Purification and Characterization of Recombinant Deinococcus radiodurans RNA Polymerase
D. M. Esyunina* and A. V. Kulbachinskiy*
Institute of Molecular Genetics, Russian Academy of Sciences, 123182 Moscow, Russia; fax: 7 (499) 196-0221; E-mail: akulb@img.ras.ru; es_dar@inbox.ru* To whom correspondence should be addressed.
Received April 13, 2015; Revision received May 15, 2015
The radioresistant bacterium Deinococcus radiodurans is one of the most interesting models for studies of cell stress resistance. Analysis of the mechanisms of gene expression in D. radiodurans revealed some specific features of the transcription apparatus that might play a role in cell resistance to DNA-damaging conditions. In particular, RNA polymerase from D. radiodurans forms unstable promoter complexes and during transcription elongation has a much higher rate of RNA cleavage than RNA polymerase from Escherichia coli. Analysis of the structure and functions of D. radiodurans RNA polymerase is complicated due to the absence of convenient genetic systems for making mutations in the RNA polymerase genes and difficulties with enzyme purification. In this work, we developed a system for expression of D. radiodurans RNA polymerase in E. coli cells. We obtained an expression vector encoding all core RNA polymerase subunits and defined optimal conditions for the expression and purification of the RNA polymerase. It was found that D. radiodurans RNA polymerase has much higher rates of RNA cleavage than E. coli RNA polymerase under a wide range of conditions, including variations in the concentration of catalytic magnesium ions and pH values of the reaction buffer. The expression system can be used for further studies of the RNA cleavage reaction and the mechanisms of transcription regulation in D. radiodurans, including analysis of mutant RNA polymerase variants.
KEY WORDS: Deinococcus radiodurans, protein expression, RNA polymerase, RNA cleavageDOI: 10.1134/S0006297915100077
Abbreviations: DTT, dithiothreitol; EC, elongation complex; IPTG, isopropyl-β-D-1-thiogalactopyranoside; RNAP, RNA polymerase.
RNA polymerase (RNAP) is a complex molecular machine that transcribes
genes. In bacteria, transcription is initiated by the RNAP holoenzyme
that contains the σ subunit responsible for promoter recognition.
Subsequent steps of transcription – elongation and
termination of RNA synthesis – can be completed in the
absence of the σ subunit by the RNAP core enzyme, which consists
of five subunits, α2ββ′ω. The
active site of RNAP is formed by the β′ and β subunits
and contains two catalytic magnesium ions that play a key role in
catalysis [1-3].
During transcription elongation, RNAP not only catalyzes RNA synthesis but can also perform endonucleolytic RNA cleavage. The RNA cleavage reaction likely plays an important role in transcription proofreading and helps to overcome pauses during transcription elongation [4-6]. RNA cleavage is preceded by backtracking of the elongation complex (EC), which results in displacement of the RNA 3′-end from the active site into the secondary RNAP channel [7, 8]. Recent biochemical and structural studies suggest a detailed molecular mechanism of RNA synthesis by bacterial RNAP [2, 3, 9], but less is known about the mechanism of RNA cleavage.
Deinococcus radiodurans is a unique bacterium that is highly resistant to ionizing radiation and other stress conditions [10]. It was shown that the cell response to such conditions involves complex changes in gene expression that are important for cell recovery and DNA repair (e.g. [11, 12]). At the same time, only a few studies of the transcription apparatus of D. radiodurans have been published to date due to difficulties in purification of RNAP and transcription factors from this bacterium and the absence of a convenient genetic system for analysis of mutations in the RNAP genes.
In comparison with E. coli RNAP, D. radiodurans RNAP forms unstable promoter complexes and poorly melts DNA around the starting point of transcription [13]. It was shown that RNAP from the phylogenetically related thermophilic bacterium Thermus aquaticus has similar properties, which are determined by both the σ subunit and the core enzyme of RNAP [13-16]. Possible roles of these features in transcription regulation and cell stress resistance remain unknown. During transcription elongation, D. radiodurans RNAP reveals a much higher level of RNA cleavage activity than E. coli RNAP, which might play a role in transcription of damaged DNA [17-19]. Analysis of these differences is important for understanding the mechanism of the RNA cleavage reaction and its functional role in transcription.
Three major approaches are currently used to purify bacterial RNAPs: purification of native RNAP from bacterial cells; reconstitution in vitro from individually expressed and purified subunits; co-expression of all RNAP genes and purification of recombinant RNAP. The advantages of the latter approach include high protein yields, the ability to obtain RNAPs of pathogenic bacteria, the absence of admixtures of transcription factors (if the expression is performed in a heterologous system), and the ability to introduce mutations in the RNAP genes. Currently, such systems are available for RNAPs from E. coli [20], T. aquaticus [21], B. subtilis [22], M. tuberculosis [23, 24], X. campestris [25], and others. The purpose of this work was to obtain a similar expression system for D. radiodurans RNAP that could be further used in studies of the transcription mechanisms in this bacterium.
