Isolation and Purification of Recombinant Serine/Threonine Protein Kinases of the Strain Bifidobacterium longum B379M and Investigation of Their Activity
M. G. Alekseeva, D. A. Mavletova, N. V. Kolchina, V. Z. Nezametdinova, and V. N. Danilenko*
Vavilov Institute of General Genetics, Russian Academy of Sciences, 119991 Moscow, Russia; fax: +7 (499) 132-8962; E-mail: valerid@rutenia.ru* To whom correspondence should be addressed.
Received June 17, 2015; Revision received July 2, 2015
Previously, we identified six serine/threonine protein kinases (STPK) of Bifidobacterium and named them Pkb1–Pkb6. In the present study, we optimized methods for isolation of the six STPK catalytic domains proteins of B. longum B379M: a method for isolation of Pkb3 and Pkb4 in native conditions, a method for isolation of Pkb5 in denaturing conditions, and a method for isolation of Pkb1, Pkb2, and Pkb6 from inclusion bodies. The dialysis conditions for the renaturation of the proteins were optimized. All of the enzymes were isolated in quantities sufficient for study of the protein activity. The proteins were homogeneous according to SDS-PAGE. The autophosphorylation ability of Pkb1, Pkb3, Pkb4, and Pkb6 was investigated for the first time. Autophosphorylation was detected only for the Pkb3 catalytic domain.
KEY WORDS: serine/threonine protein kinases, protein isolation and purification, autophosphorylationDOI: 10.1134/S0006297915100119
Abbreviations: a.a., amino acid residue; DTT, dithiothreitol; IPTG, isopropyl β-D-1-thiogalactopyranoside; PASTA, penicillin-binding protein- and serine/threonine kinase-associated domain (located in the C-terminal region of some bacterial proteins); PASTA_pknB, PASTA domain of bacterial PknB-like serine/threonine protein kinases; PMSF, phenylmethylsulfonyl fluoride; STPK, serine/threonine protein kinase; TM-region, transmembrane region.
Serine/threonine protein kinases (STPK) (EC 2.7.11.1) are enzymes that
modify other proteins by reversible phosphorylation at serine or
threonine residues within consensus sequences, thereby modulating the
activity of a broad range of enzymes and controlling specific
protein–protein interactions. Eukaryotic-type STPKs are
ubiquitously distributed among Gram-positive bacteria [1]. These enzymes are found in soil microorganisms,
pathogens, commensals, and probiotic bacteria [2].
Some bacterial genera such as Lactobacillus,
Staphylococcus, Streptococcus, and Bacillus
express one or two STPKs [2]. The highest number of
STPKs was found in the class Actinobacteria: >10 in genus
Mycobacterium [1], >20 in genus
Nocardia [2], >30 in genus
Streptomyces [3], and six STPKs were
identified in genus Bifidobacterium [4].
Reversible phosphorylation is one of the most important tools of signals transduced from the environment into bacterial cells as well as in regulating cellular functions [5-7]. STPKs and coupled serine/threonine phosphatases regulate expression of prokaryotic genes via posttranslational modification of various proteins including components of the transcription and translation systems [8, 9]. It was demonstrated that STPKs play essential role in cell growth and cell division, DNA replication [10], cell wall biosynthesis [11], biofilm production [12], sporulation and spore germination [13], virulence [5], and stress response and adaptive response [14, 15]. Inhibitors of STPKs are considered as promising antimicrobial agents [16, 17].
In recent years, increased interest has been given to examining bifidobacteria based on their probiotic properties and ability to positively influence human health [18]. Bifidobacterial strains are widely used in the food industry for obtaining fermentable nutritional and medicinal products with probiotic activity [19]. Bifidobacteria are one of the most important representatives of human microbiota, which are dominant in the gut microbiota in infants [20, 21]. Recently, a role of bifidobacteria in the “gut microbiota–brain axis” has been attracting major interest, primarily in the process of the developing nervous system and brain in children [22-25].
Until recently, data regarding examination of structure and functions exhibited by bifidobacterial STPKs were unavailable both in Russian and foreign publications. It should be noted that the experimental data on the functions of bifidobacterial genes are extremely limited because genetic manipulation systems, especially efficient methods for targeted gene mutagenesis, have not yet been established [26]. Functions of almost half of the proteins were only predicted by in silico analysis, based on homology with known proteins of other bacteria, and the others are still hypothetical proteins [27]. Similarly to other actinobacteria, it is plausible to suppose that STPK of Bifidobacterium can be elements of signal transduction networks and play major regulatory roles in bifidobacterial cells.
Earlier, we identified and characterized six STPKs in various strains of B. longum [4, 28]. Because investigation of functions of bifidobacterial protein kinases via the gene knockout is inaccessible due to low efficiency of transformation and recombination [26, 27], direct study of properties and functions of these protein kinases, search for their substrates, and determination of 3D-crystal structures is needed. In this connection, the question of obtaining target proteins in amounts sufficient for preparative analysis is raised.
The aim of the current study was to develop methods for isolation of protein kinases Pkb1-Pkb6 derived from strain B. longum B379M [29] and to determine the ability of protein kinases Pkb1, Pkb3, Pkb4, and Pkb6 for autophosphorylation in vitro.
