Four Components of the Conjugated Redox System in Organisms: Carbon, Nitrogen, Sulfur, Oxygen
E. V. Tereshina*, V. N. Laskavy, and S. I. Ivanenko
Worldwide Medical Assistance, Fuchsloch 6A, Oberwil Zug 6317, Switzerland; E-mail: winterel@mail.ru* To whom correspondence should be addressed.
Received June 29, 2015
C1 compounds participate in various metabolic processes and regulations including DNA methylation. Formaldehyde (FA), a product of methyl group oxidation, is highly cytotoxic. In the cell, there are two pathways of its utilization: assimilation and oxidation. Formaldehyde displays cytotoxicity, and therefore its oxidation is considered as detoxification. The sensitivity to the threshold concentration of FA we regard as an indication of its major role in biosystem functioning. A model of a three-component conjugated redox system is proposed in which the methyl group oxidation pathway is an archaic and conservative donor of protons and electrons, the reduction of O2 serves as an acceptor, and the arginine amino group is used for production of both urea and nitric oxide (the donor and acceptor, respectively). The fourth component of the redox system is glutathione, which maintains redox balance. The three-level system of proton donors includes the oxidation of a methyl group (first level), the oxidation of acetate in mitochondria (second level), and glucose catabolism in the pentose phosphate pathway (third level). The whole redox system is united by the sulfhydryl groups of cysteines, glutathione, thioredoxin, and α-lipoic acid. The central regulatory role in this redox system belongs to glutathione-dependent formaldehyde dehydrogenase, which controls FA binding with tetrahydrofolic acid, arginine methylation, and denitrosation of sulfhydryl groups. The conjugated redox system was formed during evolution as a union of separate redox cycles of carbon, nitrogen, sulfur, and oxygen.
KEY WORDS: redox system, formaldehyde, arginine, hydrogen peroxide, sulfhydryl groups, regulationDOI: 10.1134/S0006297915090096
Abbreviations: ADH, alcohol dehydrogenase; ADMA, asymmetric dimethylarginine; ALA, α-lipoic acid; BH2, 7,8-dihydropterin; BH4, tetrahydropterin; CBR1, carbonyl reductase 1; DHAK, dihydroacetone kinase; DHALA, dihydro-α-lipoic acid; DHFA, dihydrofolic acid; ETC, electron transport chain; FA, formaldehyde; FDH, formate dehydrogenase; FLD, formaldehyde dehydrogenase; GSNOR, GSNO reductase; MMO, methane monooxygenase; NOHA, NG-hydroxy-L-arginine; PPP, pentose phosphate pathway; ROS, reactive oxygen species; SOD, superoxide dismutase; THFA, tetrahydrofolic acid; THMP, tetrahydromethanopterin.
THE CONJUGATED REDOX SYSTEM
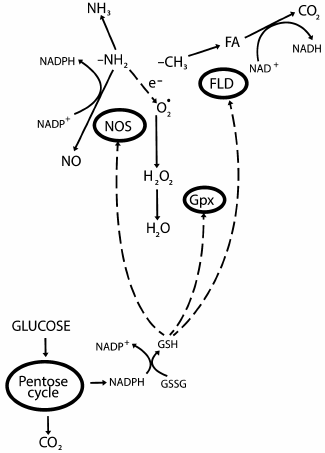
Reactive oxygen species (ROS) are highly reactive compounds that cause oxidative damage to biomolecules. Aging is associated not only with accumulation of oxidized products, but also with an increase in ROS generation that is called “oxidative stress” (OS). In particular, OS develops in systemic age-related pathologies such as atherosclerosis. A massive generation of ROS indicates a disturbance in balances in the distribution of electrons and protons where oxygen acts as the major acceptor. Transition metals are donors of electrons, and reduced compounds of carbon, nitrogen, and sulfur are donors of protons. The essence of our hypothesis is the uniting of donors and acceptors in a conjugated redox system. This system is responsible for the steady distribution of protons and electrons in the cell, maintains biosystem functioning, and influences its life duration. Thus, in Caenorhabditis elegans a slight increase in ROS content in mitochondria increases life duration, whereas a similar increase in the cytoplasm decreases it [1]. The conjugated redox system was created during evolution as the union of redox cycles of four atoms – carbon, nitrogen, sulfur, and oxygen. In this work, a model is proposed of organization of such system and of its functioning (Fig. 1).
Fig. 1. Three-component conjugated redox system: oxidation of methyl group as a donor of protons, reduction of molecular oxygen as an acceptor of protons, amino group as concurrent donor and acceptor. The transfer of protons is controlled by the fourth participant, glutathione, which receives protons from the pentose phosphate pathway (PPP).
THE METHYL GROUP IS A SOURCE OF FORMALDEHYDE
The methyl group relates to C1 compounds (methane, methyl group, methanol, formaldehyde, formic acid, carbon dioxide). These compounds participate in very different syntheses and a multiplicity of regulations of metabolic processes. The main processes are as follows: synthesis of purines and thymidine, pantothenic acid, chlorophyll and heme, metabolism of amino acids and biogenic amines, biogenesis of mitochondria and chloroplasts [2], and continual methylation–demethylation of DNA [3].
In the majority of biosystems, donors of methyl groups include methionine, serine, and glycine, whereas tetrahydrofolic acid (THFA) and S-adenosyl-L-methionine are carriers [4, 5]. In demethylation and transmethylation reactions, methyl groups are often oxidized with production of formaldehyde (FA) [6-8]. Decarboxylation of glyoxylate [9] and oxidation of xenobiotics by P-450 oxidases [10] also are sources of FA. In plants, FA is mainly generated by dissociation of 5,10-methylene-THFA and oxidation of methanol produced on demethylation of pectin [11]. Methanol and FA are considered endogenous poisons; therefore, their oxidation is usually considered as detoxification. The product of oxidation of FA (formate) is not toxic [12]. Formate causes a deceleration of Corynebacterium glutamicum growth, whereas the molybdenum-containing formate dehydrogenase (FDH) decreases this effect [1]. Functions of formate are well studied in plants, where it is a product of degradation of glyoxylate.
Formaldehyde is generated in all biosystems: in plants and animals, vertebrates and invertebrates, unicellular and multicellular organisms, bacteria and archaea. In the majority of biosystems, FA is bound with endogenous nucleophiles, glutathione, and THFA [5, 13]. FA is highly cytotoxic and, therefore, all organisms produce it in low concentrations. Physiological concentrations of FA in human blood and urine are 0.4-0.6 and 2.8-4.0 µg/ml, respectively [14]. The effect of FA on a cell depends on dose: at the concentration of 10 mM, it induces necrosis of tumor and epithelial cells in culture, at 1 mM it increases apoptosis and decreases mitosis, at 0.5 and 0.1 mM it increases proliferation [15]. The cytotoxicity manifests itself on overcoming a certain threshold of free FA concentration. High cytotoxicity and a modulating effect of FA on proliferation and apoptosis suggest that it can be in a central regulatory junction of the main processes functioning in a biosystem.
THE MOST ANCIENT DONORS OF PROTONS
Metabolism of C1 compounds was mastered by methanogenic and methanotrophic archaea at the dawn of evolution. Methanotrophic bacteria use methane as the only source of carbon. Methylotrophic bacteria can oxidize methane, methanol, methylated amines, halomethanes, sulfur-containing methylated compounds, and even methyl groups of such substrates as choline. In all methano- and methylotrophic bacteria, FA plays the central role in metabolism [16]. The metabolism of these bacteria is organized and regulated to provide non-limiting rate of FA utilization, because its accumulation leads to the instantaneous death of the biosystem [17]. During the oxidation of FA, protons and electrons are produced, which are necessary for the activity of these bacteria.
Methane is oxidized step-by-step in the chain “methane — methanol — FA — formate — carbon dioxide”, which results in the replacement of all hydrogen atoms by oxygen atoms. In all methylotrophs, the first part of the pathway to FA is realized, and they differ only in the character of FA assimilation. Based on this feature, the methylotrophs are combined in three groups: in type 1 bacteria, the cyclic ribulose monophosphate pathway is realized [18, 19], the linear serine pathway is realized in type 2 bacteria [20, 21], and in the X type bacteria, both metabolic pathways are used. The methylotrophic yeast Pichia pastoris uses the xylulose monophosphate pathway of FA assimilation [22]. Low concentrations of methane and a high content of O2 mainly promote the growth of type 1 bacteria, whereas high concentrations of methane and low content of O2 are favorable for type 2 bacteria [23]. The methanotrophic bacteria are found to have two methane monooxidases – soluble (sMMO) and membrane-bound (mMMO). They are classic monooxygenases. sMMO is found in type 2 and X methanotrophs; it contains nonheme iron, flavin adenine nucleotide, and iron–sulfur [Fe2S2] clusters and uses NADPH produced by the oxidation of FA and formate as a donor of electrons [24]. To start the oxidation of methane, these bacteria have initially to oxidize FA. mMMO is expressed in the case of the presence in the medium of a sufficient amount of copper (0.85-1.0 mmol/g), because copper induces synthesis of intracellular membranes. It seems that the enzyme itself may contain copper [25]. When mMMO is activated, the cells grow faster because they do not require NADPH limited by the oxidation of FA [26]. However, the substrate specificity of sMMO is higher than for other oxygenases.
In Gram-negative methanotrophs, methanol is oxidized to FA as a MMO product or as an exogenous substrate under the influence of periplasmic methanol dehydrogenase [27]. This enzyme is a tetramer with its subunits containing two moles of pyrrol quinoline quinone and one mole of Ca2+. Cytochrome cL acts as a specific acceptor of electrons [28]. In Gram-positive methanotrophs, methanol is oxidized by methanol dehydrogenase, which uses NAD+ as an acceptor of electrons [29]. In methylotrophs, NADP+-bound aldehyde dehydrogenase, which utilizes or does not utilize glutathione, oxidizes FA to formate, and formate is oxidized by NAD+-dependent FDH to carbon dioxide in virtually all methanotrophs [30]. Reducing syntheses occur with expenditure of protons and electrons, which are mainly released on the oxidation of FA.
Methylotrophs, which do not utilize methane, prefer the ribulose monophosphate pathway of FA assimilation associated with its concurrent oxidation to carbon dioxide. In this cycle, three moles of FA are assimilated. FA interacts with ribulose-5-phosphate with production of hexulose-6-phosphate, which isomerizes to fructose-6-phosphate. The further oxidation produces three-carbon compounds, a CO2 molecule is released, and ribulose-6-phosphate is regenerated. NAD+ or NADP+ acts as electron acceptors at two stages of FA oxidation [31]. The entrance of FA in the cycle is catalyzed by ribulose bisphosphate carboxylase [19], which promotes the assimilation of CO2 in the Calvin–Benson cycle. However, during the ribulose monophosphate cycle FA is oxidized to CO2 and assimilated, whereas during the Calvin–Benson cycle only CO2 is assimilated. However, the two cycles result similarly in production of 3-phosphoglycerate.