MATERIALS AND METHODS
The experiments were performed with standard reagents from Sigma, USA (for electrophoresis and buffer solutions), New England Biolabs, USA (enzymes for molecular cloning), and GE Healthcare, USA (chromatography columns). Molecular cloning was performed with kits from Zymo Research and Thermo Fisher Scientific (USA). DNA and RNA oligonucleotides were synthesized by Syntol and DNA Synthesis (Russia).
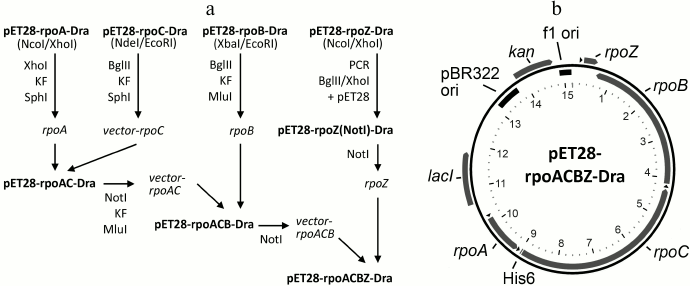
Fig.1Construction of the pET28-rpoACBZ-Dra plasmid. The scheme of the plasmid assembly is shown in Fig. 1a. The rpoA, rpoC, rpoB, and rpoZ genes of D. radiodurans were cloned into the pET28 vector between the NcoI/XhoI, NdeI/EcoRI, XbaI/EcoRI, and NcoI/XhoI sites, respectively. Plasmid pET28-rpoZ(NotI)-Dra, containing NotI sites at both sides of the rpoZ gene, was obtained by PCR amplification of rpoZ from plasmid pET28-rpoZ-Dra with primers pet28-BglII-NotI-d 5′-ATCGAGATCTGCGGCCGCCGAAATTAATACG-3′ and DraZ-XhoI-NotI-r 5′-TCACTCGAGTGCGGCCGCTCAGTCGCGTTCGCGCTCG-3′, followed by cloning of the PCR product into the BglII/XhoI sites of pET28. Plasmid pET28-rpoAC-Dra was obtained by transfer of the rpoA gene from pET28-rpoA-Dra into pET28-rpoC-Dra. For this purpose, plasmids pET28-rpoA-Dra and pET28-rpoC-Dra were cut with restriction endonucleases XhoI and BglII, respectively, followed by treatment with the Klenow enzyme. Both linear plasmids were treated with restriction endonuclease SphI, and the fragment XhoI(blunt)-SphI from pET28-rpoA-Dra, containing rpoA gene and a part of pET28, was ligated with pET28-rpoC-Dra. Plasmid pET28-rpoACB-Dra was obtained by transfer of the rpoB gene from plasmid pET28-rpoB-Dra into pET28-rpoAC-Dra. For this purpose, pET28-rpoB-Dra was treated with BglII and pET28-rpoAC-Dra was treated with NotI, followed by treatment of both plasmids with the Klenow enzyme, restriction endonuclease MluI, and ligation. At the last step, the rpoZ gene was transferred from plasmid pET28-rpoZ(NotI)-Dra into the NotI site of pET28-rpoACB-Dra. The final plasmid pET28-rpoACBZ-Dra encodes all four core RNAP subunits with a His6-tag at the N-terminus of the β′ subunit (rpoC gene).
Fig. 1. Expression plasmid pET28-rpoACBZ-Dra. a) Scheme of the plasmid assembly. KF, Klenow fragment. b) Plasmid map. The plasmid contains 15,215 bp, the numbering being shown inside (in kb). The pBR322 and f1 replication origins are shown in black. The pET28 vector encodes kanamycin resistance (kan) and lac-repressor. The rpoA, rpoB, rpoC, and rpoZ genes encode the α, β, β′, and ω subunits, respectively, of the core enzyme of D. radiodurans RNAP. Positions of the T7 RNAP promoters are shown with black triangles. The rpoC gene encodes a His6 tag at its beginning. The plasmid map was generated by the PlasMapper version 2.0 software.
