Temperature Dependence of Light-Induced Absorbance Changes Associated with Chlorophyll Photooxidation in Manganese-Depleted Core Complexes of Photosystem II
A. A. Zabelin1*, V. A. Shkuropatova1, A. Ya. Shkuropatov1, and V. A. Shuvalov1,2
1Institute of Basic Biological Problems, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; fax: +7 (4967) 33-0532; E-mail: zabelin.bio@gmail.com2Semenov Institute of Chemical Physics, Russian Academy of Sciences, 119991 Moscow, Russia; fax: +7 (495) 651-2191
* To whom correspondence should be addressed.
Received May 15, 2015; Revision received June 19, 2015
Mid-infrared (4500-1150 cm–1) absorbance changes induced by continuous illumination of Mn-depleted core complexes of photosystem II (PSII) from spinach in the presence of exogenous electron acceptors (potassium ferricyanide and silicomolybdate) were studied by FTIR difference spectroscopy in the temperature range 100-265 K. The FTIR difference spectrum for photooxidation of the chlorophyll dimer P680 was determined from the set of signals associated with oxidation of secondary electron donors (β-carotene, chlorophyll) and reduction of the primary quinone QA. On the basis of analysis of the temperature dependence of the P680+/P680 FTIR spectrum, it was concluded that frequencies of 131-keto-C=O stretching modes of neutral chlorophyll molecules PD1 and PD2, which constitute P680, are similar to each other, being located at ~1700 cm–1. This together with considerable difference between the stretching mode frequencies of keto groups of PD1+ and PD2+ cations (1724 and 1709 cm–1, respectively) is in agreement with a literature model (Okubo et al. (2007) Biochemistry, 46, 4390-4397) suggesting that the positive charge in the P680+ dimer is mainly localized on one of the two chlorophyll molecules. A partial delocalization of the charge between the PD1 and PD2 molecules in P680+ is supported by the presence of a characteristic electronic intervalence band at ~3000 cm–1. It is shown that a bleaching band at 1680 cm–1 in the P680+/P680 FTIR spectrum does not belong to P680. A possible origin of this band is discussed, taking into account the temperature dependence (100-265 K) of light-induced absorbance changes of PSII core complexes in the visible spectral region from 620 to 720 nm.
KEY WORDS: core complex of photosystem II, FTIR spectroscopy, chlorophyll, photooxidation, radical cationDOI: 10.1134/S0006297915100089
Abbreviations: ΔA, absorbance change; Car, β-carotene; Chl, chlorophyll a; ChlD1 and ChlD2, monomeric chlorophyll molecules bound to D1 and D2 polypeptides of RC; Chlz, redox-active additional chlorophyll molecule of RC; cyt b559, cytochrome b559; P680, dimer of chlorophyll molecules in RC of PSII; PD1, PD2, chlorophyll molecules composing P680; Pheo, pheophytin a; PheoD1, pheophytin bound to D1 polypeptide of RC; PSII, photosystem II; QA, primary quinone acceptor; RC, reaction center; SiMo, silicomolybdate.
Photosystem II (PSII) is a transmembrane pigment–protein complex
carrying out high-yield photochemical separation of opposite charges in
the initial stages of light energy conversion to chemical energy in
higher plants, algae, and cyanobacteria. The localization of the
positive charge formed by the separation reaction in PSII on the
P680 chlorophyll a (Chl) dimer is one of the key
stages of oxygenic photosynthesis, which generates the radical cation
P680+, a potent oxidizing agent essential for the
oxidation of water to molecular oxygen. In this regard, studies of the
electronic properties of P680+ and evaluation of
excess positive charge distribution between its two halves
(PD1 and PD2 [1-3]), an important factor significantly affecting the
redox potential of the P680+/P680 pair
[4], are of great interest. Such information can be
obtained by analyzing the value of high-frequency shift of the
131-keto-C=O stretching mode of chlorophyll during its
oxidation [5-8], measured by
light-induced Fourier transform infrared difference spectroscopy (FTIR
spectroscopy). FTIR can be used to examine vibrational properties,
structure, and molecular interactions of cofactors in their neutral as
well as radical ion states with very high sensitivity [9, 10]. Earlier, positive signals
at 1723-1725 and 1709-1711 cm–1 attributable with
131-keto C=O cation groups of PD1+ and
PD2+ were identified in light-induced FTIR
difference spectra of P680+/P680 PSII
core complexes of cyanobacteria [8, 11] and PSII membranes of higher plants [8]. However, a definite assignment of keto C=O modes
for neutral PD1 and PD2 was not determined.
According to [8], the 131-keto C=O
groups of PD1 and PD2 molecules have similar
vibrational frequencies, and a bleaching band observed in the FTIR
spectrum at ~1700 cm–1 can be attributed to both
groups. At the same time, it is proposed [11] that
absorption of the PD2 keto group corresponds to a negative
signal located in the region of lower frequencies of the FTIR spectrum
(at 1681 cm–1).
To further identify IR absorption bands derived from specific P680 and P680+ vibrations, in this study we analyzed the temperature dependence of light-induced FTIR difference spectra of P680+/P680 (4500-1150 cm–1) in PSII core complexes of higher plants in the 100-265 K temperature range. The light-induced photooxidation difference spectra for P680 were obtained for the same temperature range in the visible spectrum as well (620-720 nm).