In obligate methanotrophs, FA is assimilated through the serine pathway. In this case, FA is oxidized through a parallel pathway. For production of three-carbon compounds, two moles of FA and one mole of CO2 are necessary as well as reducing equivalents. Carbon dioxide and NADH can be produced by FA oxidation through the FA–formate–carbon dioxide pathway. Thus, in this model FA occurs at the bifurcation into two pathways: through one pathway, FA is oxidized to carbon dioxide, and through the other pathway, it is assimilated for synthesis of three-carbon compounds. Therefore, assimilation and oxidation of FA are separated.
The first reaction of the serine pathway catalyzed by hydroxymethyl transferase is the interaction of FA with glycine and generation of serine [20]. The back reaction, the synthesis of glycine from serine, which has been detected in Methylobacterium extorquens AM1 and in Methylobacterium organophilum XX [21], exists in all eukaryotes. Serine gives the amino group to glyoxylate with production of glycine and hydroxypyruvate. Resources of glyoxylate are renewed due to malate, because facultative methylotrophs do not possess isocitrate lyase, which is a key enzyme of the glyoxylate cycle. Malyl-CoA is produced in the reaction catalyzed by malate thiokinase and cleaved by lyase. These enzymes are present only in methylotrophs possessing the serine pathway of FA assimilation. Malyl-CoA degrades to acetate and glyoxylate. In connection with glyoxylate regeneration, the serine pathway also can be considered cyclic [21]. During the first stage of the evolution, glyoxylate could be a product of an immediate interaction of two one-carbon compounds, e.g. of two formate molecules [32]. Later, glyoxylate degradation to formate and CO2 was recorded. The evolution is characterized by reversal of some key metabolic pathways, which is exemplified, in particular, by the reversibility of the “glycine–serine” reaction.
Dihydroacetone synthase catalyzes the first reaction of FA uniting with D-xylulose-5-phosphate [33] in the xylulose monophosphate pathway of FA assimilation. In the methylotrophic yeast Hansenula polymorpha, the distribution of FA between oxidation and assimilation is regulated by the ATP/ADP ratio. Excess ATP stimulates dihydroacetone kinase (DHAK) and inhibits formaldehyde dehydrogenase (FLD). When ADP is accumulated, DHAK is inhibited and FLD is activated. If this mechanism does not work, FA regulates its own production by binding with methanol oxidase and decreasing its activity [34].
The catabolism and assimilation of FA are well studied in the aerobic facultative methylotrophic α-proteobacterium Methylobacillus flagellates KT, which can grow on methane and methylamine. Methanol is oxidized in the periplasm, and the resulting FA crosses the cytoplasmic membrane and interacts with two carriers: THFA and tetrahydromethanopterin (THMP) [35, 36]. Note that it interacts with THFA spontaneously and nonenzymatically even in vitro [37]. The reaction is regulated by the concentrations of the substrates. This carrier is involved in the oxidation of FA to CO2, which is associated with production of formyl derivatives participating in the biosynthesis of adenine and tRNA of formylmethionine. The folate cycle includes both FA and formate. FA interacts with THFA nonenzymatically, producing 5,10-methylene-THFA. For interaction of formate with THFA resulting in the production of 10-formyl-THFA, ATP is needed. The production of 5,10-methylene-THFA puts FA on the assimilation pathway.
The condensation of FA with THMP is catalyzed by the protein Fae, which is one of major cytoplasmic proteins of these bacteria with expression activated by FA [38]. THMP is the major carrier of C1 compounds in methanogenic and sulfate-reducing archaea and serves in them for both oxidation of these compounds to CO2 and their reduction to methane. THMP is also found in the majority of methylotrophic and methanotrophic bacteria [39]. The THMP-dependent oxidation of FA was received by M. extorquens AM1 from a more ancient precursor by a horizontal transfer. The gene encoding Fae is localized in the same cluster with enzymes oxidizing methylene-THMP to CO2 [39]. In methanotrophs, THMP is used only for oxidation of FA, whereas THFA is used only for assimilation.
In prokaryotes, there are pathways for oxidation of ammonia and hydrogen sulfide. Aminomonooxidase, which oxidizes ammonia in autotrophic bacteria, has many features in common with mMMO. The two enzymes utilize copper, and cytochrome is used as a reducer. Ammonia can be oxidized by both sMMO and mMMO and is the main donor of protons and electrons. Hydrogen sulfide can also be a donor of protons, whereas sulfides can be donors of electrons. Sulfide is oxidized to sulfur and sulfur to sulfate. A microbial community of nitrate-reducing and sulfide-oxidizing bacteria with the dominant Arcobacter sp. was found [40], as well as a community of anaerobic methanotrophs and sulfate-reducing bacteria [41]. Under the influence of cytochrome c and ferredoxin, plants can assimilate in light sulfate, which is reduced and converted to sulfhydryl groups of cysteine. Methionine is synthesized from cysteine through the pathway “O-acetyl-L-serine–cystathionine–homocysteine–methionine”, which is the same in bacteria, plants, and fungi. Cysteine with utilization of hydrogen sulfide can be synthesized even by archaea [42].
Thus, biosystems use reduced compounds of carbon, nitrogen, and sulfur as donors of protons and electrons. In methylotrophs, FA plays the central role in the pathways of C1 oxidation and assimilation. FA is used for production of C2 (acetate and glyoxylate) and C3 (phosphoglycerate) compounds and for production of reducing equivalents. We consider the methyl group oxidation pathway, which exists in all biosystems, as the primary donor of protons and electrons in the global redox cycle of carbon. During evolution conjugated redox systems were produced that combine redox cycles of nitrogen and sulfur, carbon and nitrogen, and of carbon and sulfur. The redox cycle of oxygen unites different biosystems. Thus, aerobic organisms use the pathway of oxygen reduction, whereas plants use photolysis of water. The redox cycles of four elements – carbon, nitrogen, sulfur, and oxygen — form the united conjugated redox system. The conjugated redox system functions with participation of the tripeptide glutathione (GSH), which contains glutamate (a carrier of amino groups), cysteine (a carrier of sulfhydryl groups), and glycine (a producer of methyl groups).
BRANCHED PATHWAY OF FORMALDEHYDE
Formaldehyde dehydrogenase. There are several pathways of FA oxidation, and two of them are direct – with involvement of FLD and after the condensation of FA with THMP. In some bacteria, e.g. in Pseudomonas putida [43] and Rhodococcus erythropolis UPV-1 [44], FLD does not use cofactors. In Escherichia coli [45], Bacillus subtilis [46], Arabidopsis thaliana [47], and humans [48], FLD uses glutathione as a cofactor, whereas in Mycobacterium tuberculosis and M. smegmatis [49] the cofactor is mycothiol (MSH). MSH is a functional analog of glutathione, it is a cysteinyl pseudo-disaccharide containing N-acetylcysteine, glucosamine, and myo-inositol (AcCys-GlcN-Ins) [50]. MSH-dependent FLDs are found not only in mycobacteria, but also in Amycolatopsis methanolica [51], R. erythropolis [52], and C. glutamicum [53]. The molecular weight and kinetic features of FLDs isolated from animals, invertebrates, plants, and microorganisms are virtually identical. The universal distribution and conservativeness of this enzyme suggest that FA oxidation is required in all biosystems.
Glutathione was not found in Gram-positive methylotrophic bacteria, it can be absent in anaerobic microorganisms, but it is present in virtually all aerobic organisms. Glutathione is a cofactor of alcohol dehydrogenase 3 (ADH3), which is also FLD [54]. Seven different genes encoding ADH isoforms (ADH1-ADH7) have been found in humans. All ADHs are dimers consisting of two units with the molecular weight of 40 kDa. Each dimer contains two Zn2+ ions. ADH3 is phylogenetically the most ancient form of ADH. The first duplication of the gene encoding ADH3 occurred on the level of osseous fishes (405-450 million years ago), which resulted in the appearance of ADH of class 1 (the classic liver ADH) [55]. ADH2, 4, and 5 are products of subsequent duplications. ADH2 oxidizes acetaldehyde in mitochondria and is involved in metabolism of carbonyl compounds [56].
The equal probability of reduction and oxidation of a FA molecule is its unique feature. The liver ADH1 oxidizes methanol to FA in the presence of NAD+. FA can be rapidly reduced by NADH to methanol or oxidized to formate. Among these three reactions, the reduction of FA is the fastest, and the rate of FA oxidation is higher than the rate of methanol oxidation [57]. FA spontaneously interacts nonenzymatically with glutathione with production of S-hydroxymethylglutathione. This reaction is easily reversible. It becomes irreversible when glutathione-dependent FLD (GSH-FLD) converts S-hydroxymethylglutathione to S-formylglutathione. Then a specific hydrolase hydrolyzes S-formylglutathione with production of formate, glutathione, and NADH [58]. Thus, FLD concurrently presents formate for further syntheses and NADH as a reducing equivalent. At high pH values, the enzyme can utilize NADP+. FLD is specific to FA. This enzyme can have only methylglyoxal as another substrate and S-pyruvylglutathione as a product. In addition to the oxidation of FA, GSH-FLD actively reduces S-nitrosoglutathione produced by the condensation of glutathione with nitric oxide [59].
GSH-FLD is well studied in A. thaliana and its DNA has been cloned [47]. The enzyme is expressed similarly in all organs of plants [47] and animals [60], whereas in A. thaliana it is represented by a single copy of the gene on chromosome 5 [61]. In Pichia methanolica, GSH-FLD is also encoded only by one gene, and its expression is regulated by sources of carbon and nitrogen and by such concentration of methanol that prevents an increase in the FA level in the cell not to overcome the “desired” value [62]. In baker’s yeast Saccharomyces cerevisiae, GSH-FLD expression increases following an increase in the contents of methylated compounds in the medium [63]. Generally, the expression of the enzyme depends on the amount of FA produced by the oxidation of substrates containing methyl groups. Transcription of the GSH-FLD gene is activated not only by FA, but also by compounds involved in metabolic pathways of FA, such as THFA, and by signals from reducing elements entering from the region of the glutathione system action [64]. In different biosystems, the oxidation of C1 compounds conjugated with glutathione influences such processes as photosynthesis, proliferation, and apoptosis [65, 66].