Protein purification. Escherichia coli core enzyme RNAP was expressed in E. coli strain BL21(DE3) from plasmid pVS10 (kindly provided by I. Artsimovitch) and purified as previously described [20]. Recombinant D. radiodurans RNAP was expressed in E. coli strain BL21(DE3) from plasmid pET28-rpoACBZ-Dra. Several transformant colonies from a fresh Petri dish were inoculated into 1 liter of LB broth containing 50 µg/ml kanamycin and 0.1 mM IPTG (isopropyl-β-D-1-thiogalactopyranoside) and grown at 22°C for 16 h with shaking (200 rpm) (final OD600 > 3, cell yield 7-9 g from 1 liter of culture). The cells were centrifuged (4000 rpm) and lysed in buffer A (50 mM Tris-HCl, pH 7.9, 233 mM NaCl, 2 mM EDTA, 5% glycerol, 0.2% Tween-20, 1 mM β-mercaptoethanol, 0.2 mg/ml lysozyme, and 0.1 mM phenylmethylsulfonyl fluoride) for 30 min on ice, followed by sonication (400W sonicator; Sonics & Materials Inc., USA). The lysate was centrifuged (15,000 rpm) and Polymin P (polyethyleneimine) was added to the supernatant to 0.6%. The suspension was centrifuged (13,000 rpm), and the pellet was washed with buffer TGED (10 mM Tris-HCl, pH 7.9, 0.1 mM EDTA, 0.1 mM dithiothreitol (DTT), 5% glycerol) containing 350 mM NaCl and centrifuged in the same regime. The proteins were eluted from the pellet fraction with buffer TGED containing 1 M NaCl, followed by the addition of dry ammonium sulfate (35 g per 100 ml of eluate) to the supernatant fraction. The pellet obtained after centrifugation (13,000 rpm) was dissolved in buffer TGED without NaCl and loaded onto a heparin-Sepharose column (5 ml Heparin-HiTrap; GE Healthcare) equilibrated with buffer B (20 mM Tris-HCl, pH 7.9, 5% glycerol, 100 mM NaCl). The column was successively washed with buffer B containing various NaCl concentrations: nonspecific proteins were eluted at 350 mM NaCl, D. radiodurans RNAP was eluted at 433 mM NaCl, and E. coli RNAP was eluted at 600 mM NaCl. This step removed ~90% of E. coli RNAP. Fractions containing D. radiodurans RNAP were further purified by affinity chromatography on 5 ml HiTrap Chelating Column charged with Ni2+ ions. The column was washed from unbound proteins with Ni-buffer (10 mM Tris-HCl, pH 7.9, 500 mM NaCl) containing 20 mM imidazole. Deinococcus radiodurans RNAP was eluted with Ni-buffer containing 100 mM imidazole. After dialysis against buffer containing 40 mM Tris-HCl, pH 7.9, 100 mM NaCl, 1 mM EDTA, 0.1 mM DTT, and 5% glycerol, RNAP was purified by anion-exchange chromatography on the Mono Q 5/5 HR column (1 ml; GE Healthcare). The RNAP was eluted with the salt gradient of buffer M (40 mM Tris-HCl, pH 8.1, 5% glycerol, 1 mM EDTA, 0.1 mM DTT) containing from 0 to 600 mM of NaCl. All fractions were analyzed by 8% SDS-PAGE. The proteins were concentrated with Amicon 100 ultrafiltration devices (to final concentrations of 2-10 mg/ml), supplemented with glycerol to 50% and DTT to 1 mM, and stored at –20°C (up to 1 year) or –70°C (for long-term storage). The RNAP yield was 2-10 mg from 1 liter of cell culture.
Transcription in vitro. The nucleotide addition rate and RNA cleavage by RNAP were analyzed on synthetic oligonucleotide transcription templates as described previously [17, 19]. ECs were assembled from the core enzyme of RNAP, template and nontemplate DNA oligonucleotides, and 5′-labeled RNA in transcription buffer TB-40 (40 mM Tris-HCl, pH 7.9, 40 mM NaCl). Reactions of RNA cleavage or RNA synthesis were initiated by the addition of 10 mM MgCl2 or 10 mM MgCl2 plus 1 mM NTP, respectively. Some experiments were performed with a rapid-quench flow apparatus as described previously [19]. The reactions were stopped by the addition of equal volume of stop-solution containing 8 M urea, 2× TBE buffer, and 20 mM EDTA. RNA products were separated by 23% denaturing PAGE and visualized by phosphorimaging, followed by quantification of the reacted RNA at each time point. The values of the observed reaction rate constants (kobs) were calculated using the GraFit software (Erithacus Software) in accordance to the single-exponential equation:
A = Amax·(1 – exp(–kobs·t)),
where A is the amount of RNA product, Amax is the maximal amount of RNA product, kobs is the observed first order kinetic constant, and t is the reaction time. The efficiency of the binding of magnesium ions in the RNAP active site during the RNA cleavage reaction was analyzed by measuring the observed RNA cleavage rate constants (kobs) at various MgCl2 concentrations (0.1-100 mM) at 20°C in buffer TB-40. The values of the apparent dissociation constant (Kd,app) for the magnesium ion binding were calculated in accordance to the hyperbolic equation:
kobs = kobs,max·C/(Kd,app + C),
where kobs,max is the maximal rate of RNA cleavage at saturating concentration of magnesium ions and C is magnesium concentration in the reaction. The RNA cleavage rates at different pH values of the reaction buffer were measured in buffer solutions containing Tris-HCl (for pH 7.3, 7.7, 8.0, 8.3, 8.7, 9.0, 9.3) or CAPS (3-(cyclohexylamino)-1-propanesulfonic acid)-NaOH (for pH 10) in the presence of 10 mM MgCl2 at 20°C for E. coli RNAP and 10°C for D. radiodurans RNAP.
