REVIEW: Beta-Amyloid and Tau-Protein: Structure, Interaction, and Prion-Like Properties
O. G. Tatarnikova1,2*, M. A. Orlov1, and N. V. Bobkova1*
1Institute of Cell Biophysics, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: nbobkova@mail.ru2Pushchino State Natural Research Institute, 142290 Pushchino, Moscow Region, Russia; E-mail: olga.psn@mail.ru
* To whom correspondence should be addressed.
Received October 10, 2015
During the last twenty years, molecular genetic investigations of Alzheimer’s disease (AD) have significantly broadened our knowledge of basic mechanisms of this disorder. However, still no unambiguous concept on the molecular bases of AD pathogenesis has been elaborated, which significantly impedes the development of AD therapy. In this review, we analyze issues concerning processes of generation of two proteins (β-amyloid peptide and Tau-protein) in the cell, which are believed to play the key role in AD genesis. Until recently, these agents were considered independently of each other, but in light of the latest studies, it becomes clear that it is necessary to study their interaction and combined effects. Studies of mechanisms of toxic action of these endogenous compounds, beginning from their interaction with known receptors of main neurotransmitters to specific peculiarities of functioning of signal intracellular pathways upon development of this pathology, open the way to development of new pharmaceutical substances directed concurrently on key mechanisms of interaction of toxic proteins inside the cell and on the pathways of their propagation in the extracellular space.
KEY WORDS: Alzheimer’s disease, β-amyloid, Tau-protein, APP, presenilins, prion-like mechanism of ADDOI: 10.1134/S000629791513012X
Abbreviations: Aβ, beta-amyloid peptide; AD, Alzheimer’s disease; ADAM, a desintegrin and metalloproteinase domain; AICD, amyloid precursor protein intracellular domain; APOE, apolipoprotein E; APP, amyloid precursor protein; BACE, beta-site amyloid precursor protein cleaving enzyme; CTF, C-terminal fragment; FAD, familial Alzheimer’s disease; GSM, γ-secretase modulator; LXR, liver X receptor; MAPT, microtubule-associated protein tau; NFT, neurofibrillary tangles; PART, primary age-related tauopathy; PHF, paired helical filaments; PPARγ, peroxisome proliferator-activated receptor γ; PrP, prion protein; PSEN, presenilin; RAGE, receptor for advanced glycation end products; RXR, retinoid X receptor; SNP, single-nucleotide polymorphism.
Alzheimer’s disease (AD) is a progressive neurodegenerative
disease, the most common form of dementia. The development of AD is
characterized by deterioration in patients’ condition aggravating
with years, rapid disability with a loss of the capacity for
independent living activity and, in the long run, death.
Notwithstanding the efforts of researchers and physicians during the
last thirty years, no efficient methods of AD treatment have been
developed yet.
There are millions of people over the world suffering from AD. Diagnostics, treatment, and nursing care of patients are extremely expensive for both governments and the patients themselves. Epidemiological prognoses published by the World Health Organization suggest a remarkable deterioration of the existing state in the nearest decades [1]. Now AD is not only an unsolved medico-biological problem, but also a serious social and economic trouble.
Therefore, studies of basic mechanisms of AD pathogenesis have become of great importance. The understanding of molecular bases of this pathology is required both for developing therapeutic methods and for early diagnostics that are extraordinarily impeded by the prolonged course of the asymptotic stage of the disease. The clinical stage of the disease manifestation begins when brain disturbances are so great that it is difficult to expect positive effects of medical treatment. More than a hundred years ago, A. Alzheimer, who described this disease, indicated its foremost morphological feature – senile plaques in the brain of patients that died of AD. These plaques consist mainly of β-amyloid peptide (Aβ) having molecular mass of 4 kDa and length of about 40-43 amino acid residues. Aβ is a fragment of a transmembrane precursor protein of β-amyloid (APP) found in many tissues including neuronal synapses. APP is involved in neuroplasticity, formation of synapses, and is required for survival of nerve cells [2]. Researchers paid attention to Tau-protein (microtubule-associated Tau-protein, MAPT) somewhat later than to Aβ. It was found that in AD patients, Tau-protein undergoes hyperphosphorylation, loses its normal capacity to stabilize microtubules, and aggregates inside the cell forming paired helical filaments (PHF) and neurofibrillary tangles (NFT). The number of Tau fibrillar deposits correlates with cognitive deficit. No such correlation has been found for senile plaques with Aβ. Today, combined consideration of pathogenetic processes with involvement of Aβ and Tau-protein seems to be the most promising. The available data suggest that the “pathological” oligomeric form of one of these factors can induce generation of oligomeric toxic forms of the other protein. Therewith, Aβ may trigger AD development, but after Aβ has induced pathological changes in Tau-protein, the latter affects the course of the pathology. In this case, Aβ itself is not already so important.