Thus, FA spontaneously nonenzymatically reacts with both THFA (assimilation) and glutathione (oxidation). The choice of pathway seems to depend on the presence of reduced forms of THFA and glutathione. GSH-FLD is activated at high concentrations of methylated compounds.
Formate dehydrogenase. FDH catalyzes the two-electron oxidation of formate to CO2. Different types of this enzyme are found in aerobic and anaerobic organisms. The enzyme of aerobic organisms contains two identical subunits without metals and prosthetic groups, and its molecular weight is lower. The amino acid sequence in the catalytic center of all this type of dehydrogenases is very conservative. The enzyme of anaerobic organisms contains many iron–sulfur clusters, other metals (molybdenum, tungsten, and selenium) are also present, and its molecular weight is high.
The hydrogen molecule and formate can be electron donors for E. coli. Formate is metabolized by three membrane-bound FDHs: the first participates in production of hydrogen (FDH H), the second is induced in the presence of nitrate, and the third is active under conditions of anaerobic growth [67]. These three enzymes contain in the active site selenocysteine, two molybdopterin guanidine nucleotides as cofactors, and Fe4S4 clusters [68]. FDH H is a component of the anaerobic formate-hydrogen lyase complex of E. coli. In the absence of exogenous electron acceptors, this enzyme cleaves formate into H2 and CO2. Expression of the FDH H gene is induced by formate and by the absence of electron acceptors and is inhibited by nitrates, nitrites, trimethylamine, nitric oxide, and oxygen [69]. The yeast S. cerevisiae lack alcohol dehydrogenase and do not oxidize methanol [70]. However, the yeast oxidizes FA to CO2 and contains NAD+-dependent FDH. Under the influence of NAD+-dependent FDH, formate is also oxidized in plant cells: at pH > 6 in mitochondria and at lower pH values in peroxisomes. The higher the O2 content in the medium, the higher is the activity of the enzyme [71].
In plants, FA enters into the folate cycle or is oxidized to formate and CO2, which later is assimilated in the Calvin–Benson cycle. Formate is utilized in synthesis of glyoxylate catalyzed by glyoxylate synthase, which has been found only in plants [72]. The oxidation of FA dominates over its assimilation in the folate cycle of A. thaliana, which is necessary for production of formate. Formate can be also produced from methanol, glyoxylate, and serine. In the plant cell, formate is oxidized by FDH in mitochondria and by peroxidase in peroxisomes [73]. Under stress conditions, the amount of FDH reaches 9% of all mitochondrial protein. Detailed studies on this enzyme in A. thaliana revealed that it could be carried from the cytoplasm into chloroplasts due to the presence of a signaling peptide on the N-terminus [74]. Bacteria and yeast do not possess the signaling peptide in FDH. The gene of FDH was most intensively expressed in A. thaliana when the plant was sprayed with methanol and FA, and the lowest expression was under spraying with formate and water [75], i.e. FDH was activated not by the presence of the substrate but by the FA concentration. The enzyme activity is regulated by phosphorylation, which decreases on increase in concentrations of NAD+, formate, and pyruvate [76]. The expression of FDH increases under stress conditions, in particular drought [77]. In transgenic potato with suppressed synthesis of the enzyme, drought led to an increase in contents of proline and glutamate [78]. In culture of the unicellular green microalga Chlamydomonas reinhardtii under illumination, treatment with methanol caused a fourfold higher increase in NADPH than in the dark. The total content of amino acids also increased, and their ratio changed. The amounts of glutamate, glutamine, alanine, serine, and tyrosine increased, whereas the amounts of valine and methionine decreased [79].
Thus, the oxidation of FA is associated with metabolism of C1 compounds, production of formate, the glutathione system, and metabolism of amino groups. The main role in the regulation of these directions seems to belong to fluctuations in the FA concentration.
THE AMINO GROUP IS A DOUBLE-FACED JANUS
The amino group is another participant of the conjugated redox system: it can be reduced to ammonia or oxidized to nitric oxide, nitrates, and nitrites. L-Arginine is an amino acid whose guanidine group is a donor of urea and NO. L-Arginine can bind one, two, and three FA molecules (a very rapid reaction), and production of mono-, di-, and trihydroxymethyl arginines is catalyzed by trans-methylases. Methylated derivatives of arginine are a reserve and transport form of FA, transmitting it into the folate cycle [80].
NG-Hydroxymethyl arginine is a compound involved in maintaining the “desired” endogenous concentration of FA [81]. At the same time, the methylation of arginine is a mode to control the free arginine content. L-Arginine competes with L-lysine, because at physiological pH values lysine can be methylated on the ε-amino group with production of mono-, di-, and trimethyl lysine [82]. Trimethyl lysine is a part of calmodulin [83], which is a component of NO synthases. Arginine is a substrate for arginase and NO-synthase (NOS).
On decarboxylation of glycine in mitochondria, ammonia and carbon dioxide are released. In the mitochondrial matrix, ammonia binds with carbon anhydride and ATP. The resulting carbamoyl phosphate enters the urea cycle through binding with ornithine. The detachment of the guanidine group of arginine with production of urea and ornithine is catalyzed by arginase. The reaction occurs in the cytoplasm, and then ornithine is transferred into mitochondria, where it is decarboxylated with production of polyamines or again captures carbamoyl phosphate. The elimination of ammonia is catalyzed by arginase. The majority of mammals have two arginase isomers. Arginase 1 is located in the cytoplasm, and arginase 2 in mitochondria, where it participates in the regulation of the arginine/ornithine ratio. Arginase of mammals is a trimer activated by Ca2+ and Mn2+, and in some bacteria, it is a hexamer containing two Mn2+ ions. The third product of glycine degradation is the methyl group. It binds with THFA, but can also participate in the methylation of arginine, decreasing the availability of this amino acid.
The oxidation of amino groups of arginine by molecular O2 in the presence of NADPH with production of nitric oxide and citrulline is catalyzed by three NOS isoforms: the neuronal NOS (type I; nNOS), the endothelial NOS (type III; eNOS), and the inducible NOS (type II; iNOS) [84]. The nNOS and eNOS are expressed constantly, but to display activity they require Ca2+/calmodulin. The iNOS is mainly expressed in macrophages in response to stimulation by cytokines or endotoxin, and it does not depend on Ca2+ [85]. The iNOS is localized in the cytoplasm, and eNOS through palmitic or myristic acid binds with caveolae, pits on the cytoplasmic membrane with size of 60-80 nm that are involved in endocytosis and transcytosis. The three isoforms are homodimers in which calmodulin is bound with a heme-containing oxygenase and reductase being a flavin cytochrome P-450. A tetrahydropterin (BH4) cofactor activates the heme-bound O2 molecule and gives one electron to it. The catalysis is maintained by the availability of arginine and the presence of reduced BH4.
BH4 is the redox sensor of the enzyme. Under anaerobic conditions in protonated medium, it transforms to 7,8-dihydropterin (BH2), and at neutral pH in air it gives up to 98% BH2 and small amounts of superoxide anion and hydrogen peroxide [86]. The oxidation of BH4 into BH2 is reversible. BH2 is reduced by glutathione and ascorbate. On the inactivation of BH4, electrons in NOS leave for O2, and instead of nitric oxide produce superoxide anion, which oxidizes BH4 and decreases more the NOS activity, because oxidized pterin does not function. Superoxide anion dismutates to hydrogen peroxide. Nitric oxide competes with SOD for the binding of superoxide anion and reacts with it with a rate close to the diffusion rate. Therefore, in many biosystems peroxide is mainly produced under conditions of NO insufficiency. At high concentrations, H2O2 reacts with BH4 with production of the stable end product 7,8-dihydroxanthopterin. The inhibition of eNOS by hydrogen peroxide seems to be caused by a direct interaction with BH4, because in a culture of endothelial cells H2O2 does not decrease the content of either mRNA or of the enzyme protein. In the presence of H2O2 in vascular endothelium cells, the expression of the gene of arginase 1 and its synthesis increase [87], i.e. the inhibition of NOS activates arginase. During the interaction of NO with H2O2, peroxynitrites (ONOO–) are produced, which also can oxidize BH4, but in this case glutathione and ascorbate win the competition for the substrate. The activity of NO synthase is reversibly inhibited by the nitric oxide molecule associated with nitrosation of the enzyme. A decrease in NO production and an increase in superoxide anion generation are also caused by S-glutathionylation of NO synthase. Concurrently, the level of BH4 and the ratio BH4/7,8-dihydropterin are decreased [88].
The Km and Vmax values of arginase are approximately 1000 times higher than those of NOS [89]; therefore, it binds the substrate preferentially. The availability of arginine is a limiting stage in the production of NO. When the amount of arginine is limited, iNOS transfers electrons onto O2, generating superoxide anion, or onto the NO molecule, that results in production of peroxynitrites. The production of peroxynitrites was detected on the combined activities of arginase 1 and iNOS [90]. However, a direct interaction of arginase 1 with iNOS or eNOS leads to its S-nitrosation. The intermediate product of amino group oxidation in the NOS-catalyzed reaction, NG-hydroxy-L-arginine (NOHA), inhibits the activity of arginase co-expressed in macrophages. This promotes increase in the production of NO [91]. NOHA also has its own significance as an inhibitor of ornithine decarboxylase and of polyamine production [92]. Thus, NOS, and in particular eNOS, is a kind of “switch” controlling the production of NO and H2O2, as well as elimination of ammonia. Interrelationships of arginase and NOS must be considered as a regulation of the balance of the nitrogen redox cycle in the oscillatory contour “ammonia–amino group–nitric oxide”.
Asymmetric dimethylarginine (ADMA) inhibits NO synthase [93]. ADMA is present in blood in very low concentrations, ~0.5 µM [94]. It is metabolized by the enzyme dimethylarginine dimethylaminohydrolase to L-citrulline and dimethylamine [95]. At this level, the methyl group and FA as its derivative is responsible for the regulation. The availability of arginine determined by the balance between binding with THFA of the methyl group released on decarboxylation of glycine and its oxidation to FA can be considered the first and main level of regulation. The free arginine content is finally determined by the glycine decarboxylation rate and by the presence of THFA, because it determines the amount of methylated arginine. In turn, the activity of FLD preventing the methylation of arginine is associated with the presence of reduced glutathione. Feedback type regulations are realized when nitric oxide blocks FDH, ADMA inhibits NOS, and NOHA inhibits arginase. The second level of regulation also depends on reduced glutathione, which maintains the reduced state of the redox sensor BH4 and determines the NO/H2O2 ratio. These complicated regulations are responsible for maintaining the balance of FA, NO, and H2O2 concentrations. Glutathione binds nitric oxide, reduces BH4, and is a cofactor of FLD. Moreover, it also reduces α-lipoic acid (ALA), which is a component of the catalytic center of the glycine decarboxylase and pyruvate dehydrogenase complexes in mitochondria and thus provides for the oxidation of both methyl group and acetate, i.e. the production of NADH. Preincubation of endothelial cells with ALA increased their ability to generate nitric oxide [96]. Thus, glutathione connects between themselves the pathways of methyl group oxidation, hydrogen peroxide reduction, and the production of ammonia and nitric oxide (Fig. 1).