RESULTS
Purification of recombinant D. radiodurans RNAP. Plasmid pET28-rpoACBZ-Dra (Fig. 1b) encoding the α, β, β′, and ω subunits of the core enzyme of D. radiodurans RNAP was obtained by stepwise cloning of the individual genes into the pET28 vector. In the final plasmid, all D. radiodurans RNAP genes (rpoA, rpoB, rpoC, rpoZ) are cloned under the control of the T7 phage RNAP promoters, and the β′ subunit contains an N-terminal His6-tag.
Deinococcus radiodurans RNAP was expressed in E. coli strain BL21(DE3) containing a chromosomally encoded copy of the T7 RNAP gene under the control of inducible lac-promoter. To obtain a high yield of active RNAP, the cells were grown at room temperature (22°C) in the presence of low IPTG concentration (0.1 mM). Proteins corresponding to the α, β, and β′ subunits of D. radiodurans RNAP become clearly visible in the cells under these conditions (Fig. 2, lane 1). Increase in the temperature results in overexpression of the β′ subunit and the appearance of inclusion bodies, while decrease in the temperature reduces the protein yield.
Fig. 2. Electrophoregram of protein fractions obtained at different steps of purification of D. radiodurans RNAP: 1) lysate of E. coli cells after expression of D. radiodurans RNAP; 2) eluate from polyethyleneimine before loading to heparin column; 3) heparin-Sepharose chromatography, elution of D. radiodurans RNAP after washing the column with buffer containing 433 mM NaCl; 4) elution of E. coli RNAP after washing the column with buffer containing 600 mM NaCl; 5) Ni-affinity chromatography, elution fraction; 6) protein marker (Thermo Scientific PageRuler Unstained Broad Range Protein Ladder). Molecular weights of the marker proteins (in kDa) are shown on the right. Positions of D. radiodurans RNAP subunits are shown on the left; the β and β′ subunits of E. coli RNAP, which have about the same mobility, are shown with an asterisk.
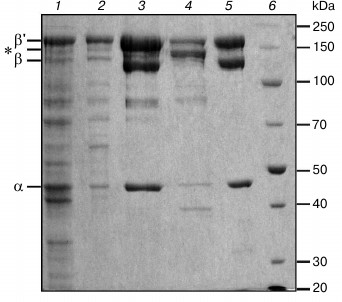
The RNAP was purified by standard methods (Fig. 2) including cell lysis (lane 1), precipitation of nucleic acids and associated proteins (including RNAP) with polyethylenimine (lane 2), heparin-Sepharose chromatography (lanes 3 and 4), Ni-affinity chromatography (lane 5), and anion-exchange chromatography on a MonoQ column. An important goal of the procedure was to remove any admixtures of E. coli RNAP from the D. radiodurans RNAP preparation. Such purification is partially achieved during heparin-Sepharose chromatography, because the two RNAPs can be eluted from the column at different salt concentrations (433 and 600 mM NaCl for D. radiodurans and E. coli RNAPs, respectively; Fig. 2, lanes 3 and 4). The remaining admixture of E. coli RNAP is removed during Ni-affinity chromatography, since only D. radiodurans RNAP has a His6-tag in its β′ subunit. The purity of the D. radiodurans RNAP preparation after the Ni-affinity chromatography is >98% (based on Coomassie staining). The final step of ion-exchange chromatography (MonoQ column) was performed to remove any possible admixtures of E. coli transcription factors, which might bind D. radiodurans RNAP and affect its activity. Comparison of RNAP samples before and after this step did not reveal significant differences in their purity and catalytic activity.
Fig.3Analysis of transcription properties of D. radiodurans RNAP. The activity of the purified RNAP was analyzed in in vitro transcription tests on RNA synthesis and RNA cleavage. The rate of nucleotide incorporation was measured in synthetic ECs obtained from oligonucleotides corresponding to the RNA–DNA hybrid and downstream DNA duplex in the EC (Fig. 3a). This allows direct measurement of the rate of nucleotide addition to the nascent RNA, excluding the step of transcription initiation.
Fig. 3. Measurement of rates of nucleotide incorporation by D. radiodurans (Dra) and E. coli (Eco) RNAPs. a) Scheme of nucleotide addition in the synthetic EC. Arrow designates the site of nucleotide incorporation into RNA. b) Rates of nucleotide incorporation. The reaction was performed at 30°C in the presence of 1 mM UTP (complementary nucleotide) or 1 mM CTP (noncomplementary nucleotide). The ratios of the incorporation rates for correct and incorrect nucleotides are shown in the last column.