MATERIALS AND METHODS
Bacterial strains, vectors, media, and culture conditions. The following strains of Escherichia coli were used in the study – BL21(DE3): F–ompT hsdSB (rB–mB–) gal dcm (DE3) (Novagen, USA) [30], and BL21(DE3) pLysS: F–ompT hsdSB (rB–mB–) gal dcm (DE3) [pLysS CamR] (Stratagene, USA) [31], plasmid pET32a (Novagen) [30] and plasmids pET32a:pkb1, pET32a:pkb2, pET32a:pkb3, pET32a:pkb4, pET32a:pkb5, and pET32a:pkb6 containing the cloned catalytic domains of serine/threonine protein kinases of the industrial probiotic strain B. longum B379M [4, 29].
Luria media (L-broth) and TB medium with chloramphenicol (34 µg/ml) were used to grow E. coli. Solid media contained 2% agar [32]. Selective growth of plasmid-containing bacteria was provided by adding ampicillin (150 µg/ml). Isolation of plasmid DNA, preparation of competent E. coli culture, and transformation were done according to standard methods [32].
Production of E. coli biomass for isolation of recombinant protein kinases’ catalytic domains. Escherichia coli containing recombinant plasmids were grown in a shaker incubator at 37°C until reaching optical density 0.6 (~2 h) followed by inducing expression of STPK genes by adding IPTG. After that, the E. coli was cultured at 28°C for 5 or 18 h followed by collection of biomass. The bacteria were sedimented by centrifuging (5000 rpm, 10 min, 4°C) and frozen at –20°C. To analyze expression, cell pellet was suspended in sample buffer (62.5 mM Tris-HCl, pH 6.8, 5% glycerol (w/v), 2% 2-mercaptoethanol, 0.1% SDS, 0.001% bromophenol blue) followed by heat-degradation at 95°C for 10 min. The protein fraction was analyzed by SDS-PAGE. The protein fraction obtained from E. coli strains containing insertion-free plasmid pET32a was used as a control sample.
Isolation of recombinant protein kinases. Recombinant proteins from E. coli lysates containing recombinant plasmids were isolated according to the QIAexpress protocol in native or denaturing conditions chromatographically by using the Ni-NTA Fast Start Kit (6) (Qiagen, USA). Protein fractions were analyzed by SDS-PAGE. Concentration of purified proteins was quantified according to the Bradford method [33]. Solutions of the isolated proteins were supplemented with glycerol (final concentration – 20%) and kept at –20°C.
Mass-spectrometric assay of purified proteins. The analysis was performed at the Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences. In-PAGE trypsinolysis for further analysis was done using peptide mass fingerprinting as earlier described [34]. Mass-spectrometry assay of tryptic hydrolysates was done by applying MALDI TOF on an Ultraflex mass spectrometer (Bruker Daltonics, Germany). Positively charged ions were quantified in reflection mode ranging within m/z 600-4000 Da. Examined proteins were identified using the Mascot search engine (MatrixScience) (http://www.matrixscience.com/home.html). The following search parameters were applied: Enzyme – Trypsin; Database – SwissProt; Taxonomy – Actinobacterium; Fixed modification – none; Variable modification – none; Missed cleavages – 1; Peptide tolerance – 100 ppm; Mass values (monoisotopic) – MH+.
In vitro quantified autophosphorylation of recombinant protein kinases. The autophosphorylation reaction was performed for 30 min at 37°C in buffer containing 25 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 5 mM MnCl2, 1 mM DTT, 1 mM EDTA, and 1 mM PMSF. The reaction was triggered by adding ATP to final concentration 10 µM (10 µCi [γ-32P]ATP; Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences), in the presence of 1 µg protein kinase. The reaction was stopped by adding SDS-PAGE sample buffer. Then, the reaction mixture was heated for 5 min at 95°C, followed by analyzing the samples by running 12.5% SDS-PAGE. For autoradiography, the gel was fixed in 50% TCA, stained with 0.1% Coomassie R250 dye, and exposed to KODAK MXG X-ray film.
In vitro autophosphorylation of protein kinases was also done using Kinase-Glo® Plus Luminescent Kinase Assay V3772 (Promega, USA). The degree of autophosphorylation was determined by quantifying the amount of ATP remaining in solution following the kinase reaction. Kinase-Glo reagent utilizing ATP as a substrate for Ultra-Glo-luciferase catalyzes luciferin oxidation accompanied by emission of photons. The activity of autophosphorylation was inversely proportional to the intensity of the luminescent signal. The autophosphorylation reaction was performed for 30 min at room temperature in buffer containing 15 mM HEPES-NaOH (pH 7.4), 20 mM NaCl, 10 mM MgCl2, 0.5 mM EDTA, 0.02% Tween 20, and 0.01% BSA. The reaction was triggered by adding ATP (final concentration 10 µM) in the presence of 5, 10, or 20 µg of the protein kinases, and it was stopped by adding an equal volume of Kinase-Glo reagent. The luminescence signal induced during the reaction was quantified using the DTX 880 Multimode Detector (Beckman Coulter, USA) for 100 msec with sensitivity mode expected high activity.