Nitric oxide participates in the regulation of O2 consumption in mitochondria because it can inhibit cytochrome c. In the presence of high concentrations of O2, the electron transport chain (ETC) electrons oxidize NO to nitrites, whereas at low concentrations of oxygen or in its absence they reduce nitrites to NO. Thus, mitochondria are involved not only in the production of methyl groups and oxygen, but also in a component of the nitrogen-including conjugated redox system. Nitric oxide causes very rapid disorders in functioning of mitochondria. It also influences the expression of peroxiredoxins and sulforedoxins and the sensitivity of peroxiredoxins to hyperoxidation [97]. The immediate participation of molecular oxygen in the oxidation of amino groups and production of hydrogen peroxide is responsible for the active regulation of angiogenesis by nitric oxide. The branched capillary network promotes improved supply of oxygen to cells. Nitric oxide also participates in insulin transport across the capillary wall in skeletal muscle, which improves the consumption of glucose and energy provision of myocytes [98].
Thus, in the conjugated redox system amino groups compete with methyl groups as donors of protons. The result of this competition depends on FLD activity, which is determined by the presence of free arginine as a substrate for production of urea and nitric oxide. It seems that the NO/H2O2 ratio must be considered as a reflection of competitive relationships between arginase and NOS. Finally, methyl group oxidation regulates the maintenance of balance in the oscillating contour “ammonia–amino group–nitric oxide”. Disorders in this complicated regulatory system are the cause of the accumulation of peroxynitrites and of the development of oxidative stress.
THE THIRD PARTICIPANT IS OXYGEN
C1 compounds are metabolized in all organisms from archaea to humans. In cells, separate stages of metabolism are compartmentalized. In the yeast Candida boidinii growing on methanol, many peroxisomes appear, which contain only two enzymes, alcohol oxidase and catalase. Catalase neutralizes H2O2 produced on oxidation of methanol to FA. The further oxidation of FA in C. boidinii occurs in the cytoplasm [99]. Methanol is also oxidized by peroxisomes in the retina of rats [100]. However, under the influence of H2O2, catalase also can oxidize FA and formate. It seems that in peroxisomes methyl groups released in the last stages of cholesterol synthesis can be fully oxidized to CO2 with involvement of hydrogen peroxide. It should be noted that insufficiency of folic acid, biotin, pantothenic acid, riboflavin, and vitamin A are considered alimentary factors decreasing catalase activity. The activity of catalase decreases on excess of methionine, tyrosine, cysteine, copper, and zinc. This set of factors suggests that catalase can be a participant of such processes as metabolism of C1 compounds, fatty acids, catecholamines, glutathione, as well of functioning of redox system components in the cytoplasm containing Cu/Zn-SOD.
In peroxisomes of liver cells, the amount of catalase is up to 40% of the total protein. In the liver cells, catalase reduces to water H2O2 that is produced in reactions catalyzed by such oxidases as urate oxidase, D-amino acid oxidases, α-oxy-acid oxidases, and on the β-oxidation of fatty acids. The flavoprotein glycine oxidase catalyzes in peroxisomes oxidative deamination of glycine with production of glyoxylate and H2O2. Then glyoxylate is oxidized in mitochondria by carboligase of glyoxylic acid to oxalate or formate and CO2 [101]. The reaction cofactor is thiamine [102]. Formate is reduced with involvement of NADPH and THFA to a formyl derivative of THFA. In plants, from glyoxylate glycine is produced, which is transferred into mitochondria. Serine is another source of glycine and 5,10-methylene-THFA. The activity of serine hydroxymethyl transferase is equally distributed between the cytoplasm and mitochondria [103]. There is a THFA-independent reaction of serine transformation into glycine with the possible production of FA [104]. 5,10-Methylene-THFA is mainly produced in mitochondria by the glycine decarboxylase system, whose catalytic center is localized in the P-peptide containing ALA. The glycine decarboxylase system is present only in mitochondria. Serine and glycine are precursors in the syntheses of chlorophyll, heme, glutathione, and tryptophan. In E. coli, 15% of all carbon atoms assimilated on oxidation of glucose goes through the serine–glycine pathway [105].
Peroxisomes and mitochondria are connected in metabolism [106]. In peroxisomes, water is produced by the enzymatic four-electron reduction of oxygen through the stage of H2O2 production and in mitochondria through the four-electron reduction of O2 in the ETC without the production of H2O2. Hydrogen peroxide in mitochondria is a product of dismutation of superoxide anion produced in complexes 1 and 3 of the ETC. The superoxide anion dismutation is the major source of H2O2 in cells. In mitochondria, the dismutation is catalyzed by Mn-SOD and by Cu/Zn-SOD in the cytoplasm. In aerobic organisms, O2 receives electrons from iron of cytochromes, flavin coenzymes, and semiquinones. Any reaction of superoxide anion in aqueous solution competes with SOD. In the absence of SOD, a spontaneous dismutation occurs [107]. In aqueous solutions, O2 is spontaneously protonated with production of H2O2 at low pH values. This reaction is very slow, and it is accelerated nearly 104 times in the presence of SOD [107]. Hydrogen peroxide is a stable compound. As differentiated from superoxide anion, it can diffuse across the cytoplasmic membrane. SOD is considered to be an antioxidant protection element capable of leveling the toxic effect of superoxide anion. However, hydrogen peroxide is necessary for various processes, including proliferation and apoptosis. It participates in the redox balance of sulfhydryl groups. Therefore, the enzymatic dismutation can be considered a producer of H2O2.
In membranes of the cytoplasmic reticulum, H2O2 is produced by cytochromes P-450, which display features of monooxygenases and participate in the degradation of xenobiotics. NADPH-dependent oxidases bound with the cytoplasmic membrane generate H2O2 in response to stimulation by growth factors or cytokines. Although microsomal enzymes and signals from growth factors are not related, the redox system regulates the folding and secretion of proteins in the endoplasmic reticulum [108].
Thus, the four-electron reduction of oxygen with production of water is realized through two pathways: first – without involvement of NADH through the stage of hydrogen peroxide generation, and second – in the ETC with involvement of NADH. The first pathway is realized on methyl group oxidation in peroxisomes and, possibly, in mitochondria (oxidation of methyl group produced on glycine decarboxylation), and the second pathway is realized on acetate oxidation in mitochondria. Methyl group and acetate are sources of protons, whereas water and urea are two forms of proton elimination from the biosystem. The carbon atom of the methyl group and acetate is eliminated as carbon dioxide. As a result, oxygen eliminates from the biosystem electrons, protons, and carbon atoms that do not participate in reducing syntheses.
SULFHYDRYL GROUPS AND SULFUR REDOX
The nonenzymatic one-electron reduction of H2O2 in the presence of transition metal ions leads to generation of hydroxyl anion (the Fenton reaction), which has destructive features. Antioxidant protection includes the elimination of peroxidation products, whereas neutralization of transition metal ions is a parameter of the regulation of superoxide anion production. This regulation is also contributed to by ALA, which is capable of chelating Cu2+ and Fe2+. The contents of other transition metals (zinc, magnesium, nickel) are also regulated [109, 110].
Toxic properties of H2O2 are manifested if its concentration is higher than a certain concentration threshold. At some concentrations, hydrogen peroxide performs regulatory functions, whereas at other concentrations it induces oxidative stress or is cytotoxic. The necessary concentration of H2O2 in cells is maintained due to balance between its generation and reduction. Under conditions of “soft” stress, H2O2 is reduced to water by glutathione peroxidase (GPx), and under “severe” stress, it is reduced by catalase. Nevertheless, at high rates of H2O2 generation in erythrocytes (10–10-10–9 mol H2O2 per mg hemoglobin per min) the GPx activity dominates, whereas catalase is more active at the low rates (10–9 mol) of H2O2 production [111]. Glutathione transferase also has features of catalase: like catalase, it has only hydrogen peroxide as a substrate. In cells, GPx is mainly localized in the cytoplasm, whereas catalase is present only in peroxisomes. The affinity of GPx for H2O2 is one-two orders of magnitude higher than the affinity of catalase. Nevertheless, catalase is an enzyme with the highest turnover and can degrade 44,000 molecules of H2O2 per second, i.e. a small amount of the enzyme is sufficient for cleavage of a great amount of hydrogen peroxide. Similarly to the case of SOD, the reaction rate is determined by diffusion and does not require energy for activation. GPx contains selenocysteine in the active center and specifically oxidizes reduced glutathione. A high level of reduced glutathione is maintained by the NADPH-dependent enzyme glutathione reductase. The activity of enzymes responsible for functioning of the glutathione system is regulated mainly in response to soft stress on the transcriptional level [112]. The transcription factor Nrf2 binds with the antioxidant-responding element (ARE) in the promoter of genes that encode, in particular, L-glutamate:L-cysteine ligase and glutathione S-transferases [113]. Nrf2 can be activated by homocysteine, which is a component of the methyl group transporting system [114].
Glutathione and proteins include cysteine whose sulfhydryl groups are involved in the redox cycle of sulfur, donating protons or being oxidized to sulfonylic acid. Dihydrolipoic acid (DHALA) also contains sulfhydryl groups. ALA is reduced to DHALA under the influence of three enzymes associated with the oxidation of NADH or NADPH: the mitochondrial NADH-dependent dihydrolipoamide dehydrogenase, NADPH-dependent thioredoxin of the cytoplasm, and glutathione reductase. Under the influence of NADPH, ALA is reduced twofold faster due to involvement of thioredoxin reductase. Thioredoxin reductase is a selenoprotein with a high molecular weight. It controls the redox status of mammalian cells together with the glutathione–glutaredoxin system (NADPH, glutathione reductase, glutathione, glutaredoxin) with which it exchanges electrons. The functioning of cytochrome P-450 and also transport of electrons in mitochondria depend on the selenium content in the organism. Due to the strong negative redox potential, DHALA can reduce glutathione and ubiquinone [115]. The reduction of ubiquinone can block the transfer of electrons from the NADH-dehydrogenase complex of the ETC. The thioredoxin system regulates the activity of many redox sensitive transcription factors [116], supplies with electrons thiol-dependent peroxidases (peroxiredoxins) to accelerate the neutralization of active forms of nitrogen and oxygen, and reduces ribonucleotide reductase and methionine sulforeductase [117, 118]. Bacteria lack the glutathione–glutaredoxin system, thioredoxin reductase in them is a low molecular weight protein, and the range of its activity is different [119].