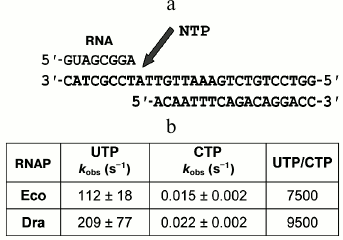
It was found that recombinant D. radiodurans RNAP does not differ from native RNAP purified from D. radiodurans cells in the rate of incorporation of UTP, which is complementary to the next template nucleotide (kobs ≈ 210 and 215 s–1, respectively, at 30°C) (Fig. 3b, [19]). Deinococcus radiodurans RNAP is also only slightly faster than E. coli RNAP in the rate of RNA synthesis (no more than 2-fold; Fig. 3b). We also measured the rates of noncomplementary nucleotide addition (for CTP) and the fidelity of RNA synthesis, which can be expressed as the ratio of the incorporation rates for correct and incorrect nucleotides. It was found that the rate of CTP addition by D. radiodurans RNAP is about 1.5-fold higher than in the case of E. coli RNAP. Therefore, these RNAPs do not differ significantly in the rates and fidelity of RNA synthesis (Fig. 3b).
To measure the rate of RNA cleavage, we used a synthetic template consisting of three oligonucleotides: template DNA strand, nontemplate DNA strand, and 15-nucleotide RNA transcript (Fig. 4a). As was shown previously, ECs obtained from synthetic oligonucleotides of such structure do not differ in their properties from natural complexes [26, 27]. EC reconstitution was performed in transcription buffer lacking divalent metal ions. The reaction was started by the addition of Mg2+, resulting in the cleavage of two nucleotides from the RNA 3′-end and appearance of a 13-nucleotide RNA product (shown with an arrow in Fig. 4a). It was found that in this EC recombinant D. radiodurans RNAP cleaves RNA 27 times faster than E. coli RNAP (kobs = 0.83 ± 0.15 and 0.03 ± 0.007 min–1, respectively, at 20°C, 10 mM MgCl2, pH 7.9). This is in agreement with previously published data on the increased rate of RNA cleavage by native RNAP purified from D. radiodurans cells [17].
Fig. 4. Analysis of RNA cleavage by D. radiodurans (Dra) and E. coli (Eco) RNAPs. a) Scheme of the synthetic EC used in the experiment. The site of RNA cleavage is indicated with an arrow. b) Dependence of RNA cleavage by D. radiodurans and E. coli RNAPs on the MgCl2 concentration. MgCl2 concentrations are shown on the x-axis; the rates of RNA cleavage (kobs, min–1) for D. radiodurans RNAP (1) and E. coli RNAP (2) at 20°C are shown on the y-axis. c) pH dependences of the RNA cleavage rates for D. radiodurans and E. coli RNAPs. pH values are shown on the x-axis; the rates of RNA cleavage (kobs, min–1) for D. radiodurans RNAP (at 10°C (1)) and E. coli RNAP (at 20°C (2)) are shown on the y-axis.
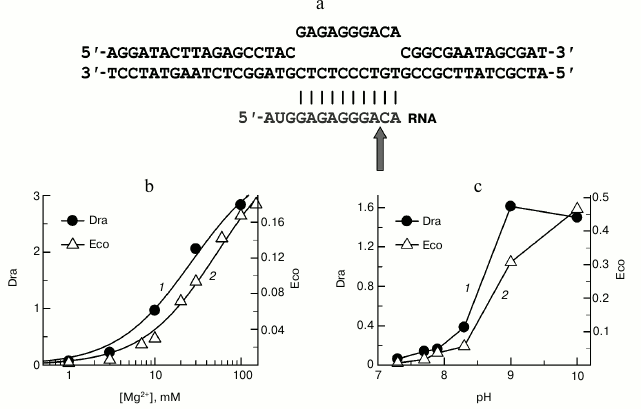
As shown previously, the rate of RNA cleavage significantly depends on the concentration of Mg2+, which plays the key role in catalysis by coordinating the substrates in the RNAP active site [28, 29]. To reveal possible difference in the binding of the catalytic ions by E. coli and D. radiodurans RNAPs, we analyzed RNA cleavage at different MgCl2 concentrations (from 0.1 to 100 mM). Deinococcus radiodurans RNAP was found to have a much higher rate of RNA cleavage at all studied MgCl2 concentrations (Fig. 4b). The concentration dependences of the RNA cleavage rates were used to calculate the apparent dissociation constants (Kd,app) for Mg2+. It was found that D. radiodurans RNAP only slightly differs from E. coli RNAP in the efficiency of Mg2+ binding (about 2-fold, Kd,app = 23 ± 5 and 55 ± 2 mM, respectively). Furthermore, significant differences between these RNAPs in the RNA cleavage rates are also observed at saturating Mg2+ concentrations (~16-fold difference in the kobs values at 100 mM MgCl2; 0.167 ± 0.004 and 2.8 ± 0.1 min–1 for E. coli and D. radiodurans RNAPs, respectively).