Bioinformatic analysis methods. Nucleotide and amino acid sequences were searched using databases available at the NCBI (http://www.ncbi.nlm.nih.gov/) and UniProt (http://www.uniprot.org/) websites.
Molecular weight and protein isoelectric points were calculated using ProtParam (http://web.expasy.org/protparam/) software.
RESULTS AND DISCUSSION
Main characteristics of genes and serine/threonine protein kinases of strain B. longum B379M. The genome of B. longum subsp. longum contains six genes encoding STPKs: four conservative genes (pkb1, pkb3, pkb5, and pkb6), one species-specific gene (pkb2), and one unique gene (pkb4) (Table 1) [4, 28]. The pkb5 and pkb6 genes are partially overlapped and form an operon. Based on high similarity of amino acid sequences of catalytic domains from bifidobacterial STPKs Pkb5 and Pkb6 as well as protein kinases PknB and PknA from Mycobacterium tuberculosis, and given a high similarity in the genetic environment around pknB-pknA and pkb5-pkb6, it can be assumed that he functions of these protein orthologs are similar and might be related to growth and division of the bacteria [4]. Gene pkb1 is located close to gene plsC1 responsible for synthesis of membrane phospholipids as well as genes coding for σ-factor of RNA polymerase and DNA-gyrase. A gene involved in signal transduction is located near the pkb3 gene.
Table 1. Characteristics of serine/threonine
protein kinases of B. longum subsp. longum
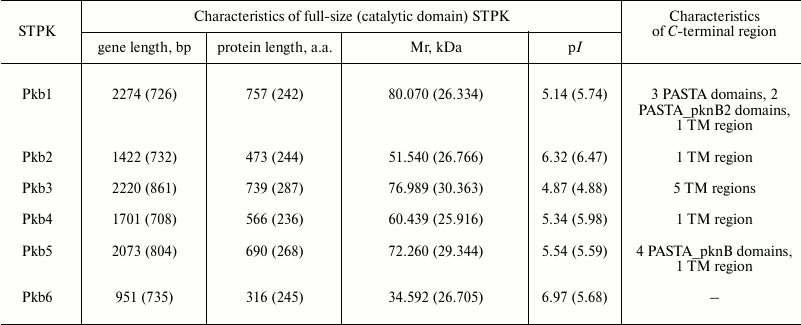
Note: The data presented in the table match those from sequenced genomes
from all strains of B. longum subsp. longum (http://www.ncbi.nlm.nih.gov/genome/?term=Bifidobacterium%20longum).
The genetic environment pkb2 (the gene encoding the domain of fibronectin and cytokine receptor; gene encoding signaling FHA-domain) as well as unusually high divergence of amino acid sequence of the catalytic domain in Pkb2 from various species of bifidobacteria suggest that this gene might be involved in their adaptation to environment. The unique gene pkb4 typical to species B. longum and B. bifidum is located next to the gene encoding the DNA-binding protein belonging to the family of transcriptional regulators involved in adaptive cell reactions [4, 28].
Earlier, catalytic domains from all six protein kinases Pkb1-Pkb6 found in strain B. longum B379M we cloned in E. coli within expression vector pET32a [4].
Optimization of conditions for culturing E. coli strains containing recombinant STPKs. Recombinant strains of E. coli containing plasmids pET32a:pkb1, pET32a:pkb2, pET32a:pkb3, pET32a:pkb4, pET32a:pkb5, and pET32a:pkb6 were grown in LB- and TB-culture media followed by induction for gene expression by adding IPTG at concentrations of 0.25, 0.5, 1.0, and 1.5 mM for 5 or 18 h. After that, the bacteria were sedimented by centrifuging (5000 rpm, 10 min, 4°C), resuspended in sample buffer, and analyzed by SDS-PAGE.
The maximum expression of all protein kinases was observed by using 1 mM IPTG applied during 18-h culture in TB-medium. The expression level of the target proteins comprised as much as 40-50% of the total cell protein. By analyzing electrophoregrams, it was found that it resulted in expression of proteins with Mr 44, 45, 48, 44, 47, and 45 kDa, which corresponded to the expected molecular weights of recombinant proteins of the catalytic domains from protein kinases summed with Mr matching the protein of the entire linker plasmid pET32a containing the thioredoxin gene (Trx). Biomass yield from the 15-ml cell culture was centrifuged (6000 rpm (4226g), 15 min, 4°C) and frozen to –20°C for subsequent isolation of the proteins.
Development of conditions for lysis of E. coli cells for isolation of catalytic domains of protein kinases from B. longum B379M. To check the ability to isolate native proteins derived from E. coli containing recombinant plasmids, the bacteria were thawed at 4°C, resuspended in 3 ml of native lysis buffer (50 mM NaH2PO4, 5 mM Tris-HCl, 300 mM NaCl, 10 mM imidazole, 1 mM PMSF, 5 mM DTT, pH 8.0) supplemented with lysozyme (1 mg/ml) and 20 mM 2-mercaptoethanol, and incubated at 4°C for 1 h. Then the cells were sonicated for 4 min, avoiding warming the suspension, and centrifuged (7500 rpm (6600g), 30 min, 4°C) for separating soluble fraction (lysate) and pellet (inclusion bodies). The final fractions were analyzed by running SDS-PAGE (Fig. 1). Based on the lysis data, strain E. coli BL21(DE3)pLysS was selected for further experiments.