MOLECULAR BASES OF ALZHEIMER’S DISEASE PATHOGENESIS
Though a great number of experimental results on morphology, biochemistry, and physiology of AD are available in the world literature, there is still no unambiguous concept on the molecular mechanisms of pathogenesis of this disease and, as a result, of adequate approaches to its treatment and prevention. One of the factors causing death of nerve cells and cognitive disturbances is pathological accumulation in brain tissue of aggregates of Aβ peptide, the main component of senile plaques, which are morphological features of AD. As a product of proteolytic cleavage of APP, Aβ has clearly expressed fibrillogenic properties, and its oligomers are toxic to nerve cells, causing their degeneration and death [3, 4]. Neurotoxicity of Aβ becomes apparent via disturbance of Ca2+ homeostasis, induction of oxidative stress, excitotoxicity, inflammation, and apoptosis. The latter effect can take place, in particular, via induction of mitochondrial pore opening, when cell death can proceed also by the mechanism of necrosis [5].
The second characteristic morphological feature of AD is disturbance of cytoskeleton in nerve cells and accumulation of neurofibrillary tangles (NFT) inside them. NFT consist mainly of insoluble filaments of hyperphosphorylated Tau-protein [6, 7]. It is speculated that during the neurodegenerative process, the first to be formed are amyloid fibrils, disturbing the functioning of nerve cells, and then neurofibrillary tangles are generated in them. The two processes lead to degeneration and death of neurons, mainly in such brain areas as the cortex and the hippocampus. Synaptic transmission is disturbed, particularly in cholinergic terminals [8]. In AD patients, the content of serotonin, noradrenalin, and other monoamines in brain tissues is reduced [9].
AD is characterized by a complex etiology. Its emergence is caused by both genetic mutations and a multitude of external factors. While the chief risk factor for sporadic AD in elderly people is their age, “familial” AD affects younger people and mainly depends on mutations in a number of genes (in the first place, presenilin-1, presenilin-2, and APP). Studies of the genetic basis of AD greatly widen our notions on the pathogenesis of the disorder, though they define first its uncommon monogenetic forms affecting separate families [10]. These diseases are characterized by mutations of the above-mentioned genes – APP, PSN-1, and PSN-2. At present, increased risk of emergence of sporadic AD is associated with enhanced expression of the apolipoprotein E gene (APOE) in brain tissues. It was demonstrated that APOE binds to Aβ and jointly with it forms senile plaques. Moreover, it was shown that expression of APOE is enhanced in AD [11]. Further investigations confirmed that carriers of one of the three APOE alleles (namely APOE ε4) run increased risk of developing AD. Thus, in heterozygote patients it increases 3-fold, whereas in homozygote ones it increases 15-fold. Meanwhile, the presence of the APOE ε2 allele, on the contrary, has a protective effect [12]. A meta-analysis of literature data shows that there are also at least 20 loci associated with pathogenesis of AD [13]. Current methods of genetic typing and mapping of single-nucleotide polymorphisms (SNP) can provide a possibility to discover other genes that increase or decrease the risk of AD, which is of great importance for improving methods of early diagnostics and development of effective drugs.