MATERIALS AND METHODS
Oxygen-evolving PSII core complexes, containing about 35 Chl molecules per reaction center (RC), were isolated from spinach PSII membrane fragments [12] as described in [13]. Chromatographically purified samples of core complexes were suspended in BTS400 buffer containing 20 mM Bis-Tris (pH 6.5), 20 mM MgCl2, 5 mM CaCl2, 75 mM MgSO4, 400 mM sucrose, and 0.03% (w/v) n-dodecyl-β-D-maltoside. The complexes contained light-reducible primary quinone acceptor QA, but they did not possess a functional secondary quinone QB [13, 14]. Redox-active β-carotene (Car) was present in the complexes [15]. Cytochrome b559 (Cyt b559) was fully oxidized in these samples [13, 14]. Manganese-depleted (Mn-depleted) PSII core complexes were obtained by incubating original complexes with NH2OH (3 mM) and Na2EDTA (3 mM) in BTS400 for 15 min in the dark followed by chromatographic purification on a Q-Sepharose column (FF). All procedures related to isolation of core complexes, extraction of manganese, and sample preparation for spectral measurements were carried out at 5°C under dim green light. The complexes were concentrated on a 30-kDa membrane (Millipore, USA) in an ultraconcentration cell under gaseous argon pressure.
Oxygen evolution rate was measured by a Clark electrode (Hansatech, UK) at 24°C for samples containing 10 µg Chl/ml illuminated by continuous saturating red light (λ > 600 nm). Potassium ferricyanide (1 mM) and 2,6-dichloro-1,4-benzoquinone (0.25 mM) were used as artificial electron acceptors. Typical O2 release rates for precursor PSII core complexes were ~1000 µmol O2/mg Chl per h; in Mn-depleted samples, no O2 release was observed. Chl concentrations were measured according to a published method [16].
Light-induced absorption changes in visible and mid-infrared spectral ranges were obtained in the presence of potassium ferricyanide and silicomolybdate (SiMo) as exogenous electron acceptors [8].
Samples for measuring FTIR spectral changes were obtained as follows. An aliquot (6 µl) of Mn-depleted PSII core complex suspension (~2.5 mg Chl/ml) in BTS400 buffer was applied to a CaF2 window, and then 1 µl of 100 mM potassium ferricyanide water solution and/or 0.6 µl of 6 mM SiMo water solution was added. The mixture was slightly dried in a stream of argon gas and covered by a second CaF2 window.
FTIR absorption spectra were recorded on an IFS66v/s infrared airless Fourier spectrometer (Bruker, Germany) with a MCT detector and a KBr beamsplitter. The spectral resolution was 4 cm–1. Sample temperature was monitored with an optical cryostat temperature controller (Specac, UK). Samples were protected from actinic effect of the He-Ne spectrophotometer laser light with a Ge filter. Another Ge filter was used for protection of the detector from red excitation light. Reversible light-induced (light-minus-relaxation) difference spectra were calculated as a difference of FTIR spectra (10 scans; acquisition interval ~4 s) measured under excitation light (λ > 600 nm; ~15 mW/cm2) and after 10-s dark relaxation of the sample. Illumination cycles were repeated hundreds of times for improving signal-to-noise ratio.
Absorption spectra in the visible spectral range were measured using an Agilent 8453 spectrophotometer (Agilent, USA) in a handmade optical cryostat, using a cuvette with a ~2-mm optical pathlength. In this case, potassium ferricyanide and SiMo solutions were added to Mn-depleted PSII core complex suspension (~100 µg Chl/ml) to the final concentration of 3 mM and 300 µM, respectively, and the resulting sample was mixed with 60% glycerol (v/v). Reversible light-induced (light-minus-relaxation) difference spectra (620-720 nm) were obtained as a difference between absorption spectra measured under actinic illumination for 7 s (λ > 600 nm; ~15 mW/cm2) and after 10 s following dark relaxation of the sample. Illumination cycles were repeated 4-16 times for improving signal-to-noise ratio.
RESULTS
Figure 1 (curve 1) shows the light-induced (light-minus-relaxation) FTIR difference spectrum of Mn-depleted PSII core complexes measured in the presence of potassium ferricyanide and SiMo mixture in the 1800-1150 cm–1 range at 265 K. The complex nature of the spectrum indicates the formation of more than one light-induced radical ion that relaxes in the dark on the time scale of our measurements. The absorbance changes observed in the 1724-1700 cm–1 range of stretching vibrations for 131-keto C=O groups of pigments indicate a contribution of signals reflecting P680 oxidation to difference spectrum 1 [8, 11]. However, it is clear that this spectrum includes absorbance changes associated to the primary quinone QA reduction, as proven by the presence of a positive band at 1478 cm–1, which was earlier attributed to stretching vibrations of semiquinone Q–A C=O groups [17, 18]. The difference spectrum 1 is therefore expected to include an IR signal at ~1724/1719 cm–1 caused by electrostatic response of a photoactive pheophytin PheoD1 133-ester C=O group to QA– formation [17, 19].
Fig. 1. Light-induced (light-minus-relaxation) FTIR difference spectra (1800-1150 cm–1) of Mn-depleted PSII core complexes measured in the presence of potassium ferricyanide and SiMo (1) and in the presence of only SiMo (2) at 265 K. Spectrum 2 is normalized to spectrum 1 by the amplitude of the band at 1478 cm–1. 3) Double difference spectrum P680+/P680 obtained by subtracting spectrum 2 from spectrum 1. The inset shows light-induced (light-minus-relaxation) FTIR difference spectrum of Mn-depleted PSII core complexes, measured in the presence of potassium ferricyanide and SiMo in stretching vibrations region of semiquinone QA– and carotenoid radical cation Car+ at 100 K.