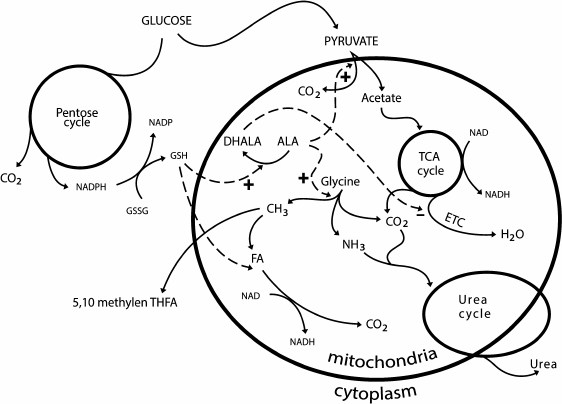
ALA is associated with the glutathione system. In low concentrations (25 and 100 µM), ALA increases the glutathione content in cells, and in high concentrations (2 mM) it can cause apoptosis. DHALA stimulates the synthesis of glutathione because it promotes the utilization of cysteine: it reduces cystine to cysteine [120]. In many cells, the extracellular cysteine availability is a limiting stage in the synthesis of glutathione [121]. ALA and oxidized glutathione are reduced by NADPH, which is generated during the pentose phosphate pathway (PPP). Glucose donating protons and electrons in the PPP provides through ALA and glutathione the conjugation between the oxidation of methyl groups and acetate: glutathione is a cofactor of FLD, and ALA is a cofactor of the pyruvate dehydrogenase and glycine decarboxylase complexes (Fig. 2).
Fig. 2. Three-level system of proton donors. The first level – methyl group oxidation; the second level – oxidation of acetate; the third level – oxidation of glucose in the pentose phosphate pathway.
The redox system regulates signal transmission and metabolic pathways. Nitric oxide regulates using two pathways – through S-nitrosation of intracellular proteins and directly because NO reversibly binds with metals in the active centers of enzymes, including iron of heme and of iron–sulfur clusters [122]. In proteins, tyrosines are nitrosated [123]. S-Nitrosation of cysteine residues regulates the structure of proteins and their functions [124]. For S-nitrosation, one-electron oxidation of the primary complex between NO and thiols is required. The presence in the medium of oxidizers, oxygen, or transition metals is also necessary. S-Nitrosation can also occur under anaerobic conditions [125]. In the presence of oxygen, endogenous nitric oxide causes S-nitrosation of thiols in proteins (SNO) and glutathione (GSNO) that is mediated by production of such active forms, as N2O3, NO2, and thiyl radicals [126]. GSNO is the richest source of NO in the cell [127]. It induces direct generation of S-nitrosothiols in proteins and realizes trans-nitrosation. S-Nitrosothiols are stable at 37°C and pH 7.4. The presence of transition metals destroys them to NO and disulfides, and they are hydrolyzed with production of highly reactive sulfonic acid, which, in turn, binds either with free glutathione or with the nearest cysteine by disulfide bonds [128]. Among proteins subjected to nitrosation, there are enzymes participating in glycolysis and oxidative phosphorylation.
The degradation of nitrosothiols is catalyzed by NADH-dependent GSNO reductase (GSNOR) which denitrosates GSNO, by carbonyl reductase 1 (CBR1), and thioredoxin reductase. GSNOR is the major enzyme responsible for control of NO metabolism and S-nitrosation. An increase in its expression results in denitrosation of many proteins. GSNOR is a FLD [129, 130]. FA promotes the reduction of GSNO at low NADH/NAD+ ratio in the cytoplasm. GSNOR and NADPH-dependent CBR1 catalyze the transfer of hydrogen from NADPH to GSNO with production of GSNHOH, which spontaneously transforms to GSONH2 [131]. The reduction of GSNO can result in production of sulfonamide and a product of its hydrolysis, glutathione-sulfonylic acid, which are inhibitors of glutathione transferase.
Three enzymes involved in the metabolism of cysteine, cystathione-synthase, cystathione-γ-lyase, and 3-mercaptopyruvate sulfur transferase, together with cysteine aminotransferase can generate hydrogen sulfide, which is another signaling molecule [132]. It modulates the activity of neurons, contraction of smooth muscles, and secretion of insulin [133]. An excess of NO inhibits the activity of cystathione-synthase, and FA decreases the expression of this enzyme, which lowers the production of hydrogen sulfide [134]. The combined action of FA and NO is confirmed by a significant influence of ADMA on the effect caused by FA. Treatment of PC12 line cells with FA causes production of nitric oxide, inhibits the expression of cystathione-synthase, and decreases the viability and endogenous production of hydrogen sulfide [135].
Thus, sulfhydryl groups are involved in the regulation of the intracellular content of hydrogen peroxide and of water production in the ETC and are responsible for the connection between the three levels of proton generation – methyl group oxidation, acetate oxidation, and glucose degradation in the PPP, and together with nitric oxide regulate the signal interactions in the cell.
ONE FOR ALL AND ALL FOR ONE
The redox cycle of carbon “methane – carbon dioxide – methane”, which is realized by methanotrophic bacteria and methanogenic archaea, underlies the metabolic pathways of C1 compounds. During this cycle, four electrons and four protons are released and utilized. In the center of the cycle there is a FA molecule that is equally capable of being oxidized or reduced and is used as a source of reducing equivalents and for assimilation. The pathway of the successive oxidation of methane is the most ancient donor of protons and electrons in biosystems. Methane and oxygen present a balanced donor–acceptor pair because methane as a donor gives and O2 as an acceptor takes four protons and four electrons. Carbon dioxide and water are products of this interaction. In the next stage, water becomes a donor of protons and reduces CO2 with production of glucose and release of O2. This is the closing of oxygen turnover. Glucose gives protons in the PPP and is oxidized to acetate, which is a more capacious source of protons than the methyl group. Finally, methyl group, acetate, and glucose present a three-level system of proton donors (Fig. 2).
Ammonia, sulfur hydroxide, and water are the other most ancient proton donors in addition to methane. Methane forms with them a united system: CH4–NH3–SH2–H2O. Nitrogen and sulfur have their own redox cycles. In aerobic organisms, the redox cycles of nitrogen and sulfur are components of the redox system that joins carbon and oxygen. The basic conjugated redox system consists of three main elements: the oxidation of methyl group as the donor of protons and electrons, molecular oxygen as an acceptor, whereas the amino group is a donor and acceptor concurrently. The redox balance of sulfhydryl groups of proteins, glutathione, thioredoxin, and ALA makes possible the functioning of the redox system and the general regulation of the main physiological functions of the cell, such as signal transmission, energy conversion, proliferation, and apoptosis. The basic regulation in the redox system is realized by GSH-FLD, which is also a GSNOR. Another important junction of the regulation is localized in NOS. The expression of GSH-FDH and the activity of NOS depend on the FA concentration. In the conjugated redox system, the balance of FA, H2O2, and NO concentrations is maintained strictly, as well as the redox balance of sulfur. This is also contributed to by redox sensors – transcriptional factors [136].
The glutathione system joins in a united whole not only the three-component conjugated redox system but also the three-level system of donors of protons. Glutathione reduces in mitochondria ALA, which is involved in the production of methyl groups (the glycine decarboxylase complex) and acetate (the pyruvate dehydrogenase complex). Similarly to FA, acetate is oxidized or assimilated. Acetate is oxidized by O2 in mitochondria or is used for synthesis of fatty acids (hydrocarbons). The oxidation of methyl groups and of amino groups is associated with the reduction of O2 to water through the stage of hydrogen peroxide production, whereas the oxidation of acetate is associated with the reduction of O2 to water in the ETC of mitochondria. Energy released in the ETC and stored in ATP is expended in particular for synthesis of fatty acids from acetate. Fatty acids are the final acceptors of protons. The accumulation of fatty acids in the biosystem leads to attenuation of functions of the conjugated redox system and to development of oxidative stress.
The glutathione system is now considered as a component of antioxidant protection. We think that glutathione joins in the united complex the functioning of the conjugated redox system, acetate oxidation in the Krebs cycle, and glucose oxidation in PPP. In the model of the conjugated redox system a new, not described earlier, role of the methyl group is revealed as a donor of protons besides the THFA-dependent oxidation pathway. This pathway is not related with energy production as ATP, but exists for stabilizing pH value.
REFERENCES
1.Witthoff, S., Muhlroth, A., Marienhagen, J., and
Bott, M. (2013) C1 metabolism in Corynebacterium glutamicum: an
endogenous pathway for oxidation of methanol to carbon dioxide,
Appl. Environ. Microbiol., 79, 6974-6983.
2.Van Wilder, V., De Brouwer, V., Loizeau, K.,
Gambonnet, B., Albrieux, C., Van Der Straeten, D., Lambert, W. E.,
Douce, R., Block, M. A., Rebeille, F., and Ravanel, S. (2009) C1
metabolism and chlorophyll synthesis: the Mg-protoporphyrin IX
methyltransferase activity is dependent on the folate status, New
Phytologist, 182, 137-145.
3.Yamagata, Y., Szabo, P., Szuts, D., Bacquet, C.,
Aranyi, T., and Paldi, A. (2012) Rapid turnover of DNA methylation in
human cells, Epigenetics, 7, 141-145.
4.Hanson, A. D., and Roje, S. (2001) One-carbon
metabolism in higher plants, Annu. Rev. Plant Physiol. Plant Mol.
Biol., 52, 119-137.
5.Cossins, E. A. (2000) The fascinating world of
folate and one-carbon metabolism, Can. J. Bot., 78,
691-708.
6.Sardi, E., and Tyihak, E. (1994) Simple
determination of formaldehyde in dimedone adduct form in biological
samples by high performance liquid chromatography, Biomed.
Chromatogr., 8, 313-314.
7.Huszti, Z., and Tyihak, E. (1986) Formation of
formaldehyde from S-adenosyl-L-[methyl-3H] methionine during enzymic
transmethylation of histamine, FEBS Lett., 209,
362-366.
8.Handler, P., Bernheim, M. L. C., and Klein, J. R.
(1941) The oxidative demethylation of sarcosine to glycine, J. Biol.
Chem., 138, 211-218.
9.Jia, G., Yang, C. G., Yang, S., Jian, X., Yi, C.,
Zhou, Z., and He, C. (2008) Oxidative demethylation of 3-methylthymine
and 3-methyluracil in single-stranded DNA and RNA by mouse and human
FTO, FEBS Lett., 582, 3313-3319.