Until recently, the accumulation of Aβ fibrils was regarded as an irreversible process, but now data have become available that demonstrate Aβ release from brain tissue through perivascular pathways [14], and different enzymes cleave it proteolytically [15, 16]. According to up-to-date notions, Aβ metabolism is a dynamic process depending on various factors of different nature – internal (genetic, cellular, vascular) and external, for example, hypoxia and stress. These factors can affect both generation and accumulation of Aβ and its degradation and release. The cause of higher frequency of AD in aged people is disturbance in the supply of the brain with oxygen during ischemia and insults. Due to this, studies are conducted on its effects on the processes of Aβ generation as well as on the activity and expression of amyloid-forming and amyloid-degrading enzymes [15, 17, 18]. In addition to doubtless theoretical importance – establishing molecular mechanisms of pathology development – these studies can contribute to developing new approaches for AD therapy and prevention.
GENERATION OF BETA-AMYLOIDS
As stated above, Aβ is a product of fragmentation of a precursor protein (APP) – a type I transmembrane protein (Figs. 1 and 2); its extracellular N-terminal domain (sAPP) can be subject to two independent mechanisms of proteolysis [19]. Enzymes involved in Aβ cleavage are called secretases. During the more common mode of proteolysis [20], APP is first affected by α-secretase, more exactly, one or several representatives of the ADAM (“a disintegrin and metalloproteinase domain”) family of proteins including ADAM10, ADAM17, ADAM9, and ADAM19 [21-23]. ADAM10 is the predominant form of α-secretase in the brain, and recently two uncommon mutations of ADAM10 were identified as a factor predisposing to an early form of AD [24]. However, this discovery should still be confirmed. This mode of proteolysis involving α-secretase is called non-amyloidogenic, because fragmentation of the APP molecule occurs near the external surface of the membrane between amino acid residues within the Aβ sequence that prevents subsequent formation of an amyloid peptide molecule. An alternative, less common cleavage reaction is catalyzed by β-secretase (BACE) breaking APP near the N-terminus of the corresponding Aβ domain located close to the external side of the membrane, at a distance of 16 amino acid residues from the site of cleavage by α-secretase (Fig. 1).
Fig. 1. Schematic representation of a full-size APP showing the position of the transmembrane domain (indicated by an oval) and sites of cleavage by α-, β-, and γ-secretases leading to generation of correspondingly soluble sAPPα and APP-CTFα; soluble sAPPβ and APP-CTFβ; Aβ intracellular amyloid domain (AICD) (from [19]).
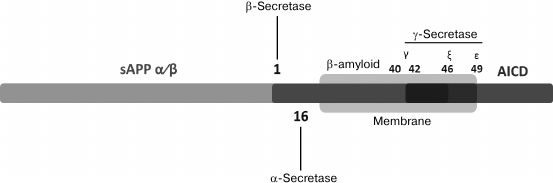
Fig. 2. Schematic representation of processing pathways of APP – non-amyloidogenic (with the involvement of α-secretase and endosome return to the cytoplasmic membrane) and amyloidogenic (cuts made by β-secretase and γ-secretase) (from [19]).