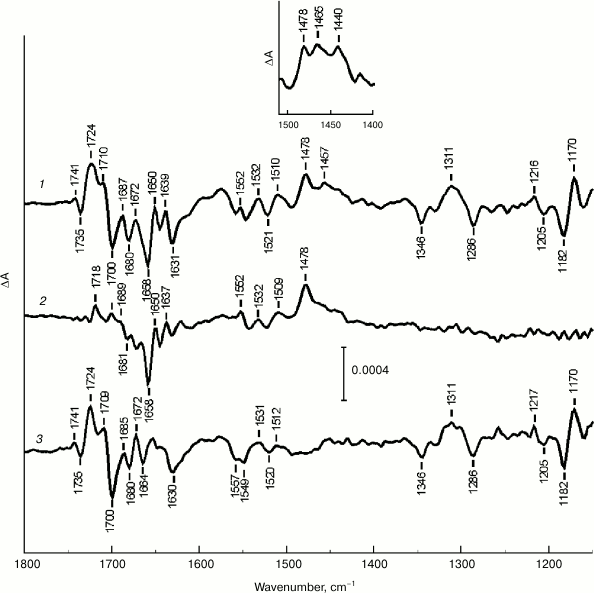
Figure 1 (curve 2) shows light-induced (light-minus-relaxation) FTIR spectrum of Mn-depleted PSII core complexes measured with the addition of only SiMo as an exogenous electron acceptor to be significantly simpler than difference spectrum 1, especially in the range of pigment keto group stretching vibrations as well as in the low frequency range (≤1420 cm–1). Judging by a distinct positive band at 1478 cm–1, difference spectrum 2 has a predominant contribution of absorbance changes attributed to QA– formation [17, 18], while P680+/P680 signals are practically absent.
Figure 1 (curve 3) shows a double difference spectrum obtained by subtracting difference spectrum 2 from difference spectrum 1, after their normalization by the QA– band amplitude at 1478 cm–1. Spectrum 3 is characterized by a set of specific signals reflecting P680 photooxidation and is in agreement with P680+/P680 light-induced FTIR difference spectra of PSII core complexes from cyanobacteria at 265 K [8] and 250 K [11], as well as spinach PSII membranes at 265 K [8]. In the stretching vibrations range for Chl keto carbonyl groups in spectrum 3, two marker positive bands at 1724 and 1709 cm–1 are well defined. They are attributed to 131-keto-C=O stretching modes of PD1+ and PD2+, respectively, which are shifted to higher frequency on cation formation [8, 11, 20]. The corresponding intense negative band of neutral P680 is located at 1700 cm–1. At the same time, according to previously obtained data [8, 11], another prominent negative band is located at 1680 cm–1. In the frequency range of chlorin macrocycle skeletal vibrations (1600-1150 cm–1) in spectrum 3, there is a discernible set of positive and negative peaks, including those at 1311(+), 1170(+), 1346(–), 1286(–), and 1182(–) cm–1 (“+” and “–” indicate absorbance change signs), corresponding to P680+ and P680, respectively [8].
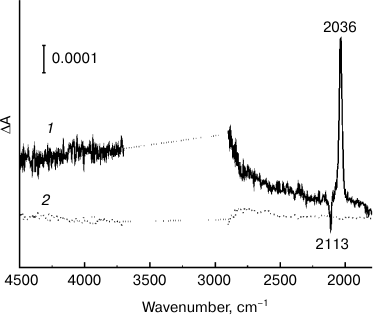
Another indication that a double difference spectrum 3 (Fig. 1) reflects Chl photooxidation in P680 dimer is the presence of a broad positive IR band with a maximum at ~3000 cm–1 (Fig. 2, curve 1), attributed to a low-energy electronic transition connected to transfer of a positive charge (a “hole”) between two halves of a dimeric radical cation [21, 22]. This transition is a unique characteristic of dimeric structure of a primary electron donor, indicating a partial charge delocalization in P680+ [8, 21, 22]. The signal in the ~3700-2900 cm–1 range (Fig. 2) was saturated due to high absorbance of water in the sample. The peaks at 2113 and 2136 cm–1 are caused by reduction of ferricyanide to ferrocyanide. Earlier [8], a similar band at ~3000 cm–1 was detected for cyanobacterial PSII core complexes and spinach PSII membranes. The absence of such a band in a light-induced FTIR spectrum for PSII core complexes with the addition of only SiMo (Fig. 2, curve 2) shows that P680+/P680 signals are not detected in this case.
Fig. 2. Light-induced (light-minus-relaxation) FTIR difference spectra of Mn-depleted PSII core complexes measured in the 4500-1800 cm–1 range at 265 K. Spectra 1 and 2 represent high-frequency ranges of spectra 3 and 2 shown in Fig. 1, respectively. Peaks at 2113 and 2036 cm–1 are caused by the ferricyanide/ferrocyanide conversion. The range of ~3700-2900 cm–1 is saturated due to considerable absorption of the sample and water.
We cannot exclude the possibility that the double difference spectrum 3 (Fig. 1) contains absorbance changes due to oxidation of antenna chlorophylls and/or redox-active chlorophyll ChlZ. In particular, differential signals at 1727(+)/1699(–) and 1713(+)/1687-1684(–) cm–1 were detected earlier for ChlZ+/ChlZ [23]. However, the contribution of these signals to spectrum 3 (Fig. 1) is apparently insignificant compared to absorbance changes connected to P680+/P680.