Another factor that might determine the difference in the RNA cleavage rates between E. coli and D. radiodurans RNAPs are differences in pH-dependence of the reaction, since RNA cleavage by bacterial RNAPs is significantly activated at increased pH values [9, 30-32]. However, our experiments revealed similar pH-dependences of the RNA cleavage rates for the two RNAPs in the pH range from 7.0 to 10.0 (Fig. 4c). At pH values above 8.0-8.5, a significant increase in the RNA cleavage rates was observed for RNAPs from both E. coli and D. radiodurans. It should be noted that the experiment with D. radiodurans RNAP was performed at lower temperature than in the case of E. coli RNAP (10 and 20°C, respectively), which was necessary to measure the very high cleavage rates at increased pH values. Nevertheless, even under these conditions the rates of RNA cleavage by D. radiodurans RNAP were much higher (4-5-fold) than the rates of E. coli RNAP over the whole pH range.
Therefore, the observed differences between E. coli and D. radiodurans RNAPs in the rates of RNA cleavage only slightly depend on the differences in the binding of catalytic Mg2+ and the pH-dependence of the reaction.
DISCUSSION
Development of the expression system for recombinant D. radiodurans RNAP is an important step in the analysis of the transcription apparatus of this bacterium, since it yields highly purified RNAP and its mutant variants for in vitro studies. In this work, we prepared a vector for the expression of the core enzyme of D. radiodurans RNAP in E. coli cells. This plasmid is analogous to the previously described plasmid used for expression of RNAP from the related bacterium T. aquaticus [21]. Analysis of the expression conditions showed that under “mild” induction conditions (0.1 mM IPTG concentration, room temperature) it is possible to obtain a sufficiently high protein yield (up to 10 mg of purified protein from 1 liter of cell culture). Purification of D. radiodurans RNAP by standard methods (polyethyleneimine precipitation, heparin-Sepharose chromatography, Ni-affinity chromatography) removes most admixtures of E. coli RNAP and other proteins. Additional anion-exchange chromatography does not change the transcriptional activity of the purified RNAP but decreases the overall protein yield. In general, the method yields RNAP without admixtures of D. radiodurans or E. coli transcription factors.
Analysis of the properties of recombinant RNAP in in vitro transcription experiments revealed no significant differences from native RNAP purified directly from D. radiodurans cells [13, 17-19]. The measurement of the rates of incorporation of correct and incorrect nucleotides in synthetic ECs demonstrated that D. radiodurans RNAP is about 1.5-2.0 times faster than E. coli RNAP and does not differ from the latter in the fidelity of RNA synthesis. This is in agreement with previously published data showing that these RNAPs do not differ significantly in the average elongation rates [13, 17].
At the same time, D. radiodurans RNAP has a much higher rate of RNA cleavage at the elongation step of transcription (this work and [17]). The reaction of RNA cleavage occurs in backtracked ECs, which can form after misincorporation of incorrect nucleotides and in the presence of various roadblocks on the DNA template [6, 33]. In such cases, RNA cleavage can help to correct transcription errors and reactivate arrested ECs. Thereby, the increased rate of RNA cleavage by D. radiodurans RNAP might have an important role under stress conditions, including those that lead to a high level of DNA damage. It should be noted that the reaction of RNA cleavage by bacterial RNAP can also be stimulated by specialized Gre factors [28, 29], and the expression of the GreA factor in D. radiodurans cells is induced after irradiation [11].
The binding of Mg2+ in the RNAP active site during RNA cleavage is much weaker than during nucleotide incorporation, which may have a regulatory role [26, 28, 29]. Therefore, a possible reason for the increased reaction rate in the case of D. radiodurans RNAP could be the more efficient binding of magnesium ions. However, our experiments demonstrated that E. coli and D. radiodurans RNAPs have similar dependences of the reaction rates on magnesium concentration, and D. radiodurans RNAP has a much higher cleavage rate even at saturating Mg2+ concentrations.
The binding of magnesium ions in the active site depends on their interactions with aspartic acid residues of the NADFDGD motif of the RNAP β′ subunit (residues 458-464 in E. coli numbering); this motif is identical in all RNAPs. However, substitution of an adjacent nonconserved residue in E. coli RNAP with the corresponding residue from D. radiodurans RNAP (A455E) was shown to increase the RNA cleavage rate about 2-fold [17]. It can be proposed that this substitution might somehow affect magnesium ion binding (in accordance with the observed small differences in the binding of these ions by D. radiodurans and E. coli RNAPs; see Fig. 4b), but this hypothesis requires further experimental study.