Fig. 1. Electrophoregram of protein fractions from E. coli BL21(DE3)pLysS containing plasmids pET32a:pkb1-pET32a:pkb6. Lanes: 1-3) Pkb1; 4-6) Pkb2; 7-9) Pkb3; 10-12) Pkb4; 13-15) Pkb5; 16-18) Pkb6; 1, 4, 7, 10, 13, 16) control samples; 2, 5, 8, 11, 14, 17) lysates; 3, 6, 9, 12, 15, 18) pellets (inclusion bodies); M, molecular weight protein marker SM0441 (Fermentas, Lithuania).
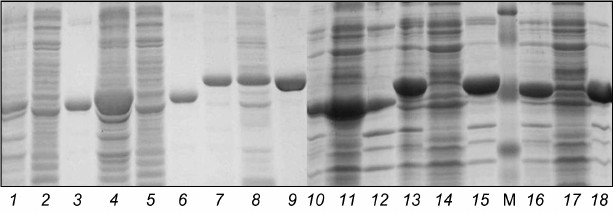
Analysis of isolated fractions containing Pkb3 and Pkb4 proteins revealed that the target proteins were found both in the soluble fraction and in the inclusion bodies, at approximately equal ratio, thereby providing the opportunity to isolate these proteins under native conditions. Examining fractions of the isolated proteins Pkb1, Pkb2, Pkb5, and Pkb6 showed that the target protein was entirely found in the pellet (enriched in cells in the form of insoluble inclusion bodies), and these proteins might be isolated only under denaturing conditions with subsequent protein refolding.
Isolation of catalytic domains of protein kinases Pkb3 and Pkb4 from soluble fraction. To increase solubility of the target proteins Pkb3 and Pkb4, conditions used for culturing strains E. coli containing recombinant plasmids pET32a:pkb3 and pET32a:pkb4 were modified: induction of IPTG-triggered expression was done on lowering temperature from 28 to 26°C, and propagation time was shortened to 5 h. Under these conditions, more than 70% of the target protein was in the soluble fraction.
Strains of E. coli containing recombinant plasmids were cultured under optimized conditions in 250 ml of culture medium. Proteins were isolated using the Ni-NTA Fast Start Kit (6). All consequent steps of protein isolation were performed at 4°C. The pellet was resuspended in 15 ml of native lysis buffer supplemented with lysozyme and 2-mercaptoethanol and incubated for 1 h at 4°C. Then, the fraction was sonicated for 4 min avoiding warming of the suspension, and centrifuged (7500 rpm (6600g), 30 min, 4°C) to sediment cell debris. The lysates were placed on a column pre-equilibrated with lysis buffer: for this, the column was washed three times with washing buffer (50 mM NaH2PO4, 5 mM Tris-HCl, 300 mM NaCl, 50 mM imidazole, 1 mM PMSF, 5 mM DTT, pH 8.0). Bound proteins were twice eluted from the column by applying 300 mM imidazole-containing buffer.
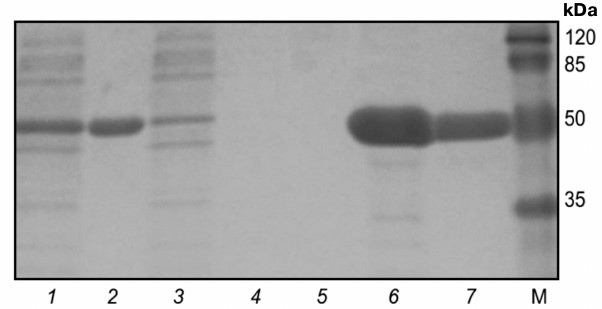
Electrophoregram of stepwise isolation and purification of catalytic domain from protein kinase Pkb3 is shown on Fig. 2. A 250-ml culture medium yielded 11.6 ± 1.5 and 28 ± 3 mg of Pkb3 and Pkb4, respectively, with purity ≥98%. Mass-spectrometric analysis confirmed that the isolated proteins were catalytic domains of protein kinase Pkb3 and Pkb4 derived from B. longum.
Fig. 2. Electrophoregram of stepwise isolation and purification of catalytic domain from protein kinase Pkb3. Lanes: 1) lysate; 2) pellet; 3) flow-through; 4) wash-1; 5) wash-2; 6) eluate-1; 7) eluate-2; M, molecular weight protein marker SM0441 (Fermentas, Lithuania).