10.Prather, C. W., and Sisler, E. C. (1972) Glycine
and glyoxylate decarboxylation in Nicotiana rustica roots,
Phytochemistry, 11, 1637-1647.
11.Clejan, L. A., and Cederbaum, A. I. (1993)
Stimulation by paraquat of microsomal and cytochrome
P-450-dependent oxidation of glycerol to formaldehyde,
Biochem. J., 295, 781-786.
12.Fall, R., and Benson, A. A. (1996) Leaf methanol:
the simplest natural product from plants, Trends Plant Sci.,
1, 296-301.
13.Case, G. L., and Benevenga, N. J. (1977)
Significance of formate as an intermediate in the oxidation of
methionine, S-methyl-L-cysteine and sarcosine methyl carbons to
CO2 in the rat, J. Nutr., 107, 1665-1676.
14.Chen, L., Chan, S. Y., and Cossins, E. A. (1997)
Distribution of folate derivatives and enzymes for synthesis of
10-formyltetrahydrofolate in cytosolic and mitochondrial fractions of
pea leaves, Plant Physiol., 115, 299-309.
15.Trezl, L., Rusznak, I., Tyihak, E., Szarvas, T.,
and Szende, B. (1983) Spontaneous Nε-methylation and
Nε-formylation reactions between L-lysine and
formaldehyde inhibited by L-ascorbic acid, Biochem. J.,
214, 289-292.
16.Szende, B., and Tyihak, E. (2010) Effect of
formaldehyde on cell proliferation and death, Cell Biol.
Int., 34, 1273-1282.
17.Yurimoto, H., Kato, N., and Sakai, Y. (2005)
Assimilation, dissimilation, and detoxification of formaldehyde, a
central metabolic intermediate of methylotrophic metabolism, Chem.
Rec., 5, 367-375.
18.Lidstrom, M. E. (1992) The aerobic methylotrophic
bacteria, in The Prokaryotes (Balows, A., Truper, H. G.,
Dworkin, M., Harder, W., and Schleifer, K.-H., eds.) Springer-Verlag,
N. Y., pp. 431-445.
19.Kato, N., Yurimoto, H., and Thauer, R. K. (2006)
The physiological role of the ribulose monophosphate pathway in
bacteria and archaea, Biosci. Biotechnol. Biochem., 70,
10-21.
20.Stolzenberger, J., Lindner, S. N., and Wendisch,
V. F. (2013) The methylotrophic Bacillus methanolicus MGA3
possesses two distinct fructose 1,6-bisphosphate aldolases,
Microbiology, 159, 1770-1781.
21.Quayle, J. R. (1980) Microbial assimilation of C1
compounds. The thirteenth CIBA medal lecture, Biochem. Soc.
Trans., 8, 1-10.
22.Smejkalova, H., Erb, T. J., and Fuchs, G. (2010)
Methanol assimilation in Methylobacterium extorquens AM1:
demonstration of all enzymes and their regulation, PLoS One,
5, e13001.
23.Luers, G. H., Advani, R., Wenzel, T., and
Subramani, S. (1998) The Pichia pastoris dihydroxyacetone kinase
is a PTS1-containing, but cytosolic, protein that is essential for
growth on methanol, Yeast, 14, 759-771.
24.Amaral, J. A., and Knowles, R. (1995) Growth of
methanotrophs in methane and oxygen counter gradients, FEMS
Microbiol. Lett., 126, 215-220.
25.Lipscomb, J. D. (1994) Biochemistry of the
soluble methane monoxygenase, Annu. Rev. Microbiol., 48,
371-399.
26.Dalton, H. (1992) Methane oxidation by
methanotrophs: physiological and mechanistic implications, in
Methane and Methanol Utilizers (Murrell, J. C., and Dalton, H.,
eds.) Plenum Press, N. Y., pp. 85-114.
27.Prior, S. D., and Dalton, H. (1985) The effect of
copper ions on membrane content and methane monooxygenase activity in
methanol-grown cells of Methylococcus capsulatus (Bath), J.
Gen. Microbiol., 131, 155-163.
28.Wolf, H. J., and Hanson, R. S. (1978) Alcohol
dehydrogenase from Methylobacterium organophilum, Appl.
Environ. Microbiol., 36, 105-114.
29.Anthony, C. (1992) The structure of bacterial
quinoprotein dehydrogenases, Int. J. Biochem., 24,
29-39.
30.Dijkhuizen, L., Levering, P. R., and de Vries, G.
E. (1992) The physiology and biochemistry of aerobic methanol-utilizing
Gram-negative and Gram-positive bacteria, in Methane and Methanol
Oxidizers (Murrell, J. C., and Dalton, H., eds.) Plenum Press, N.
Y., pp. 149-181.
31.Hanson, R. S., and Hanson, T. E. (1996)
Methanotrophic bacteria, Microbiol. Rev., 60,
439-471.
32.Whittenbury, R., and Krieg, N. R. (1984)
Methylococcacea fam. nov., in Bergeyʼs Manual of Systematic
Bacteriology (Krieg, N. R., and Holt, J. G., eds.) Vol. 1, pp.
256-262.
33.Hurd, Ch. D. (1938) Pyrolysis of Carbon
Compounds [in Russian], Glavnaya Redaktsiya Khimicheskoi
Literatury, Moscow-Leningrad.
34.Kato, N., Omori, Y., Tani, Y., and Ogata, K.
(1976) Alcohol oxidases of Kloeckera sp. and Hansenula
polymorpha. Catalytic properties and subunit structures, Eur. J.
Biochem., 64, 341-350.
35.Kato, N., Higuchi, T., Sakazawa, C., Nishizawa,
T., Tani, Y., and Yamada, H. (1982) Purification and properties of a
transketolase responsible for formaldehyde fixation in a
methanol-utilizing yeast, Candida boidinii (Kloeckera
sp.) No. 2201, Biochim. Biophys. Acta, 715, 143-150.
36.Chistoserdova, L., Gomelsky, L., Vorholt, J. A.,
Gomelsky, M., Tsygankov, Y. D., Thauer, R. K., and Lidstrom, M. E.
(2000) Analysis of two formaldehyde oxidation pathways in
Methylobacillus flagellates KT, a ribulose monophosphate cycle
methylotroph, Microbiology, 146, 233-238.
37.Chistoserdova, L., Vorholt, J. A., Thauer, R. K.,
and Lidstrom, M. E. (1998) C1 transfer enzymes and coenzymes linking
methylotrophic bacteria and methanogenic archaea, Science,
281, 99-102.
38.Kallen, R. G., and Jencks, W. P. (1966) The
mechanism of the condensation of formaldehyde with tetrahydrofolic
acid, J. Biol. Chem., 241, 5851-5863.
39.Vorholt, J. A., Chistoserdova, L., Stolyar, S.
M., Thauer, R. K., and Lidstrom, M. E. (1999) Distribution of
tetrahydromethanopterin-dependent enzymes in methylotrophic bacteria
and phylogeny of methylene tetrahydromethanopterin cyclohydrolases,
J. Bacteriol., 181, 5750-5757.
40.Vorholt, J. A., Chistoserdova, L., Lidstrom, M.
E., and Thauer, R. K. (1998) The NADP-dependent methylene
tetrahydromethanopterin dehydrogenase in Methylobacterium extorquens
AM1, J. Bacteriol., 180, 5351-5356.
41.De Gusseme, B., De Schryver, P., De Cooman, M.,
Verbeken, K., Boeckx, P., Verstraete, W., and Boon, N. (2009)
Nitrate-reducing, sulfide-oxidizing bacteria as microbial oxidants for
rapid biological sulfide removal, FEMS Microbiol. Ecol.,
67, 151-161.
42.Orphan, V. J., Turk, K. A., Green, A. M., and
House, C. H. (2009) Patterns of 15N assimilation and growth
of methanotrophic ANME-2 archaea and sulfate-reducing bacteria within
structured syntrophic consortia revealed by FISH-SIMS, Environ.
Microbiol., 11, 1777-1791.
43.Hell, R., and Wirtz, M. (2008) Metabolism of
cysteine in plants and phototrophic bacteria, in Sulfur Metabolism
in Phototrophic Organisms (Hell, R., Dahl, C., and Leustek, T.,
eds.) Springer, Dordrecht, Netherlands, pp. 61-94.
44.Tsuru, D., Oda, N., Matsuo, Y., Ishikawa, S.,
Ito, K., and Yoshimoto, T. (1997) Glutathione-independent formaldehyde
dehydrogenase from Pseudomonas putida: survey of functional
groups with special regard for cysteine residues, Biosci. Biotecnol.
Biochem., 61, 1354-1357.
45.Jaureguibeitia, A., Saa, L., Llama, M. J., and
Serra, J. L. (2007) Purification, characterization and cloning of
aldehyde dehydrogenase from Rhodococcus erythropolis UPV-1,
Appl. Microbiol. Biotechnol., 73, 1073-1086.
46.Gutheil, W. G., Kasimoglu, E., and Nicholson, P.
C. (1997) Induction of glutathione-dependent formaldehyde dehydrogenase
activity in Escherichia coli and Hemophilus influenza,
Biochem. Biophys. Res. Commun., 238, 693-696.
47.Fang, Z., Roberts, A. A., Weidman, K., Sharma, S.
V., Claiborne, A., Hamilton, C. J., and Dos Santos, P. C. (2013)
Cross-functionalities of Bacillus deacetylases involved in
bacillithiol biosynthesis and bacillithiol-S-conjugate detoxification
pathways, Biochem. J., 454, 239-247.
48.Martinez, M. C., Achkor, H., Persson, B.,
Fernandez, M. R., Shafqat, J., Farres, J., Jornvall, H., and Pares, X.
(1996) Arabidopsis formaldehyde dehydrogenase. Molecular
properties of plant class III alcohol dehydrogenase provide further
insights into the origins, structure and function of plant class P and
liver class I alcohol dehydrogenases, Eur. J. Biochem.,
241, 849-857.
49.Yang, Z. N., Bosron, W. F., and Hurley, T. D.
(1997) Structure of human chi chi alcohol dehydrogenase: a
glutathione-dependent formaldehyde dehydrogenase, J. Mol. Biol.,
265, 330-343.
50.Vogt, R. N., Steenkamp, D. J., Zheng, R., and
Blanchard, J. S. (2003) The metabolism of nitrosothiols in the
Mycobacteria: identification and characterization of
S-nitrosomycothiol reductase, Biochem. J., 374,
657-666.