Summarizing these data, the double difference spectrum 3 (Fig. 1) can be concluded to represent sufficiently “pure” FTIR spectrum of P680+/P680 for spinach PSII core complexes.
Figure 3 compares the double difference FTIR spectra of P680+/P680 spinach PSII core complexes, calculated as described above and normalized by differential signal amplitude at 1724/1700 cm–1, in the 1750-1670 cm–1 frequency range at several chosen temperatures in the 100-265 K interval. Measurements at temperatures above 265 K were not conducted for this study due to core complex lability and a potential for their degradation under the relatively prolonged illumination used. It should be noted that at temperatures ≤180 K the FTIR spectra measured in the presence of potassium ferricyanide and SiMo mixture (Fig. 1, insert) and in the presence of only SiMo (data not shown) also demonstrated peaks of Car+ radical cation at ~1465 and ~1440 cm–1 [24], which were mostly subtracted when calculating respective double difference spectra.
Fig. 3. P680+/P680 FTIR double difference spectra of Mn-depleted PSII core complexes in the range of 131-keto-C=O stretching modes at selected temperatures: 1) 100; 2) 180; 3) 230; 4) 250; 5) 265 K. The spectra are normalized by the differential signal amplitude at 1724/1700 cm–1 (normalization coefficients are shown in parentheses). The differential signal amplitude at 1724/1700 cm–1 at 265 K was 6·10–4 absorbance units.
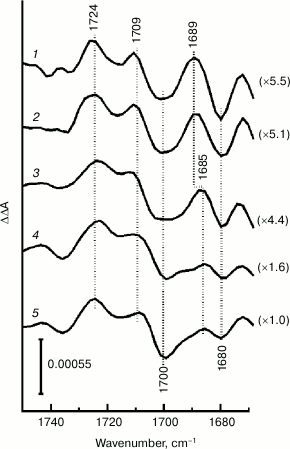
Figure 3 shows that frequency position of bands as well as the overall shape of the FTIR spectrum in the stretching vibrations range for pigment 131-keto C=O groups is mostly preserved after lowering sample temperature, indicating that P680 photooxidation is a major contributor to absorbance changes at all temperatures studied. However, P680+/P680 signal intensities in the 1724-1700 cm–1 range in the measured double difference spectra depended on temperature significantly: they decreased several-fold during the transition from 265 to ~230 K (Fig. 3, curves 3-5) and further changed little at lower temperatures. Sample temperature decrease was also accompanied by a significant intensity decrease in ferricyanide/ferrocyanide differential signal at 2113/2036 cm–1 (data not shown). It is plausible that at temperatures lower than ~230 K (Fig. 3, curves 1 and 2) a “freezing” of molecular diffusion processes occurred in the samples, accompanied by a decrease in efficiency of electron transfer from PheoD1– or QA– to exogenous ferricyanide. This, in turn, led to a decrease in the amount of photo-accumulated P680+ and to a decrease in amplitudes of corresponding IR signals under constant illumination conditions. To improve the representation of low intensity signals detected at low temperatures, double difference spectra on Fig. 3 were normalized by P680+/P680 signal amplitude at 1724/1700 cm–1 (normalizing coefficient are given in parentheses).
As shown in Fig. 3, the negative band at 1680 cm–1 is present in P680+/P680 FTIR spectra at all temperatures studied. This band might be a part of a high-frequency shift with a corresponding positive peak located at 1689 cm–1 at low temperatures (curves 1 and 2) and slightly shifted to 1685 cm–1 at temperatures higher than ~230 K (curves 3-5). Comparing normalized P680+/P680 spectra (Fig. 3) suggests that signal intensity at 1689/1680 cm–1 is comparable to differential signal intensity at 1724/1700 cm–1 at low temperatures, but it is significantly decreased during transition from ~230 to 265 K. This indicates a different effect of temperature on IR signals for these frequency ranges.
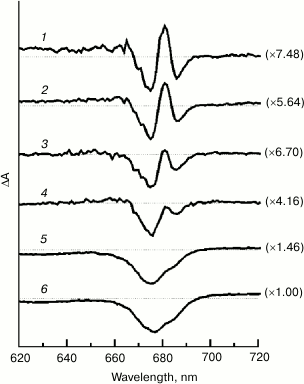
Figure 4 shows light-induced (light-minus-relaxation) electronic absorption difference spectra for Mn-depleted PSII core complexes measured in the presence of potassium ferricyanide and SiMo in the Qy spectral range (620-720 nm) in the temperature range from 100 to 265 K. The spectra are normalized at 675-677 nm (normalizing coefficients are shown in parentheses). The figure indicates that difference spectra shape significantly depends on temperature. At low temperatures (from 100 to ~200 K; curves 1-3), the spectra are characterized by bleaching bands at 675 and 686 nm and a positive peak at 681 nm. At temperatures above ~200 K (Fig. 4, curves 4-6), the positive peak at 681 nm and the negative peak at 686 nm undergo a significant amplitude decrease and do not appear on difference spectra, the dominant feature of which is a wide bleaching band at 675-677 nm with a weak shoulder on its long-wavelength slope. The positive signal at λ ≥ 690 nm corresponds to absorption of light-generated chlorophyll radical cation. Structured difference spectra were measured earlier at cryogenic temperatures for the P680+QA– state in cyanobacterial PSII core complexes [25, 26] and spinach membrane fragments [25], as well as for reversible light-induced absorbance changes in spinach PSII core complexes in the presence of SiMo [27]. A similar effect of temperature on P680+QA–/P680QA spectral shape was also observed for cyanobacterial PSII core complexes [25]. However, the temperature dependence of absorbance changes for plant PSII core complexes was apparently not studied.