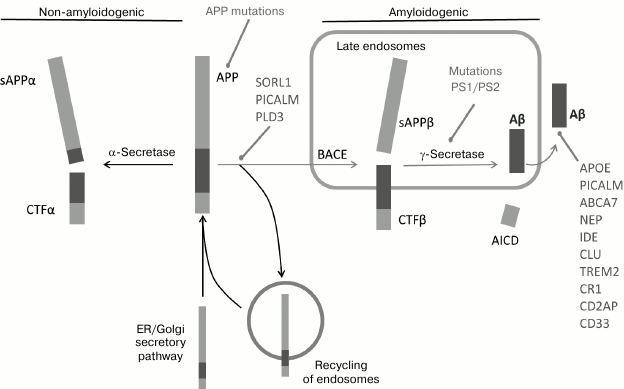
APP and β-secretase get into the cell in independent ways of endocytosis and meet in clathrin vesicles or in early endosomes, where the full-size APP is cleaved by β-secretase with involvement of such proteins as PICALM, BIN1, and CD2AP [25]. After endocytosis, APP is directed to specific compartments inside the cell due to the work of intracellular receptors of vesicle transport and grading. Interactions with these receptors and with SORL1 determine whether the full-size APP (holoprotein) will be redirected to the retromeric complex or will continue moving to mature endosomes [26-29]. The retromeric complex is a conservative protein complex that is assembled on endosomes and returns to the Golgi complex a number of physiologically important proteins, including SORL1. The C-terminal fragment (CTF) of APP bound to the membrane that appeared because of action of either α- or β-secretase is further subjected to secondary intramembrane endoproteolysis by γ-secretase, realized by the connected to the membrane multimeric protein complex called a presenilin or γ-secretase complex. This presenilin complex consists of four proteins [30]: presenilin 1 or presenilin 2 [31], nicastrin, and also aph-1 and pen-2 [32]. The products of APP-CTF cutting by γ-secretase are subsequently released from the plasmatic membrane into the intracellular space and into the extracellular space in the case of Aβ generated in the BACE pathway and p3 from the α-secretase pathway (Fig. 2).
KEY STATEMENTS OF THE AMYLOID HYPOTHESIS
After extraction of Aβ from brain tissues of deceased AD patients and incomplete establishment of its primary structure, George Glenner advanced a hypothesis that stated that AD could be a result of accumulation of misfolded β-structural proteins as occurs in systemic amyloidosis [33]. This hypothesis has been widely, though not universally, acknowledged by specialists due to a number of subsequent studies (mentioned below) that support concepts on the central role of Aβ in AD pathogenesis. However, it should be emphasized that only accumulation of Aβ is insufficient for generation of AD pathology – this requires also accumulation of deposits of Tau-protein that plays a significant and possibly decisive role in mechanisms of subsequent stages of the pathology.
The importance of Aβ in AD genesis is inter alia confirmed by the fact that a small percentage of pathology cases are associated with inherence of the disease in autosomal dominant manner. Familial Alzheimer’s disease (FAD) patients are carriers of mutations in one of the genes encoding APP, presenilin 1 (PSEN1) [31], or presenilin 2 (PSEN2) (Fig. 3). Products of expression of the three genes interact closely upon Aβ formation, and AD mutations in these genes associated with FAD affect peptide generation and its biology with some differences in mechanisms. With FAD, some mutations in genes encoding PS1, PS2, and APP switch the generation of the peptide from the Aβ40 form to Aβ42 with propensity for aggregation [34]. Other mutations increase the overall generation of Aβ by (i) providing a larger number of substrates – APPP (also upon APP duplication) [35]; (ii) facilitating the accessibility of APP for BACE (for example, the so-called Swedish mutation of APP – KM760/671NL [36]); (iii) enhancing capacity of Aβ for aggregation (for example, the Arctic mutation – APP E693G [37]).
Fig. 3. Scheme of the cascade of the complement system indicating genes of inherent immune system associated with risk of AD development (from [19]).
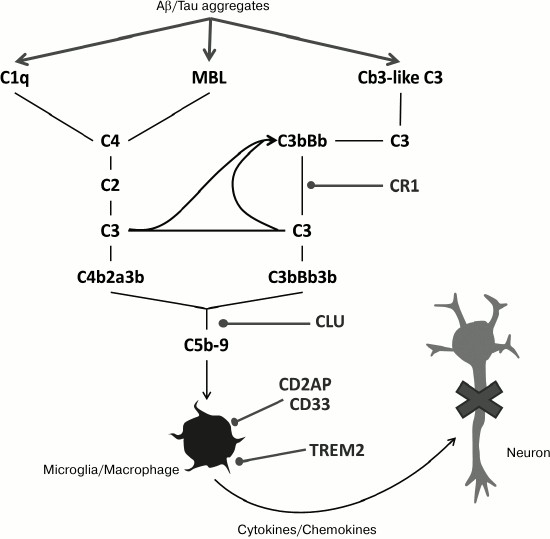
Though most monogenetic autosomal dominant forms of FAD develop much earlier than in the case of the more common later sporadic AD, both forms of the disease have very close clinical and neuropathological features, which leads to a proposal on the general character of molecular mechanisms of their pathogenesis. The similarity between familial and sporadic pathologies was confirmed by reports that several genes most likely involved in pathogenesis of late-stage AD are also involved in APP processing (Figs. 2 and 3). By now, data on genetic investigations have made it possible to compile a list of putative genes, in their common and uncommon versions (by their coding or noncoding sequences), associated with a small increase in the risk of development of AD [38-43]. Bioinformatics methods have approximated classification of these genes by their functional categories, defined as genes encoding proteins involved in (i) vesicle transport, (ii) lipid metabolism, and (iii) inflammatory processes. It is important that some of these genes evidently affect the processing of APP and Aβ formation.