51.Garg, A., Soni, B., Singh Paliya, B., Verma, S.,
and Jadaun, V. (2013) Low-molecular-weight thiols: glutathione (GSH),
mycothiol (MSH) potential antioxidant compound from actinobacteria,
J. App. Pharm. Sci., 3, 117-120.
52.Van Ophem, P. W., Van Beeumen, J., and Duine, J.
A. (1992) NAD linked, factor-dependent formaldehyde dehydrogenase or
trimeric, zinc-containing, long-chain alcohol dehydrogenase from
Amycolatopsis methanolica, Eur. J. Biochem., 206,
511-518.
53.Eggeling, L., and Sahm, H. (1985) The
formaldehyde dehydrogenase of Rhodococcus erythropolis, a
trimeric enzyme requiring a cofactor and active with alcohols, Eur.
J. Biochem., 150, 129-134.
54.Lessmeier, L., Hoefener, M., and Wendisch, V. F.
(2013) Formaldehyde degradation in Corynebacterium glutamicum
involves acetaldehyde dehydrogenase and mycothiol-dependent
formaldehyde dehydrogenase, Microbiology, 159,
2651-2662.
55.Holmquist, B., and Vallee, B. L. (1991) Human
liver class III alcohol and glutathione-dependent formaldehyde
dehydrogenase are the same enzyme, Biochem. Biophys. Res.
Commun., 178, 1371.
56.Danielsson, O., and Jornvall, H. (1992)
“Enzymogenesis”: classical liver alcohol dehydrogenase
origin from the glutathione-dependent formaldehyde dehydrogenase line,
Proc. Natl. Acad. Sci. USA, 89, 9247-9251.
57.Gong, D., Zhang, H., and Hu, S. (2013)
Mitochondrial aldehyde dehydrogenase 2 activation and cardioprotection,
J. Mol. Cell. Cardiol., 55, 58-63.
58.Pocker, Y., and Li, H. (1991) Kinetics and
mechanism of methanol and formaldehyde interconversion and formaldehyde
oxidation catalyzed by liver alcohol dehydrogenase, Adv. Exp. Med.
Biol., 284, 315-325.
59.Neben, I., Sahm, H., and Kula, M.-R. (1980)
Studies on an enzyme, S-formylglutathione hydrolase, of the
dissimilatory pathway of methanol in Candida boidinii,
Biochim. Biophys. Acta, 614, 81-91.
60.Sakamoto, A., Ueda, M., and Morikawa, H. (2002)
Arabidopsis glutathione-dependent formaldehyde dehydrogenase is
an S-nitrosoglutathione reductase, FEBS Lett., 515,
20-24.
61.Smith, M. (1988) Molecular genetic studies on
alcohol and aldehyde dehydrogenase: individual variation, gene mapping
and analysis of regulation, Biochem. Soc. Trans., 16,
227-230.
62.Dolferus, R., Osterman, J. C., Peacock, W. J.,
and Dennis, E. S. (1997) Cloning of the Arabidopsis and rice
formaldehyde dehydrogenase genes: implications of the origin of plant
ADH enzymes, Genetics, 146, 1131-1141.
63.Nakagawa, T., Ito, T., Fujimura, Sh., Chikui, M.,
Mizumura, T., Miyaji, T., Yurimoto, H., Kato, N., Sakai, Y., and
Tomizuka, N. (2004) Molecular characterization of the
glutathione-dependent formaldehyde dehydrogenase gene FLD1 from the
methylotrophic yeast Pichia methanolica, Yeast,
21, 445-453.
64.Wehner, E., Rao, E., and Brendel, M. (1993)
Molecular structure and genetic regulation of SFA, a gene responsible
for resistance to formaldehyde in Saccharomyces cerevisiae, and
characterization of its protein product, Mol. Gen. Genet.,
237, 351-358.
65.Barber, R. D., and Donohue, T. J. (1998) Pathways
for transcriptional activation of a glutathione-dependent formaldehyde
dehydrogenase gene, J. Mol. Biol., 280, 775-784.
66.Marchetti, P., Decaudin, D., Macho, A., Zamzami,
N., Hirsch, T., Susin, S. A., and Kroemer, G. (1997) Redox regulation
of apoptosis: impact of thiol oxidation status on mitochondrial
function, Eur. J. Immunol., 27, 289-296.
67.Sen, C. K., and Packer, L. (1996) Antioxidant and
redox regulation of gene transcription, FASEB J., 10,
709-720.
68.Soboh, B., Pinske, C., Kuhns, M., Waclawek, M.,
Ihling, C., Trchounian, K., Trchounian, A., Sinz, A., and Sawers, G.
(2011) The respiratory molybdo-selenoprotein formate dehydrogenases of
Escherichia coli have hydrogen: benzyl viologen oxidoreductase
activity, BMC Microbiol., 11, 173-184.
69.Boyington, J. C., Gladyshev, V. N., Khangulov, S.
V., Stadtman, T. C., and Sun, P. D. (1997) Crystal structure of formate
dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine,
and Fe4S4 cluster, Science, 275,
1305-1308.
70.Abaibou, H., Giordano, G., and
Mandrand-Berthelot, M. A. (1997) Suppression of Escherichia coli
formate hydrogenlyase activity by trimethylamine N-oxide is due to
drainage of the inducer formate, Microbiology, 143,
2657-2664.
71.Distel, B., Veenhuis, M., and Tabak, H. F. (1987)
Import of alcohol oxidase into peroxisomes of Saccharomyces
cerevisiae, EMBO J., 6, 3111-3116.
72.Van den Berg, M. A., and Steensma, H. Y. (1997)
Expression cassettes for formaldehyde and fluoroacetate resistance, two
dominant markers in Saccharomyces cerevisiae, Yeast,
13, 551-561.
73.Janave, M. T., Ramaswami, N. K., and Nair, P. M.
(1993) Purification and characterization of glyoxylate synthetase from
greening potato-tuber chloroplasts, Eur. J. Biochem.,
214, 889-896.
74.Halliwell, B. (1974) Oxidation of formate by
peroxisomes and mitochondria from spinach leaves, Biochem. J.,
138, 77-85.
75.Olson, B. J., Skavdahl, M., Ramberg, H.,
Osterman, J. C., and Markwell, J. (2000) Formate dehydrogenase in
Arabidopsis thaliana: characterization and possible targeting to
the chloroplast, Plant Sci., 159, 205-212.
76.Fukusaki, E., Ikeda, T., Shiraishi, T.,
Nishikawa, T., and Kobayashi, A. (2000) Formate dehydrogenase gene of
Arabidopsis thaliana is induced by formaldehyde and not by
formic acid, J. Biosci. Bioeng., 90, 691-693.
77.Bykova, N. V., Stensballe, A., Egsgaard, H.,
Jensen, O. N., and Moller, I. M. (2003) Phosphorylation of formate
dehydrogenase in potato tuber mitochondria, J. Biol. Chem.,
278, 26021-26030.
78.David, P., Colas des Francs-Small, C., Sevignac,
M., Thareau, V., Macadre, C., Langin, T., and Geffroy, V. (2010) Three
highly similar formate dehydrogenase genes located in a cluster of
NBS-LRR are differentially expressed under biotic and abiotic stresses
in Phaseolus vulgaris, Theor. Appl. Genet., 121,
87-103.
79.Ambard-Bretteville, F., Sorin, C., Rebeille, F.,
Hourton-Cabassa, C., and Colas des Francs-Small, C. (2003) Repression
of formate dehydrogenase in Solanum tuberosum increases
steady-state levels of formate and accelerates the accumulation of
proline in response to osmotic stress, Plant Mol. Biol.,
52, 26021-26030.
80.Stepanov, S. S., and Zolotareva, E. K. (2013)
Influence of methanol on the contents of NAD(P)H, free amino acids, and
protein in the cells of Chlamydomonas reinhardtii, Ukr.
Biokhim. Zh., 85, 82-89.
81.Szende, B., Tyihak, E., and Trezl, L. (2001) Role
of arginine and its methylated derivatives in cancer biology and
treatment, Cancer Cell Int., 1, 3-10.
82.Hullan, L., Trezl, L., Szarvas, T., and Csiba, A.
(1998) The hydrazine derivative aminoguanidine inhibits the reaction of
tetrahydrofolic acid with hydroxymethylarginine biomolecule, Acta
Biol. Hung., 49, 265-273.
83.Tyihak, E., Trezl, L., and Rusznak, I. (1980)
Spontaneous Nε-methylation of L-lysine by
formaldehyde, Pharmazie, 35, 18-20.
84.Trezl, L., Hullan, L., Jaszay, Z. M., Szarvas,
T., Petnehazy, I., Szende, B., Bocsi, J., Takats, Z., Vekey, K., and
Toke, L. (2003) Antagonistic reactions of arginine and lysine against
formaldehyde and their relation to cell proliferation, apoptosis,
folate cycle and photosynthesis, Mol. Cell. Biochem.,
244, 167-176.
85.Mayer, B., and Werner, E. R. (1995) In search of
a function for tetrahydrobiopterin in the biosynthesis of nitric oxide,
Naunyn Schmied. Arch. Pharmacol., 351, 453-463.
86.Alp, N. J., and Channon, K. M. (2004) Regulation
of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular
disease, Arterioscler. Thromb. Vasc. Biol., 24,
413-420.
87.Biondi, R., Ambrosio, G., De Pascali, F., Tritto,
I., Capodicasa, E., Druhan, L. J., Hemann, C., and Zweier, J. L. (2012)
HPLC analysis of tetrahydrobiopterin and its pteridine derivatives
using sequential electrochemical and fluorimetric detection:
application to tetrahydrobiopterin autoxidation and chemical oxidation,
Arch. Biochem. Biophys., 520, 7-16.
88.Kuo, L., and Hein, T. W. (2013) Vasomotor
regulation of coronary microcirculation by oxidative stress: role of
arginase, Front. Immunol., 4, 237-250.
89.Crabtree, M. J., Brixey, R., Batchelor, H., Hale,
A. B., and Channon, K. M. (2013) Integrated redox sensor and effector
functions for tetrahydrobiopterin- and glutathionylation-dependent
endothelial nitric oxide synthase uncoupling, J. Biol. Chem.,
288, 561-569.
90.Wu, G., and Morris, S. M., Jr. (1998) Arginine
metabolism: nitric oxide and beyond, Biochem. J., 336,
1-17.
91.Bronte, V., and Zanovello, P. (2005) Regulation
of immune responses by L-arginine metabolism, Nat. Rev. Immunol., 5,
641-654.
92.Hecker, M., Nematollahi, H., Hey, C., Busse, R.,
and Racke, K. (1995) Inhibition of arginase by
NG-hydroxy-L-arginine in alveolar macrophages: implications
for the utilization of L-arginine for nitric oxide synthesis, FEBS
Lett., 359, 251-254.