Isolation of catalytic domains of protein kinases Pkb1, Pkb2, Pkb5, and Pkb6 from insoluble fraction. The target proteins were isolated under denaturing conditions. For refining conditions for lysis of strains of E. coli containing recombinant plasmids pET32a:pkb1, pET32a:pkb2, pET32a:pkb5, and pET32a:pkb6, the bacteria were grown being induced with 1 mM IPTG in 15 ml TB-medium containing chloramphenicol and ampicillin. After that, the bacteria were centrifuged (5000 rpm (2935g), 10 min, 4°C), resuspended in 3 ml of denaturing lysis buffer (8 M urea, 100 mM Na2HPO4, 10 mM Tris-HCl, pH 8.0), and incubated at room temperature for 1 h. However, after separating the final lysate into soluble and insoluble fractions, all target proteins were found in the pellet, thereby significantly hindering their further purification and renaturation. The simplest way to increase efficacy of the lysis procedure was to extend exposure time of lysis up to ≥3 h followed by sonication. Under these conditions, the target proteins went into the soluble fraction without binding to Ni-agarose column. Owing to this, the concentration of urea was lowered to 6 M in all buffer solutions. Cells centrifuged from 250-ml culture medium were resuspended in 15 ml of denaturing lysis buffer (6 M urea, 100 mM Na2HPO4, 10 mM Tris-HCl, pH 8.0) and incubated at room temperature for 2.5 h followed by sonication for 4 min and centrifugation (7500 rpm (6600g), 30 min, 4°C). Then the lysate was placed on a column pre-equilibrated with lysis buffer containing 6 M urea. The columns were washed three times with denaturing wash buffer (6 M urea, 100 mM Na2HPO4, 10 mM Tris-HCl, 50 mM imidazole, pH 5.3). Bound proteins were eluted twice from the columns using denaturing buffer containing 6 M urea and 300 mM imidazole.
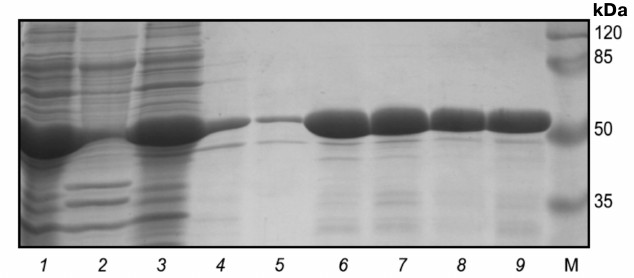
A pure catalytic domain of protein kinase Pkb5 was isolated under the above noted conditions. After that, the purified protein was restored using 2-mercaptoethanol and renatured by dialyzing against buffer (1.5 M urea, 50 mM Tris-HCl, 200 mM NaCl, 10% glycerol, pH 8.0). The yield of Pkb5 from 250 ml of culture medium was 10 ± 1 mg, purity ≥98% (Fig. 3). Mass-spectrometric analysis confirmed that the isolated protein was the catalytic domain of protein kinase Pkb5 derived from B. longum.
Fig. 3. Electrophoregram of stepwise isolation and purification of catalytic domain from protein kinase Pkb5 under denaturing conditions. Lanes: 1) lysate; 2) pellet; 3) flow-through; 4) wash-1; 5) wash-2; 6) eluate-1; 7) eluate-2; 8) eluate-3; 9) post-dialysis sample; M, molecular weight protein marker SM0441 (Fermentas, Lithuania).
Catalytic domains of protein kinase Pkb1, Pkb2, and Pkb6 failed to be isolated under denaturing conditions by applying the method used to isolate Pkb5. Therefore, another method was developed to isolate Pkb1, Pkb2, and Pkb6 from inclusion bodies. In particular, the bacteria were resuspended in 15 ml of lysis buffer supplemented with lysozyme, 2-mercaptoethanol, and 0.3% Triton X-100 (to increase solubility) for 1 h followed by 4-min sonication and centrifugation (7500 rpm (6600g), 30 min, 4°C). The inclusion bodies were twice washed with 10 ml of lysis buffer supplemented with 0.5% Triton X-100, sonicated for 2 min, and centrifuged (7500 rpm (6600g), 30 min, 4°C). Then the inclusion bodies were resuspended in 15 ml of denaturing lysis buffer supplemented with 6 M urea, incubated at room temperature for 30 min, and centrifuged (see above), and the lysate was applied to a column pre-equilibrated with denaturing lysis buffer. The column was washed three times with denaturing wash buffer (6 M urea, 100 mM Na2HPO4, 10 mM Tris-HCl, 50 mM imidazole, pH 5.3). Bound proteins were eluted twice from the column using denaturing buffer containing 6 M urea and 300 mM imidazole. Then the target proteins were reduced using 2-mercaptoethanol and renatured by dialysis (see above).
Electrophoregram of stepwise isolation and purification of catalytic domain from protein kinase Pkb2 is shown on Fig. 4. The yield of Pkb1, Pkb2, and Pkb6 obtained from 250-ml culture media was 8.0 ± 1.0, 8.5 ± 1.3, and 8.5 ± 1.8 mg, respectively, with purity ≥98%. Mass-spectrometric analysis confirmed that the isolated proteins were the catalytic domains of protein kinases Pkb1, Pkb2, and Pkb6 of B. longum.
Fig. 4. Electrophoregram of stepwise isolation and purification of catalytic domain from protein kinase Pkb2 derived from inclusion bodies. Lanes: 1) pellet; 2) lysate; 3) flow-through; 4) wash-1; 5) wash-2; 6) eluate-1; 7) eluate-2; M, molecular weight protein marker SM0441 (Fermentas, Lithuania).