The rate of RNA cleavage by RNAP significantly depends on the pH value of the reaction buffer, which probably affects ionization of certain groups in the enzyme active site [9, 30-32]. In the case of E. coli and T. aquaticus RNAPs, this reaction is activated at pH ≥ 8.5. We demonstrated that D. radiodurans RNAP has a similar pH dependence, and the RNA cleavage rate at all studied pH values is much higher than the rate of E. coli RNAP. Thus, the observed differences are not explained by differences in ionization of functional groups of the active site involved in the cleavage reaction. These differences might be explained by specific features of the active site of D. radiodurans RNAP and its contacts with nucleic acids during the cleavage reaction. Analysis of these structural features will be an important goal of future studies.
In conclusion, we have developed a system for expression of recombinant D. radiodurans RNAP that allows analysis of detailed mechanisms of catalysis of various reactions in the active site of this RNAP, including analysis of mutant enzyme variants. This system can also be used in studies of regulation of gene expression by transcription factors from D. radiodurans.
This work was supported by the Russian Science Foundation grant 14-14-01074.
REFERENCES
1.Haugen, S. P., Ross, W., and Gourse, R. L. (2008)
Advances in bacterial promoter recognition and its control by factors
that do not bind DNA, Nat. Rev. Microbiol., 6,
507-519.
2.Nudler, E. (2009) RNA polymerase active center: the
molecular engine of transcription, Annu. Rev. Biochem.,
78, 335-361.
3.Pupov, D. V., and Kulbachinskiy, A. V. (2010)
Structural dynamics of the active center of multisubunit RNA
polymerases during RNA synthesis and proofreading, Mol. Biol.
(Moscow), 44, 573-590.
4.Sydow, J. F., and Cramer, P. (2009) RNA polymerase
fidelity and transcriptional proofreading, Curr. Opin. Struct.
Biol., 19, 732-739.
5.Gordon, A. J., Halliday, J. A., Blankschien, M. D.,
Burns, P. A., Yatagai, F., and Herman, C. (2009) Transcriptional
infidelity promotes heritable phenotypic change in a bistable gene
network, PLoS Biol., 7, e44.
6.Nudler, E. (2012) RNA polymerase backtracking in
gene regulation and genome instability, Cell, 149,
1438-1445.
7.Cheung, A. C., and Cramer, P. (2011) Structural
basis of RNA polymerase II backtracking, arrest and reactivation,
Nature, 471, 249-253.
8.Sekine, S., Murayama, Y., Svetlov, V., Nudler, E.,
and Yokoyama, S. (2015) The ratcheted and ratchetable structural states
of RNA polymerase underlie multiple transcriptional functions, Mol.
Cell, 57, 408-421.
9.Sosunova, E., Sosunov, V., Epshtein, V., Nikiforov,
V., and Mustaev, A. (2013) Control of transcriptional fidelity by
active center tuning as derived from RNA polymerase endonuclease
reaction, J. Biol. Chem., 288, 6688-6703.
10.Slade, D., and Radman, M. (2011) Oxidative stress
resistance in Deinococcus radiodurans, Microbiol. Mol. Biol.
Rev., 75, 133-191.
11.Liu, Y., Zhou, J., Omelchenko, M. V., Beliaev, A.
S., Venkateswaran, A., Stair, J., Wu, L., Thompson, D. K., Xu, D.,
Rogozin, I. B., Gaidamakova, E. K., Zhai, M., Makarova, K. S., Koonin,
E. V., and Daly, M. J. (2003) Transcriptome dynamics of Deinococcus
radiodurans recovering from ionizing radiation, Proc. Natl.
Acad. Sci. USA, 100, 4191-4196.
12.Luan, H., Meng, N., Fu, J., Chen, X., Xu, X.,
Feng, Q., Jiang, H., Dai, J., Yuan, X., Lu, Y., Roberts, A. A., Luo,
X., Chen, M., Xu, S., Li, J., Hamilton, C. J., Fang, C., and Wang, J.
(2014) Genome-wide transcriptome and antioxidant analyses on
gamma-irradiated phases of deinococcus radiodurans R1, PloS One,
9, e85649.
13.Kulbachinskiy, A., Bass, I., Bogdanova, E.,
Goldfarb, A., and Nikiforov, V. (2004) Cold sensitivity of thermophilic
and mesophilic RNA polymerases, J. Bacteriol., 186,
7818-7820.
14.Barinova, N., Zhilina, E., Bass, I., Nikiforov,
V., and Kulbachinskiy, A. (2008) Lineage-specific amino acid
substitutions in region 2 of the RNA polymerase sigma subunit affect
the temperature of promoter opening, J. Bacteriol., 190,
3088-3092.