Fig. 4. Light-induced (light-minus-relaxation) electronic (620-720 nm) absorbance difference spectra for Mn-depleted PSII core complexes measured in the presence of potassium ferricyanide and SiMo at selected temperatures: 1) 100; 2) 150; 3) 180; 4) 230; 5) 250; 6) 265 K. Horizontal dotted lines indicate the baselines. The spectra are normalized at 675-677 nm (normalization coefficients are shown in parentheses). The amplitude of bleaching at 677 nm for the spectrum measured at 265 K was 2·10–2 absorbance units.
DISCUSSION
Excitation of PSII by light quanta is known to induce fast electron transfer in the active branch of RC cofactors with successive formation of charge-separated states P680+PheoD1– and P680+QA– (see review [28]). Monomeric chlorophyll ChlD1 located in the active branch between P680 dimer and pheophytin PheoD1 [1-3] is also involved in light-induced electron transfer as a primary electron donor [29-32] or a primary acceptor [25, 31, 33]. When PSII Mn cluster is not functional and Cyt b559 is in its oxidized state, Car and ChlZ molecules can serve as secondary electron donors for P680+, competing with low quantum yield with charge recombination in the P680+QA– state [34, 35]. In the presence of exogenous electron acceptor capable of certain effectiveness in electron extraction from light-generated PheoD1– and/or QA–, a light-induced accumulation of redox states is possible for Mn-depleted PSII samples. These states include P680+, Car+, ChlZ+, QA–, as well as reduced exogenous electron acceptors.
In this study, reversible absorbance changes induced by constant illumination of Mn-depleted samples of spinach PSII core complex in the presence of exogenous electron acceptors, potassium ferricyanide, and silicomolybdate were measured in the mid-infrared spectral range. The study focused on isolating the FTIR P680+/P680 spectrum from a set of other light-induced signals and analyzing its temperature dependence in the 100-265 K range. Simultaneously, absorbance changes in the visible spectrum range for the same temperature interval were studied.
The most notable feature of P680+/P680 FTIR spectra temperature dependence (Fig. 3) is a prominent difference in P680+/P680 differential signal amplitude at 1724/1700 cm–1, and signal intensity at 1689/1680 cm–1 in relation to temperature, indicating a different nature of these signals. This fact makes it hardly probable to attribute the bleaching at 1680 cm–1 to P680 dimer and indicates that stretching vibrations bands for 131-keto C=O groups in PD1 and PD2 molecules in neutral states are not resolved in the IR spectrum. The data apparently agree with the following assumption [8]: the keto groups of neutral P680 do not form hydrogen bonds, and both absorb at ~1700 cm–1. Based on the analysis of PSII core complex crystal structure, a hypothesis was proposed earlier [11] that D2-Ser282 may indirectly (through a water molecule) form a hydrogen bond with the PD2 chlorophyll keto group, shifting its absorbance to lower frequency (up to ~1680 cm–1) compared to the absorbance of a corresponding band in the PD1 molecule located in a less polar environment. The proposed hydrogen bond might however not be strong enough to cause noticeable changes in PD1 and PD2 vibrational properties.
Earlier [8] the presence of two P680+ positive peaks (at 1724 and 1709 cm–1 on Fig. 1) and a single negative P680 peak at 1700 cm–1 in the P680+/P680 FTIR spectrum was interpreted according to a model assuming that the positive charge in the P680+ cation is largely (70-80%) localized on one of the two Chl molecules. The nonequivalence of vibrational shifts to higher frequency for PD1+ and PD2+ might also be partly due to differences in electrostatic interactions of the formed radical cations with their protein environment, as observed for Rhodobacter (Rba.) sphaeroides RC [36]. The intervalence band at ~3000 cm–1 (Fig. 2, spectrum 1 [8]) reflects partial delocalization of positive charge between two Chl molecules in P680+ [21, 22]. The preferential localization of PSII positive charge on the PD1 chlorophyll was also expected from a comparison of shifts of absorbance bands to higher frequency for keto groups of (bacterio)chlorophylls in FTIR spectra measured for PSII core complexes of Synechocystis sp. PCC 6803 and Rba. sphaeroides RC [11]. According to calculations based on density function theory [4], significant charge localization on PD1 chlorophyll [37] is one of the important factors determining the high positive redox potential of P680 essential for water oxidation in PSII. The fact that keto group vibrational frequencies of PD1 and PD2 molecules do not change significantly with temperature in neutral and radical cation states (Fig. 3) suggests that P680+ electronic structure (asymmetric charge distribution) in PSII core complexes is preserved in the 100-265 K interval.
If the bleaching IR-band at 1680 cm–1 and, therefore, the differential signal at 1689/1680 cm–1 (Figs. 1 and 3) cannot be attributed to P680, it raises a question about their origins. Earlier, the negative peak at 1681 cm–1 was detected in a QA–/QA FTIR difference spectrum of primary quinone acceptor reduction [18]. This peak was supposed to be caused by protein carbonyl stretching mode (amide mode I) of PSII [18]. Figure 1 (curve 2) shows the negative change at 1681 cm–1 to be also visible in the PSII core complex FTIR spectrum measured in the presence of only SiMo, when the main contribution is made by QA–/QA signals. Differential signal at 1689/1680 cm–1 in P680+/P680 FTIR spectra (Fig. 3) can be supposed to reflect changes in the amide I band caused by conformational rearrangements of the surrounding protein during P680+ formation. However, such an explanation would be difficult to conform to different temperature influence on this signal and the signal at 1724/1700 cm–1 belonging to P680+/P680 (Fig. 3).