Investigation of autophosphorylation of isolated proteins Pkb1, Pkb3, Pkb4, and Pkb6. The autophosphorylation reaction is the first step in functioning of many bacterial STPKs [35-37]. Earlier, we demonstrated that two STPKs (Pkb2 and Pkb5) [4] of the six kinases examined in the current study are capable of autophosphorylation. Here, we investigated the potential for autophosphorylation in the four remaining STPKs by applying two approaches: using [γ-32P]ATP and the luminescence reaction. In particular, proteins were incubated with [γ-32P]ATP in buffer solution followed by SDS-PAGE (Fig. 5a). To detect radiolabeled proteins, the SDS-PAGE gel was exposed to X-ray KODAK MXG film (100sh) (Fig. 5b).
Fig. 5. Autophosphorylation of catalytic domains of protein kinases quantified by using [γ-32P]ATP. a) Electrophoregram of autophosphorylated protein kinases: 1) Pkb1; 2) Pkb3; 3) Pkb4; 4) Pkb6; M, molecular weight protein marker SM0441 (Fermentas, Lithuania). b) Autoradiograph of autophosphorylated protein kinases.

The data demonstrate that the catalytic domain of protein kinase Pkb3 is capable of autophosphorylation; however, the catalytic domains from Pkb1, Pkb4, and Pkb6 were found to lack the ability to autophosphorylate under these conditions.
Autophosphorylation of proteins Pkb1, Pkb3, Pkb4, and Pkb6 was also examined by measuring intensity of the luminescence reaction corresponding to the amount of ATP remaining after the reaction. The degree of autophosphorylation per 5 µg of protein kinase catalytic domain Pkb3 was 25%, per 10 µg – 43%, per 20 µg – 70%, thus revealing a linear dose-dependent relationship (Table 2). During the autophosphorylation reaction of the catalytic domains of protein kinases Pkb1, Pkb4, and Pkb6, the amount of ATP remaining after the reaction did not differ from that in the control sample.
Table 2. Investigation of
autophosphorylation of protein kinase Pkb3

Note: Average data from four independent repeats ± SD are
presented.
The ability to autophosphorylate was found only in the catalytic domain of protein kinase Pkb3. In summary, our earlier data [4] and those obtained during this study demonstrate that only three of six STPKs are capable of autophosphorylation: Pkb2, Pkb3, and Pkb5.
The properties of mycobacterial STPKs were investigated most thoroughly. Strains of M. tuberculosis contain 11 serine/threonine protein kinases of eukaryotic type. All protein kinases from M. tuberculosis were cloned in E. coli (catalytic domain and/or full-size protein): an individualized approach was used to select the kind of vector plasmid for each protein kinase including optimized conditions for its expression, expression strain, developed isolation technique either under native or denaturing conditions, and optimized conditions for their renaturation. All protein kinases derived from M. tuberculosis were shown to undergo autophosphorylation [35-41].
In the current work we optimized conditions for culturing E. coli strains for further isolation of protein kinases and also developed methods for isolation of preparative amounts of proteins of B. longum six protein kinases’ catalytic domains: a method for isolation of native proteins Pkb3 and Pkb4, a method for isolating protein Pkb5 in denatured conditions, and a method for isolating proteins Pkb1, Pkb2, and Pkb6 from inclusion bodies. In addition, a dialysis procedure for renaturation of proteins isolated from insoluble fraction was optimized.
The data of the present study on optimizing methods for isolation of preparative amounts of catalytic domains of six protein kinases of B. longum open new opportunities in investigating structure and functions of serine/threonine protein kinases from actinobacteria as well as creating their 3D crystal structure and looking for substrates.
REFERENCES
1.Pereira, S. F., Goss, L., and Dworkin, J. (2011)
Eukaryote-like serine/threonine kinases and phosphatases in
bacteria, Microbiol. Mol. Biol. Rev., 75, 192-212.
2.Zakharevich, N. V., Osolodkin, D. I., Artamonova,
I. I., Palyulin, V. A., Zefirov, N. S., and Danilenko, V. N. (2012)
Signatures of the ATP binding pocket as a basis for structural
classification of the serine/threonine protein kinases of gram-positive
bacteria, Proteins, 80, 1363-1376.
3.Petrickova, K., and Petricek, M. (2003)
Eukaryotic-type protein kinases in Streptomyces coelicolor:
variations on a common theme, Microbiology, 149,
1609-1621.
4.Nezametdinova, V. Z., Zakharevich, N. V.,
Alekseeva, M. G., Averina, O. V., Mavletova, D. A., and Danilenko, V.
N. (2014) Identification and characterization of the serine/threonine
protein kinases in Bifidobacterium, Arch. Microbiol.,
196, 125-136.
5.Chao, J., Wong, D., Zheng, X., Poirier, V., Bach,
H., Hmama, Z., and Av-Gay, Y. (2010) Protein kinase and phosphatase
signaling in Mycobacterium tuberculosis physiology and
pathogenesis, Biochim. Biophys. Acta, 1804, 620-627.
6.Ruggiero, A., De Simone, P., Smaldone, G.,
Squeglia, F., and Berisio, R. (2012) Bacterial cell division regulation
by ser/thr kinases: a structural perspective, Curr. Protein Pept.