Thus, changes in SORL1, PICALM, ABCA7, ADAM10, and PLD3 are associated both with increase in Aβ generation due to changes in intracellular processing and/or with the capture of Aβ (Figs. 2 and 3) [38, 44].
Some other genes connected with the risk of AD may be involved in the pathological response to accumulation of extracellular aggregates of incorrectly folded Aβ, and in particular, in immune response to the generation of these aggregates (Fig. 3). For example, the changes both in coding and noncoding sequences of the clusterin gene (CLU) [45], encoding a molecular chaperone, are associated with enhanced risk of development of late AD. Though the effect of these mutations associated with AD has not been reliably established on the molecular level, it is clear that clusterin binds Aβ oligomers and prevents their further aggregation [46]. In this way, coding (uncommon versions of the sequence itself and common insertions in it) and noncoding parts of the gene encoding receptor 1 of the complement system (CR1) are connected with the development of late-stage AD [43, 47-49]. Therefore, though on the molecular level the effects of these mutations on the release of Aβ aggregates and the immune response to peptide have not been studied, it looks likely that these mutations can modulate activation of inherent immunity in response to the formation of aggregates [50-52]. Changes in microglia activity in response to the presence of Aβ aggregates are also a remarkable feature of AD neuropathology. Recently, it was reported that CD33 [43, 53], TREM2 [39, 54], and TREML2 [55] are associated with changed probability of late-stage AD development (Fig. 3).
The presumptive protective effect of uncommon missense TREML2 is to be confirmed. By the results of sequential meta-analyses of the data from 74,046 test subjects, CD33 has not been shown to be important on the level of the whole genome [42]. These genes are supposed to be able to modulate directly and indirectly the microglial response to Aβ, decreasing the capture of the peptide by microglia cells and affecting its subsequent activation [56, 57].
Criticism of the “amyloid cascade” hypothesis. Due to the apparent significant role of intracellular Tau-protein aggregates in AD pathogenesis, there are two lines of criticism pointed at the amyloid cascade hypothesis. The first is based on that fact that the NFT content correlates well with the severity of the pathology [58]. For senile plaques, no such correlation is observed [59]. In addition, quite often plaques can be found in the brain of healthy elderly people. A more serious criticism is based on comparative studies of Tau-protein pathology in patients of different age. Some results of these studies demonstrate that most early-stage pathological changes in AD may be the accumulation of Tau-protein aggregates in neurons of the temporal cortex and olfactory bulbs, which becomes more expressed and propagates over the brain tissues in more elderly people [60]. Therefore, it was assumed that initial pathological changes in AD consist of accumulation of Tau-protein aggregates, which then spread over the brain. In studies of the amyloid cascade, a method of neurovisualization of biomarkers in liquor was developed that allows measurements of the content of accumulated Aβ and Tau-protein in brain tissues (both in patients with AD symptoms and without them) [61-65]. This method made it possible to observe the correlation of these cognitive, neurovisualization, and neuropathological studies and propose another interpretation of the events. In particular, evidences have appeared that the available data on pathogenesis reflect the course of two different processes: (i) age-related neurodegeneration of the hippocampus with the development of tauopathy without Aβ pathology (the primary age-related tauopathy – PART), and (ii) the disease independent of and associated with Aβ that begins from the early accumulation of Aβ in the neurocortex, occurring at the preclinical stage, followed by Tau-protein accumulation, inflammatory process, and cognitive disturbances in accord with the amyloid cascade hypothesis [66, 67].