In this respect, the fact that a wide bleaching band at 675-677 nm observed in the Qy range of electronic difference spectra at temperatures above ~200 K (Fig. 4, curves 4-6) is defined at lower temperatures as a complicated structured signal with negative bands at 675 and 686 nm and a positive peak at 681 nm, is of interest. While a detailed assignment of these spectral features to particular pigment cofactors is debatable [25-27], absorbance changes observed in this spectral range at cryogenic temperatures are supposed to include a bleaching band resulting from P680 photooxidation and P680+-induced electrochromic shift of a nearby monomeric Chl absorbance band [25, 26]. The differential signal at 1689/1680 cm–1 (Fig. 3) might represent a vibrational analog of electrochromic shift present in electronic difference spectra (Fig. 4). The charge on P680+ can be supposed to have an electrostatic effect on the vibrational mode of a 131-keto C=O group in one of the RC monomeric chlorophylls (ChlD1 or ChlD2), shifting the frequency of this mode from ~1680 to ~1689 cm–1. The abovementioned lack of correlation in differential signal behavior at 1724/1700 cm–1 (reflecting the amount of P680+ detected) and at 1689/1680 cm–1 in response to sample temperature change from ~230 to 265 K might be explained in this case by an increase in effective dielectric protein constant of the protein at temperatures above ~200 K (see [38] for references and further discussion), that would lead to a partial screening of electrostatic interactions and to a decrease in electrochromic shift at elevated temperatures.
Another interpretation is that the signal at 1689/1680 cm–1 reflects a high-frequency stretching mode shift for the 131-keto C=O group of the ChlD1 molecule due to formation of the ChlD1+ cation. Indeed, recent electrostatic calculations [27] showed that, at low temperatures, the light-generated positively charged hole, which was initially localized on P680+ in PSII, might be (partially) transferred to the ChlD1 molecule due to a shift in the redox potential of the ChlD1+/ChlD1 pair, compared to the P680+/P680 potential in the field of QA–. At room temperature, the charge redistribution effect becomes minimal due to QA– field screening by the molecular environment (pigment, protein, water) reorientation, and the hole is localized only on P680+ as a result. A decrease in differential signal amplitude at 1689/1680 cm–1 compared to signal amplitude at 1724/1700 cm–1 on increasing temperature (Fig. 3) is consistent with this interpretation. The observed high-frequency shift of the band at 1680 cm–1 (9 cm–1), corresponding to the ChlD1+/ChlD1 pair in this model, would correspond to a similar shift between the bands at 1700 and 1709 cm–1 during P680 oxidation (Fig. 3 [8]).
Earlier, on the basis of the IR spectrum for chlorophyll triplet state formation in isolated spinach PSII RC (D1–D2–cyt b559 complexes) the band at 1668-1670 cm–1 was assigned to the 131-keto C=O group of ChlD1 [39]. At the same time, the results of femtosecond IR measurements suggested that the keto group band of ChlD1 in isolated RC is located at 1687 cm–1, shifting to 1697 cm–1 during ChlD1+ cation formation [29]. During the analysis of femtosecond IR measurements on PSII core complexes from wild-type Synechocystis sp. PCC 6803 cells [11], preference was given to the assumption that the absorption of the ChlD1 keto group was at ~1670 cm–1 [39]. With this assignment, the differential signal at 1689/1680 cm–1 in the P680+/P680 FTIR spectra of PSII core complexes (Fig. 3) might be connected to the 131-keto C=O stretching mode of monomeric chlorophyll ChlD2 located in the inactive cofactor branch of the PSII RC [1-3]. However, it should be noted that spectral properties of RC D1–D2–cyt b559 complexes might undergo some changes [26, 40, 41], possibly due to a deletion of integral antenna CP43 and CP47 polypeptides. Currently, vibrational properties of the triplet-carrying ChlD1 molecule [26] in more intact PSII core complexes are apparently not defined. Therefore, the data obtained in this study and information from the literature do not include the possibility of attributing the signal at 1689/1680 cm–1 to the ChlD1 molecule as well. Further research will be needed to make more definite conclusions.
This study was conducted with financial support of the Russian Foundation for Basic Research (project No. 13-00-40297-K), the Program of Fundamental Studies of the Russian Academy of Sciences “Molecular and Cell Biology”, and the Program of the President of the Russian Federation (project No. SS-4771.2014.4). Results presented on Fig. 4 were obtained with the support of the Russian Science Foundation (project No. 14-14-00789).
REFERENCES
1.Zouni, A., Witt, H. T., Kern, J., Fromme, P.,
Krauss, N., Saenger, W., and Orth, P. (2001) Crystal structure of
photosystem II from Synechococcus elongatus at 3.8 Å
resolution, Nature, 409, 739-743.
2.Ferreira, K. N., Iverson, T. M., Maghlaoui, K.,
Barber, J., and Iwata, S. (2004) Architecture of the photosynthetic
oxygen-evolving center, Science, 303, 1831-1838.
3.Umena, Y., Kawakami, K., Shen, J. R., and Kamiya,
N. (2011) Crystal structure of oxygen-evolving photosystem II at a
resolution of 1.9 Å, Nature, 473, 55-61.
4.Takahashi, R., Hasegawa K., and Noguchi, T. (2008)
Effect of charge distribution over a chlorophyll dimer on the redox
potential of P680 in photosystem II as studied by density functional
theory calculations, Biochemistry, 47, 6289-6291.