Sci., 13, 756-766.
7.Alber, T. (2009) Signaling mechanisms of the
Mycobacterium tuberculosis receptor ser/thr protein kinases,
Curr. Opin. Struct. Biol., 19, 650-657.
8.Kobir, A., Shi, L., Boskovic, A., Grangeasse, C.,
Franjevic, D., and Mijakovic, I. (2011) Protein phosphorylation in
bacterial signal transduction, Biochim. Biophys. Acta,
1810, 989-994.
9.Burnside, K., and Rajagopal, L. (2012) Regulation
of prokaryotic gene expression by eukaryotic-like enzymes, Curr.
Opin. Microbiol., 15, 125-131.
10.Fernandez, P., Saint-Joanis, B., Barilone, N.,
Jackson, M., Gicquel, B., Cole, S. T., and Alzari, P. M. (2006) The
ser/thr protein kinase PknB is essential for sustaining mycobacterial
growth, J. Bacteriol., 188, 7778-7784.
11.Gomez-Velasco, A., Bach, H., Rana, A. K., Cox, L.
R., Bhatt, A., Besra, G. S., and Av-Gay, Y. (2013) Disruption of the
serine/threonine protein kinase H affects phthiocerol dimycocerosates
synthesis in Mycobacterium tuberculosis, Microbiology,
159, 726-736.
12.Reck, M., Rutz, K., Kunze, B., Tomasch, J.,
Surapaneni, S. K., Schulz, S., and Wagner-Dobler, I. (2011) The biofilm
inhibitor carolacton disturbs membrane integrity and cell division of
Streptococcus mutans through the serine/threonine protein kinase
PknB, J. Bacteriol., 193, 5692-5706.
13.Shah, I. M., Laaberki, M.-H., Popham, D. L.,
and Dworkin, J. (2008) A eukaryotic-like ser/thr kinase signals
bacteria to exit dormancy in response to peptidoglycan fragments,
Cell, 135, 486-496.
14.Kumar, D., and Narayanan, S. (2012) PknE, a
serine/threonine kinase of Mycobacterium tuberculosis modulates
multiple apoptotic paradigms, Infect. Genet. Evol., 12,
737-747.
15.Kumar, D., Palaniyandi, K., Challu, V. K., Kumar,
P., and Narayanan, S. (2013) PknE, a serine/threonine protein kinase
from Mycobacterium tuberculosis has a role in adaptive
responses, Arch. Microbiol., 195, 75-80.
16.Lougheed, K. E., Osborne, S. A., Saxty, B.,
Whalley, D., Chapman, T., Bouloc, N., Chugh, J., Nott, T. J., Patel,
D., Spivey, V. L., Kettleborough, C. A., Bryans, J. S., Taylor, D. L.,
Smerdon, S. J., and Buxton, R. S. (2011) Effective inhibitors of the
essential kinase PknB and their potential as anti-mycobacterial agents,
Tuberculosis (Edinburgh), 91, 277-286.
17.Danilenko, V. N., Osolodkin, D. I., Lakatosh, S.
A., Preobrazhenskaya, M. N., and Shtil, A. A. (2011) Bacterial
eukaryotic type serine-threonine protein kinases: from structural
biology to targeted anti-infective drug design, Curr. Top. Med.
Chem., 11, 1352-1369.
18.Ventura, M., OʼFlaherty, S., Claesson, M.
J., Turroni, F., Klaenhammer, T. R., van Sinderen, D., and
OʼToole, P. W. (2009) Genome-scale analyses of health-promoting
bacteria: probiogenomics, Nat. Rev. Microbiol., 7,
61-71.
19.Reid, G., Younes, J. A., Van der Mei, H. C.,
Gloor, G. B., Knight, R., and Busscher, H. J. (2011) Microbiota
restoration: natural and supplemented recovery of human microbial
communities, Nat. Rev. Microbiol., 9, 27-38.
20.Avershina, E., Storro, O., Oien, T., Johnsen, R.,
Wilson, R., Egeland, T., and Rudi, K. (2013) Bifidobacterial succession
and correlation networks in a large unselected cohort of mothers and
their children, Appl. Environ. Microbiol., 79,
497-507.
21.Turroni, F., Peano, C., Pass, D. A., Foroni, E.,
Severgnini, M., Claesson, M. J., Kerr, C., Hourihane, J., Murray, D.,
Fuligni, F., Gueimonde, M., Margolles, A., De Bellis, G.,
OʼToole, P. W., van Sinderen, D., Marchesi, J. R., and Ventura,
M. (2012) Diversity of bifidobacteria within the infant gut microbiota,
PLoS One, 7, e36957.
22.Borre, Y. E., OʼKeeffe, G. W., Clarke, G.,
Stanton, C., Dinan, T. G., and Cryan, J. F. (2014) Microbiota and
neurodevelopmental windows: implications for brain disorders,
Trends. Mol. Med., 20, 509-518.
