrophobic proteins is involved in
aerial hyphae formation in Streptomyces coelicolor by forming
amyloid-like fibrils, Genes Dev., 17, 1714-1726.
179.Bieler, S., Estrada, L., Lagos, R., Baeza, M.,
Castilla, J., and Soto, C. (2005) Amyloid formation modulates the
biological activity of a bacterial protein, J. Biol. Chem.,
280, 26880-26885.
180.Oh, J., Kim, J.-G., Jeon, E., Yoo, C.-H., Moon,
J. S., Rhee, S., and Hwang, I. (2007) Amyloidogenesis of type
III-dependent hairpins from plant pathogenic bacteria, J. Biol.
Chem., 282, 13601-13609.
181.Alteri, C. J., Xicohtencatl-Cortes, J., Hess,
S., Caballero-Olin, G., Giron, J. A., and Friedman, R. L. (2007)
Mycobacterium tuberculosis produces pili during human infection,
PNAS, 104, 5145-5150.
182.Wang, R., Braughton, K. R., Kretschmer, D.,
Bach, T.-H. L., Queck, S. Y., Li, M., Kennedy, A. D., Dorward, D. W.,
Klebanoff, S. J., Peschel, A., DeLeo, F. R., and Otto, M. (2007)
Identification of novel cytolytic peptides as key virulence
determinants for community-associated MRSA, Nat. Med.,
13, 1510-1514.
183.Dueholm, M. S., Petersen, S. V., Sonderkaer,
M., Larsen, P., Christiansen, G., Hein, K. L., Enghild, J. J., Nielsen,
J. L., Nielsen, K. L., Nielsen, P. H., and Otzen, D. E. (2010)
Functional amyloid in Pseudomonas, Mol. Microbiol.,
77, 1009-1020.
184.Romero, D., Aguilar, C., Losick, R., and
Kolter, R. (2010) Amyloid fibers provide structural integrity to
Bacillus subtilis biofilms, PNAS, 107,
2230-2234.
185.Majumdar, A., Cesario, W. C., White-Grindley,
E., Jiang, H., Ren, F., Khan, M. R., Li, L., Choi, E. M., Kannan, K.,
Guo, F., Unruh, J., Slaughter, B., and Si, K. (2012) Critical role of
amyloid-like oligomers of Drosophila Orb2 in the persistence of
memory, Cell, 148, 515-529.
186.Bavdek, A., Kostanjsek, R., Antonini, V.,
Lakey, J. H., Dalla Serra, M., Gilbert, R. J. C., and Anderluh, G.
(2012) pH dependence of listeriolysin O aggregation and pore-forming
ability, FEBS J., 279, 126-141.
187.Gossler-Schofberger, R., Hesser, G., Reif, M.
M., Friedmann, J., Duscher, B., Toca-Herrera, J. L., Oostenbrink, C.,
and Jilek, A. (2012) A stereochemical switch in the aDrs model system,
a candidate for a functional amyloid, Arch. Biochem. Biophys.,
522, 100-106.
188.Li, J., McQuade, T., Siemer, A. B.,
Napetschnig, J., Moriwaki, K., Hsiao, Y. S., Damko, E., Moquin, D.,
Walz, T., McDermott, A., Chan, F. K., and Wu, H. (2012) The RIP1/RIP3
necrosome forms a functional amyloid signaling complex required for
programmed necrosis, Cell, 150, 339-350.
189.Guyonnet, B., Egge, N., and Cornwall, G. A.
(2014) Functional amyloids in the mouse sperm acrosome, Mol. Cell.
Biol., 34, 2624-2634.
190.Lacroute, F. J. (1971) Non-Mendelian mutation
allowing ureidosuccinic acid uptake in yeast, J. Bacteriol.,
106, 519-522.
191.Cox, B. S. (1965) Psi, a cytoplasmic suppressor
of supersuppressors in yeast, Heredity, 20, 505-521.
192.Roberts, B. T., and Wickner, R. B. (2003)
Heritable activity: a prion that propagates by covalent
autoactivation, Genes Dev., 17, 2083-2087.
193.Du, Z., Park, K. W., Yu, H., Fan, Q., and Li,
L. (2008) Newly identified prion linked to the chromatin-remodeling
factor Swi1 in Saccharomyces cerevisiae, Nat. Genet.,
40, 460-465.
194.Patel, B. K., Gavin-Smyth, J., and Liebman, S.
W. (2009) The yeast global transcriptional co-repressor protein Cyc8
can propagate as a prion, Nat. Cell. Biol., 11,
344-349.
195.Brown, J. C., and Lindquist, S. (2009)
Genes. Dev., 23, 2320-2332.
196.Rogoza, T., Goginashvili, A., Rodionova, S.,
Ivanov, M., Viktorovskaya, O., Rubel, A., Volkov, K., and Mironova, L.
(2010) Non-Mendelian determinant [ISP+] in yeast is a
nuclear-residing prion form of the global transcriptional regulator
Sfp1, PNAS, 107, 10573-10577.
197.Saifitdinova, A. F., Nizhnikov, A. A., Lada, A.
G., Rubel, A. A., Magomedova, Z. M., Ignatova, V. V., Inge-Vechtomov,
S. G., and Galkin, A. P. (2010) [NSI+]: a novel non-Mendelian
nonsense suppressor determinant in Saccharomyces cerevisiae,
Curr. Genet., 56, 467-478.
198.Nizhnikov, A. A., Magomedova, Z. M., Rubel, A.
A., Kondrashkina, A. M., Inge-Vechtomov, S. G., and Galkin, A. P.
(2012) [NSI+] determinant has a pleiotropic phenotypic
manifestation that is modulated by SUP35, SUP45, and
VTS1 genes, Curr. Genet., 58, 35-47.
199.Nizhnikov, A. A., Magomedova, Z. M.,
Saifitdinova, A. F., Inge-Vechtomov, S. G., and Galkin, A. P. (2012)
Identification of genes encoding potentially amyloidogenic proteins
that take part in the regulation of nonsense suppression in yeast
Saccharomyces cerevisiae, Russ. J. Genet. Appl. Res.,
2, 398-404.
200.Nizhnikov, A. A., Kondrashkina, A. M., and
Galkin, A. P. (2013) Interactions of [NSI+] prion-like
determinant with SUP35 and VTS1 genes in Saccharomyces
cerevisiae, Russ. J. Genet., 49, 1004-1012.
201.Suzuki, G., Shimazu, N., and Tanaka, M. (2012)
A yeast prion, Mod5, promotes acquired drug resistance and cell
survival under environmental stress, Science, 336,
355-359.