5.Mattioli, T. A., Hoffmann, A., Robert, B.,
Schrader, B., and Lutz, M. (1991) Primary donor structure and
interactions in bacterial reaction centers from near-infrared Fourier
transform resonance Raman spectroscopy, Biochemistry, 30,
4648-4654.
6.Morita, E. H., Hayashi, H., and Tasumi, M. (1993)
Temperature dependence of the light-induced infrared difference spectra
of chromatophores and reaction centers from photosynthetic bacteria,
Biochim. Biophys. Acta, 1142, 146-154.
7.Noguchi, T., Kusumoto, N., Inoue, Y., and Sakurai,
H. (1996) Electronic and vibrational structure of the radical cation of
P840 in the putative homodimeric reaction center from Chlorobium
tepidum as studied by FTIR spectroscopy, Biochemistry,
35, 15428-15435.
8.Okubo, T., Tomo, T., Sugiura, M., and Noguchi, T.
(2007) Perturbation of the structure of P680 and the charge
distribution on its radical cation in isolated reaction center
complexes of photosystem II as revealed by Fourier transform infrared
spectroscopy, Biochemistry, 46, 4390-4397.
9.Lutz, M., and Mäntele, W. (1991) in
Chlorophylls (Scheer, H., ed.) CRC Press, Boca Raton, Florida,
pp. 855-902.
10.Noguchi, T., and Berthomieu, C. (2005) in
Photosystem II: the Light-Driven Water/Plastoquinone Oxidoreductase
(Wydrzynski, T., and Satoh, K., eds.) Vol. 22, Springer, Dordrecht,
pp. 367-387.
11.Di Donato, M., Cohen, R. O., Diner, B. A.,
Breton, J., Van Grondelle, R., and Groot, M. L. (2008) Primary charge
separation in the photosystem II core from Synechocystis: a
comparison of femtosecond visible/mid-infrared pump-probe spectra of
wild type and two P680 mutants, Biophys. J., 94,
4783-4795.
12.Berthold, D. A., Babcock, G. T., and Yocum, C. F.
(1981) A highly resolved, oxygen-evolving photosystem II preparation
from spinach thylakoid membranes, FEBS Lett., 134,
231-234.
13.Van Leeuwen, P. J., Nieveen, M. C., Van de Meent,
E. J., Dekker, J. P., and Van Gorkom, H. J. (1991) Rapid and simple
isolation of pure photosystem II core and reaction center particles
from spinach, Photosynth. Res., 28, 149-153.
14.Smith, P. J., Peterson, S., Masters, V. M.,
Wydrzynski, T., Styring, S., Krausz, E., and Pace, R. J. (2002)
Magneto-optical measurements of the pigments in fully active
photosystem II core complexes from plants, Biochemistry,
41, 1981-1989.
15.Vishnev, M. I., Zabelin, A. A., Shkuropatova, V.
A., Yanyushin, M. F., Shuvalov, V. A., and Shkuropatov, A. Y. (2013)
Chemical modification of photosystem II core complex pigments with
sodium borohydride, Biochemistry (Moscow), 78,
377-384.
16.Arnon, D. I. (1949) Copper enzymes in isolated
chloroplasts. Polyphenol oxidase in Beta vulgaris, Plant
Physiol., 24, 1-15.
17.Berthomieu, C., Nabedryk, E., Mäntele, W.,
and Breton, J. (1990) Characterization by FTIR spectroscopy of the
photoreduction of the primary quinone acceptor QA in
photosystem II, FEBS Lett., 269, 363-367.
18.Hienerwadel, R., Boussac, A., Breton, J., and
Berthomieu, C. (1996) Fourier transform infrared difference study of
tyrosine D oxidation and plastoquinone QA reduction in
photosystem II, Biochemistry, 35, 15447-15460.
19.Dejonghe, D., Andrianambinintsoa, S., Berger, G.,
and Breton, J. (1998) in Photosynthesis: Mechanisms and Effects
(Garab, G., ed.) Vol. 2, Kluwer Academic Publishers, Dordrecht, pp.
1121-1124.
20.Breton, J., Hienerwadel, R., and Nabedryk, E.
(1997) in Spectroscopy of Biological Molecules (Carmona, P.,
Navarro, R., and Hernanz, A., eds.) Kluwer Academic Publishers,
Dordrecht, pp. 101-102.
21.Breton, J., Nabedryk, E., and Parson, W. W.
(1992) A new infrared electronic transition of the oxidized primary
electron donor in bacterial reaction centers: a way to assess resonance
interactions between the bacteriochlorophylls, Biochemistry,
31, 7503-7510.
22.Reimers, J. R., and Hush, N. S. (2003) Modeling
the bacterial photosynthetic reaction center. VII. Full simulation of
the intervalence hole-transfer absorption spectrum of the special-pair
radical cation, J. Chem. Phys., 119, 3262-3277.
23.Kitajima, Y., and Noguchi, T. (2006)
Photooxidation pathway of chlorophyll Z in photosystem II as studied by
Fourier transform infrared spectroscopy, Biochemistry,
45, 1938-1945.
24.Noguchi, T., Mitsuka, T., and Inoue, Y. (1994)
Fourier transform infrared spectrum of the radical cation of
β-carotene photoinduced in photosystem II, FEBS Lett.,
356, 179-182.