23.Diaz Heijtz, R., Wang, S., Anuar, F., Qian, Y.,
Bjorkholm, B., Samuelsson, A., Hibberd, M. L., Forssberg, H., and
Pettersson, S. (2011) Normal gut microbiota modulates brain development
and behavior, Proc. Natl. Acad. Sci. USA, 108,
3047-3052.
24.Savignac, H. M., Tramullas, M., Kiely, B., Dinan,
T. G., and Cryan, J. F. (2015) Bifidobacteria modulate cognitive
processes in an anxious mouse strain, Behav. Brain Res.,
287, 59-72.
25.Hidalgo-Cantabrana, C., Lopez, P., Gueimonde, M.,
de los Reyes-Gavilan, C. G., Suarez, A., Margolles, A., and
Ruas-Madiedo, P. (2012) Immune modulation capability of
exopolysaccharides synthesized by lactic acid bacteria and
bifidobacteria, Probiotics Antimicrob. Prot., 4,
227-237.
26.Hirayama, Y., Sakanaka, M., Fukuma, H., Murayama,
H., Kano, Y., Fukiya, S., and Yokota, A. (2012) Development of a
double-crossover markerless gene deletion system in Bifidobacterium
longum: functional analysis of the α-galactosidase gene for
raffinose assimilation, Appl. Environ. Microbiol., 78,
4984.
27.Sakaguchi, K., He, J., Tani, S., Kano, Y., and
Suzuki, T. (2012) A targeted gene knockout method using a newly
constructed temperature-sensitive plasmid mediated homologous
recombination in Bifidobacterium longum, Appl. Microbiol.
Biotechnol., 95, 499-509.
28.Zakharevich, N. V., Averina, O. V., Klimina, K.
M., Kudryavtseva, A. V., Kasianov, A. S., Makeev, V. J., and Danilenko,
V. N. (2015) Complete genome sequence of Bifidobacterium longum
GT15: identification and characterization of unique and global
regulatory genes, Microb. Ecol., in press.
29.Averina, O. V., Nezametdinova, V. Z., Alekseeva,
M. G., and Danilenko, V. N. (2012) Genetic instability of probiotic
characteristics in the Bifidobacterium longum subsp. longum
B379M strain during cultivation and maintenance, Russ. J.
Genet., 48, 1103-1111.
30.Mierendorf, R., Yeager, K., and Novy, R. (1994)
Innovations, Newsletter Novagen, 1, 1-3.
31.Calik, P., and Levent, H. (2009) Effects of
pretreated beet molasses on benzaldehyde lyase production by
recombinant Escherichia coli BL21(DE3)pLySs, J. Appl.
Microbiol., 107, 1536-1541.
32.Sambrook, J., Fritsch, E. E., and Maniatis, T.
(1989) Molecular Cloning: a Laboratory Manual, Cold Spring
Harbor Laboratory Press.
33.Bradford, M. M. (1976) A rapid and sensitive
method for the quantitation of microgram quantities of protein
utilizing the principle of protein–dye binding, Anal.
Biochem., 72, 248-254.
34.Shevchenko, A., Tomas, H., Havlis, J., Olsen J.
V., and Mann, M. (2006) In-gel digestion for mass spectrometric
characterization of proteins and proteomes, Nat. Protoc.,
1, 2856-2860.
35.Damle, N. P., and Mohanty, D. (2014) Mechanism of
autophosphorylation of mycobacterial PknB explored by molecular
dynamics simulations, Biochemistry, 53, 4715-4726.
36.Molle, V., Brown, A. K., Besra, G. S., Cozzone,
A. J., and Kremer, L. (2006) The condensing activities of the
Mycobacterium tuberculosis type II fatty acid synthase are
differentially regulated by phosphorylation, J. Biol. Chem.,
281, 30094-30103.
37.Thakur, M., Chaba, R., Mondal, A. K., and
Chakraborti, P. K. (2008) Interdomain interaction reconstitutes the
functionality of PknA, a eukaryotic type Ser/Thr kinase from
Mycobacterium tuberculosis, J. Biol. Chem., 283,
8023-8033.
38.Duran, R., Villarino, A., Bellinzoni, M.,
Wehenkel, A., Fernandez, P., Boitel, B., Cole, S. T., Alzari, P. M.,
and Cervenansky, C. (2005) Conserved autophosphorylation pattern in
activation loops and juxtamembrane regions of Mycobacterium
tuberculosis Ser/Thr protein kinases, Biochem. Biophys. Res.
Commun., 333, 858-867.
39.Lakshminarayan, H., Narayanan, S., Bach, H.,
Sundaram, K. G., and Av-Gay, Y. (2008) Molecular cloning and
biochemical characterization of a serine threonine protein kinase,
PknL, from Mycobacterium tuberculosis, Protein Express
Purif., 58, 309-317.
40.Gopalaswamy, R., Narayanan, P. R., and Narayanan,
S. (2004) Cloning, overexpression, and characterization of a
serine/threonine protein kinase pknI from Mycobacterium
tuberculosis H37Rv, Protein Express Purif., 36,
82-89.
41.Av-Gay, Y., Jamil, S., and Drews, S. J. (1999)
Expression and characterization of the Mycobacterium
tuberculosis serine/threonine protein kinase PknB, Infect.
Immun., 67, 5676-5682.