15.Miropolskaya, N., Ignatov, A., Bass, I., Zhilina,
E., Pupov, D., and Kulbachinskiy, A. (2012) Distinct functions of
regions 1.1 and 1.2 of RNA polymerase sigma subunits from
Escherichia coli and Thermus aquaticus in transcription
initiation, J. Biol. Chem., 287, 23779-23789.
16.Mekler, V., Minakhin, L., Kuznedelov, K.,
Mukhamedyarov, D., and Severinov, K. (2012) RNA
polymerase–promoter interactions determining different stability
of the Escherichia coli and Thermus aquaticus
transcription initiation complexes, Nucleic Acids Res.,
40, 11352-11362.
17.Pupov, D. V., Barinova, N. A., and Kulbachinskiy,
A. V. (2008) Analysis of RNA cleavage by RNA polymerases from
Escherichia coli and Deinococcus radiodurans,
Biochemistry (Moscow), 73, 725-729.
18.Miropolskaya, N., Artsimovitch, I., Klimasauskas,
S., Nikiforov, V., and Kulbachinskiy, A. (2009) Allosteric control of
catalysis by the F loop of RNA polymerase, Proc. Natl. Acad. Sci.
USA, 106, 18942-18947.
19.Miropolskaya, N., Esyunina, D., Klimasauskas, S.,
Nikiforov, V., Artsimovitch, I., and Kulbachinskiy, A. (2014) Interplay
between the trigger loop and the F loop during RNA polymerase
catalysis, Nucleic Acids Res., 42, 544-552.
20.Svetlov, V., and Artsimovitch, I. (2015)
Purification of bacterial RNA polymerase: tools and protocols,
Methods Mol. Biol., 1276, 13-29.
21.Kuznedelov, K., Minakhin, L., and Severinov, K.
(2003) Preparation and characterization of recombinant Thermus
aquaticus RNA polymerase, Methods Enzymol., 370,
94-108.
22.Yang, X., and Lewis, P. J. (2008) Overproduction
and purification of recombinant Bacillus subtilis RNA
polymerase, Protein Expr. Purif., 59, 86-93.
23.Banerjee, R., Rudra, P., Prajapati, R. K.,
Sengupta, S., and Mukhopadhyay, J. (2014) Optimization of recombinant
Mycobacterium tuberculosis RNA polymerase expression and
purification, Tuberculosis, 94, 397-404.
24.Hu, Y., Morichaud, Z., Perumal, A. S.,
Roquet-Baneres, F., and Brodolin, K. (2014) Mycobacterium RbpA
cooperates with the stress-response sigmaB subunit of RNA polymerase in
promoter DNA unwinding, Nucleic Acids Res., 42,
10399-10408.
25.Cheng, C. Y., Yu, Y. J., and Yang, M. T. (2010)
Coexpression of omega subunit in E. coli is required for the
maintenance of enzymatic activity of Xanthomonas campestris pv.
campestris RNA polymerase, Protein Expr. Purif.,
69, 91-98.
26.Sosunov, V., Zorov, S., Sosunova, E., Nikolaev,
A., Zakeyeva, I., Bass, I., Goldfarb, A., Nikiforov, V., Severinov, K.,
and Mustaev, A. (2005) The involvement of the aspartate triad of the
active center in all catalytic activities of multisubunit RNA
polymerase, Nucleic Acids Res., 33, 4202-4211.
27.Zenkin, N., Yuzenkova, Y., and Severinov, K.
(2006) Transcript-assisted transcriptional proofreading,
Science, 313, 518-520.
28.Laptenko, O., Lee, J., Lomakin, I., and Borukhov,
S. (2003) Transcript cleavage factors GreA and GreB act as transient
catalytic components of RNA polymerase, EMBO J., 22,
6322-6334.
29.Sosunova, E., Sosunov, V., Kozlov, M., Nikiforov,
V., Goldfarb, A., and Mustaev, A. (2003) Donation of catalytic residues
to RNA polymerase active center by transcription factor Gre, Proc.
Natl. Acad. Sci. USA, 100, 15469-15474.
30.Orlova, M., Newlands, J., Das, A., Goldfarb, A.,
and Borukhov, S. (1995) Intrinsic transcript cleavage activity of RNA
polymerase, Proc. Natl. Acad. Sci. USA, 92,
4596-4600.
31.Sosunov, V., Sosunova, E., Mustaev, A., Bass, I.,
Nikiforov, V., and Goldfarb, A. (2003) Unified two-metal mechanism of
RNA synthesis and degradation by RNA polymerase, EMBO J.,
22, 2234-2244.
32.Yuzenkova, Y., and Zenkin, N. (2010) Central role
of the RNA polymerase trigger loop in intrinsic RNA hydrolysis,
Proc. Natl. Acad. Sci. USA, 107, 10878-10883.
33.McGlynn, P., Savery, N. J., and Dillingham, M. S.
(2012) The conflict between DNA replication and transcription, Mol.
Microbiol., 85, 12-20.