93.Pervin, S., Singh, R., and Chaudhuri, G. (2008)
Nitric oxide, Nω-hydroxy-L-arginine and breast cancer,
Nitric Oxide, 19, 103-106.
94.Mohan, S., and Benson, C. (2014) Understanding
the metabolic possibilities for L-arginine tolerance development,
Austin J. Nutr. Food Sci., 2, 1015-1022.
95.Vallance, P., Leone, A., Calver, A., Collier, J.,
and Moncada, S. (1992) Accumulation of an endogenous inhibitor of
nitric oxide synthesis in chronic renal failure, Lancet,
339, 572-575.
96.Janssen, W., Pullamsetti, S. S., Cooke, J.,
Weissmann, N., Guenther, A., and Schermuly, R. T. (2013) The role of
dimethylarginine dimethylaminohydrolase (DDAH) in pulmonary
fibrosis, J. Pathol., 229, 242-249.
97.Jones, W., Li, X., Qu, Z. C., Perriot, L.,
Whitesell, R. R., and May, J. M. (2002) Uptake, recycling, and
antioxidant actions of α-lipoic acid in endothelial cells,
Free Radic. Biol. Med., 33, 83-93.
98.Poole, L. B., and Nelson, K. J. (2008)
Discovering mechanisms of signaling-mediated cysteine oxidation,
Curr. Opin. Chem. Biol., 12, 18-24.
99.Russo, I., Del Mese, P., Doronzo, G., Mattiello,
L., Viretto, M., Bosia, A., Anfossi, G., and Trovati, M. (2008)
Resistance to the nitric oxide/cyclic guanosine
5′-monophosphate/protein kinase G pathway in vascular smooth
muscle cells from the obese Zucker rat, a classical animal model of
insulin resistance: role of oxidative stress, Endocrinology,
149, 1480-1489.
100.Roggenkamp, R., Sahm, H., Hinkelmann, W., and
Wagner, F. (1975) Alcohol oxidase and catalase in peroxisomes of
methanol-grown Candida boidinii, Eur. J. Biochem.,
59, 231-236.
101.Garner, C. D., Lee, E. W., Terzo, T. S., and
Louis-Ferdinand, R. T. (1995) Role of retinal metabolism in
methanol-induced retinal toxicity, J. Toxicol. Environ. Health,
44, 43-56.
102.Danpure, C. J., Purkiss, P., Jennings, P. R.,
and Watts, R. W. (1986) Mitochondrial damage and the subcellular
distribution of 2-oxoglutarate: glyoxylate carboligase in normal human
and rat liver and in the liver of a patient with primary hyperoxaluria
type I, Clin. Sci. (London), 70, 417-423.
103.Nemeria, N., Binshtein, E., Patel, H.,
Balakrishnan, A., Vered, I., Shaanan, B., Barak, Z., Chipman, D., and
Jordan, F. (2012) Glyoxylate carboligase: a unique thiamin
diphosphate-dependent enzyme that can cycle between the
4′-aminopyrimidinium and 1′,4′-iminopyrimidine
tautomeric forms in the absence of the conserved glutamate,
Biochemistry, 51, 7940-7952.
104.Appling, D. R. (1991) Compartmentation of
folate-mediated one-carbon metabolism in eukaryotes, FASEB J.,
5, 2645-2651.
105.Chiba, Y., Terada, T., Kameya, M., Shimizu, K.,
Arai, H., Ishii, M., and Igarashi, Y. (2012) Mechanism for
folate-independent aldolase reaction catalyzed by serine
hydroxymethyltransferase, FEBS J., 279, 504-514.
106.Wilson, R. L., Steiert, P. S., and Stauffer, G.
V. (1993) Positive regulation of the Escherichia coli glycine
cleavage enzyme system, J. Bacteriol., 175, 902-904.
107.Bonekamp, N. A., Islinger, M., Lazaro, M. G.,
and Schrader, M. (2013) Cytochemical detection of peroxisomes and
mitochondria, Methods Mol. Biol., 931, 467-482.
108.Fridovich, I. (1995) Superoxide radical and
superoxide dismutases, Annu. Rev. Biochem., 64,
97-112.
109.Santos, C. X., Tanaka, L. Y., Wosniak, J., and
Laurindo, F. R. (2009) Mechanisms and implications of reactive oxygen
species generation during the unfolded protein response: roles of
endoplasmic reticulum oxidoreductases, mitochondrial electron
transport, and NADPH oxidase, Antioxid. Redox Signal.,
11, 2409-2427.
110.Lee, J. W., and Helmann, J. D. (2006)
Biochemical characterization of the structural Zn2+ site in
the Bacillus subtilis peroxide sensor PerR, J. Biol.
Chem., 281, 23567-23578.
111.Lee, J. W., and Helmann, J. D. (2007)
Functional specialization within the Fur family of metalloregulators,
Biometals, 20, 485-499.
112.Lushchak, V. I. (2012) Glutathione homeostasis
and functions: potential targets for medical interventions, J. Amino
Acids, DOI: 10.1155/2012/736837.
113.Lu, S. C. (2009) Regulation of glutathione
synthesis, Mol. Asp. Med., 30, 42-59.
114.Matouskova, P., Bartikova, H., Bousova, I.,
Levorova, L., Szotakova, B., and Skalova, L. (2015) Drug-metabolizing
and antioxidant enzymes in monosodium L-glutamate obese mice, Drug
Metab. Dispos., 43, 258-265.
115.Mani, M., Khaghani, S., Gol Mohammadi, T.,
Zamani, Z., Azadmanesh, K., Meshkani, R., Pasalar, P., and Mostafavi,
E. (2013) Activation of Nrf2-antioxidant response element mediated
glutamate cysteine ligase expression in hepatoma cell line by
homocysteine, Hepat. Mon., 13, e8394.
116.Nohl, H., and Gille, L. (1998) Evaluation of
the antioxidant capacity of ubiquinol and dihydrolipoic acid, Z.
Naturforsch. C, 53, 250-253.
117.Hawkes, H. J., Karlenius, T. C., and Tonissen,
K. F. (2014) Regulation of the human thioredoxin gene promoter and its
key substrates: a study of functional and putative regulatory elements,
Biochim. Biophys. Acta, 1840, 303-314.
118.Sengupta, R., and Holmgren, A. (2012) The role
of thioredoxin in the regulation of cellular processes by
S-nitrosylation, Biochim. Biophys. Acta, 1820,
689-700.
119.Sengupta, R., and Holmgren, A. (2013)
Thioredoxin and thioredoxin reductase in relation to reversible
S-nitrosylation, Antioxid. Redox Signal., 18,
259-269.
120.Lu, J., and Holmgren, A. (2014) The thioredoxin
antioxidant system, Free Radic. Biol. Med., 66,
75-87.
121.Busse, E., Zimmer, G., Schopohl, B., and
Kornhuber, B. (1992) Influence of alpha-lipoic acid on intracellular
glutathione in vitro and in vivo,
Arzneimittelforschung, 42, 829-831.
122.Han, D., Handelman, G., Marcocci, L., Sen, C.
K., Roy, S., Kobuchi, H., Tritschler, H. J., Flohe, L., and Packer, L.
(1997) Lipoic acid increases de novo synthesis of cellular
glutathione by improving cystine utilization, Biofactors,
6, 321-338.
123.Cruz-Ramos, H., Crack, J., Wu, G., Hughes, M.
N., Scott, C., Thomson, A. J., Green, J., and Poole, R. K. (2002) NO
sensing by FNR: regulation of the Escherichia coli
NO-detoxifying flavohaemoglobin, Hmp, EMBO J., 21,
3235-3244.
124.Beckman, J. S., and Koppenol, W. H. (1996)
Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and
ugly, Am. J. Physiol., 271, 1424-1237.
125.Gould, N., Doulias, P.-Th., Tenopoulou, M.,
Raju, K., and Ischiropoulos, H. (2013) Regulation of protein function
and signaling by reversible cysteine S-nitrosylation, J.
Biol. Chem., 288, 26473-26479.
126.Seth, D., Hausladen, A., Wang, Y. J., and
Stamler, J. S. (2012) Endogenous protein S-nitrosylation in E.
coli: regulation by OxyR, Science, 336, 470-473.
127.Schrammel, A., Gorren, A. C., Schmidt, K.,
Pfeiffer, S., and Mayer, B. (2003) S-nitrosation of glutathione by
nitric oxide, peroxynitrite, and NO/superoxide, Free Radic. Biol.
Med., 34, 1078-1088.
128.Corpas, F. J., Alche, J. D., and Barroso, J. B.
(2013) Current overview of S-nitrosoglutathione (GSNO) in higher
plants, Front. Plant Sci., 4, 126-132.
129.Poole, L. B., Karplus, P. A., and Claiborne, A.
(2004) Protein sulfenic acids in redox signaling, Annu. Rev.
Pharmacol. Toxicol., 44, 325-347.
130.Haqqani, A. S., Do, S. K., and Birnboim, H. C.
(2003) The role of a formaldehyde dehydrogenase-glutathione pathway in
protein S-nitrosation in mammalian cells, Nitric Oxide,
9, 172-181.
131.Hou, Q., Jiang, H., Zhang, X., Guo, C., Huang,
B., Wang, P., Wang, T., Wu, K., Li, J., Gong, Z., Du, L., Liu, Y., Liu,
L., and Chen, C. (2011) Nitric oxide metabolism controlled by
formaldehyde dehydrogenase (fdh, homolog of mammalian GSNOR) plays a
crucial role in visual pattern memory in Drosophila, Nitric
Oxide, 24, 17-24.
132.Smith, B. C., and Marletta, M. A. (2012)
Mechanisms of S-nitrosothiol formation and selectivity in nitric oxide
signaling, Curr. Opin. Chem. Biol., 16, 498-506.
133.Kimura, H. (2010) Hydrogen sulfide: from brain
to gut, Antioxid. Redox Signal., 12, 1111-1123.
134.Kimura, H. (2011) Hydrogen sulfide: its
production, release and functions, Amino Acids, 41,
113-121.
135.Tang, X. Q., Fan, H. R., Zhou, C. F., Zhuang,
Y. Y., Zhang, P., Gu, H. F., and Hu, B. (2013) A novel mechanism of
formaldehyde neurotoxicity: inhibition of hydrogen sulfide generation
by promoting overproduction of nitric oxide, PLoS One, 8,
e54829.
136.Brigelius-Flohe, R., and Flohe, L. (2011) Basic
principles and emerging concepts in the redox control of transcription
factors, Antioxid. Redox Signal., 15, 2335-2381.