25.Hillmann, B., Brettel, K., Van Mieghem, F.,
Kamlowski, A., Rutherford, A. W., and Schlodder, E. (1995) Charge
recombination reactions in photosystem II. 2. Transient absorbance
difference spectra and their temperature dependence,
Biochemistry, 34, 4814-4827.
26.Schlodder, E., Renger, T., Raszewski, G.,
Coleman, W. J., Nixon, P. J., Cohen, R. O., and Diner, B. A. (2008)
Site-directed mutations at D1-Thr179 of photosystem II in
Synechocystis sp. PCC 6803 modify the spectroscopic properties
of the accessory chlorophyll in the D1-branch of the reaction center,
Biochemistry, 47, 3143-3154.
27.Shelaev, I. V., Gostev, F. E., Vishnev, M. I.,
Shkuropatov, A. Ya., Ptushenko, V. V., Mamedov, M. D., Sarkisov, O. M.,
Nadtochenko, V. A., Semenov, A. Yu., and Shuvalov, V. A. (2011)
P680 (PD1PD2) and ChlD1 as
alternative electron donors in photosystem II core complexes and
isolated reaction centers, J. Photochem. Photobiol. B,
104, 44-50.
28.Renger, G., and Holzwarth, A. R. (2005) in
Photosystem II: The Light-Driven Water/Plastoquinone
Oxidoreductase (Wydrzynski, T., and Satoh, K., eds.) Vol. 22,
Springer, Dordrecht, pp. 139-175.
29.Groot, M. L., Pawlowicz, N. P., Van Wilderen, L.
J., Breton, J., Van Stokkum, I. H., and Van Grondelle, R. (2005)
Initial electron donor and acceptor in isolated photosystem II reaction
centers identified with femtosecond mid-IR spectroscopy, Proc. Natl.
Acad. Sci. USA, 102, 13087-13092.
30.Holzwarth, A. R., Muller, M. G., Reus, M.,
Nowaczyk, M., Sander, J., and Rogner, M. (2006) Kinetics and mechanism
of electron transfer in intact photosystem II and in the isolated
reaction center: pheophytin is the primary electron acceptor, Proc.
Natl. Acad. Sci. USA, 103, 6895-6900.
31.Romero, E., Van Stokkum, I. H., Novoderezhkin, V.
I., Dekker, J. P., and Van Grondelle, R. (2010) Two different charge
separation pathways in photosystem II, Biochemistry, 49,
4300-4307.
32.Novoderezhkin, V. I., Romero, E., Dekker, J. P.,
and Van Grondelle, R. (2011) Multiple charge-separation pathways in
photosystem II: modeling of transient absorption kinetics, Chem.
Phys. Chem., 12, 681-688.
33.Shelaev, I. V., Gostev, F. E., Nadtochenko, V.
A., Shkuropatov, A. Ya., Zabelin, A. A., Mamedov, M. D., Semenov, A.
Ya., Sarkisov, O. M., and Shuvalov, V. A. (2008) Primary light energy
conversion in tetrameric chlorophyll structure of photosystem II and
bacterial reaction centers. II. Femto- and picosecond charge separation
in PSII D1/D2/Cyt b559 complex, Photosynth. Res.,
98, 95-103.
34.Faller, P., Fufezan, C., and Rutherford, A. W.
(2005) in Photosystem II: the Light-Driven Water/Plastoquinone
Oxidoreductase (Wydrzynski, T., and Satoh, K., eds.) Vol. 22,
Springer, Dordrecht, pp. 347-365.
35.Shinopoulos, K. E., and Brudvig, G. W. (2012)
Cytochrome b559 and cyclic electron transfer within
photosystem II, Biochim. Biophys. Acta, 1817, 66-75.
36.Johnson, E. T., Müh, F., Nabedryk, E.,
Williams, J. C., Allen, J. P., Lubitz, W., Breton, J., and Parson, W.
W. (2002) Electronic and vibronic coupling of the special pair of
bacteriochlorophylls in photosynthetic reaction centers from wild-type
and mutant strains of Rhodobacter sphaeroides, J. Phys. Chem.
B, 106, 11859-11869.
37.Diner, B. A., Schlodder, E., Nixon, P. J.,
Coleman, W. J., Rappaport, F., Lavergne, J., Vermaas, W. F. J., and
Chisholm, D. A. (2001) Site-directed mutations at D1-His198 and
D2-His197 of photosystem II in Synechocystis PCC 6803: sites of
primary charge separation and cation and triplet stabilization,
Biochemistry, 40, 9265-9281.
38.Raszewski, G., Diner, B. A., Schlodder, E., and
Renger, T. (2008) Spectroscopic properties of reaction center pigments
in photosystem II core complexes: revision of the multimer model,
Biophys. J., 95, 105-119.
39.Noguchi, T., Tomo, T., and Kato, C. (2001)
Triplet formation on a monomeric chlorophyll in the photosystem II
reaction center as studied by time-resolved infrared spectroscopy,
Biochemistry, 40, 2176-2185.
40.Hughes, J. L., Prince, B. J., Krausz, E., Smith,
P. J., Pace, R. J., and Riesen, H. (2004) Highly efficient spectral
hole-burning in oxygen-evolving photosystem II preparations, J.
Phys. Chem. B, 108, 10428-10439.
41.Acharya, K., Neupane, B., Zazubovich, V., Sayre,
R. T., Picorel, R., Seibert, M., and Jankowiak, R. (2012) Site energies
of active and inactive pheophytins in the reaction center of
photosystem II from Chlamydomonas reinhardtii, J. Phys. Chem.
B, 116, 3890-3899.