INTERACTION OF BETA-AMYLOID AND TAU-PROTEIN
AD has been traditionally considered as a disorder proceeding with parallel incorrect folding (misfolding) and aggregation of two different proteins, key factors of pathogenesis, existing independently of each other: β-amyloid peptides, which are proteolytic fragments of transmembrane precursor protein of beta-amyloid (APP), and Tau-protein normally existing in neurons (in small amounts in axons) – the protein associated with microtubules. However, studies performed during recent years have revealed numerous functional interactions between Aβ and Tau-protein that impeach the correctness of the dominant hypothesis of “amyloid cascade” in AD pathogenesis. Moreover, the character of the spread of toxic Aβ and Tau-protein subjected to misfolding has considerable similarity with the spread of toxic incorrectly folded forms of the constitutive prion protein PrPsc. In addition, the misfolded Aβ induces misfolding of Tau-protein in vitro due to direct intermolecular interaction. It is proposed that in a pathological conformation Aβ is a matrix leading to misfolding of Tau-protein in vivo [68].
As mentioned, AD is a slowly progressing neurodegenerative disease characterized by misfolding, aggregation, and toxicity of Aβ and Tau-protein in brain tissue [69]. Aggregated Aβ is deposited extracellularly as tightly packed fibrils forming amyloid (senile) plaques. Upon aggregation, Tau-protein also generates tightly packed filaments, but in contrast to amyloid plaques, they are accumulated intracellularly in affected neurons, forming neurofibrillary tangles (NFT). The term “paired helical filaments” (PHF) is frequently used to designate separate filaments of Tau-protein in NFT.
During the latest decade, it has been confirmed that soluble oligomer forms of Aβ play the key role in AD pathogenesis. As compared to the fibrillar form, the level of soluble Aβ oligomers correlates better with neurotoxicity revealed in vivo – for neural cultures, they are much more toxic than fibrillar Aβ [70-75].
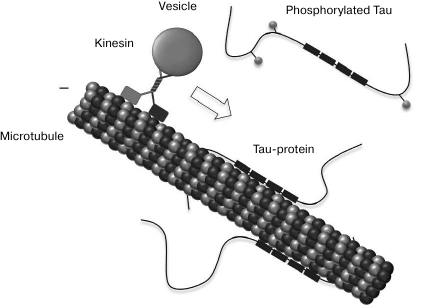
Tau-protein was discovered about 40 year ago as a microtubule-associated Tau-protein (MAPT) that stimulates tubulin polymerization [76], but its presence in NFT was demonstrated only a decade later [77-79]. It should be noted that in addition to the nonspecific function as a factor stimulating the assembling of microtubules, the only known specific function of Tau-protein consists in slowing of the motion of microtubule-associated kinesin motor proteins (as well as loads associated with them) along microtubules (Fig. 4) [80, 81].
Fig. 4. Tau-protein functioning. Tau-protein stabilizes microtubules via four tubulin-binding domains in the case of its full-size isoform. The association of Tau-protein with microtubules is maintained because of the coordinated action of kinases and phosphatases. Tau-protein phosphorylation regulates its capacity to bind to microtubules and affects the axon transport. Tau-protein can inhibit the kinesin-aided (+)-terminus-directed transport of vesicles along microtubules (from [81]).
Historically, in spite of a wide range of neurodegenerative diseases designated as non-Alzheimer’s tauopathies characterized by accumulation of PHF in the brain, in studying AD much less attention was paid to Tau-protein as compared to Aβ. These pathologies can be caused by tens of Tau-protein mutations [81]. In PHF, Tau-protein is abnormally phosphorylated in tens of sites [82], and some of them can be revealed in vivo both in AD patients and in transgenic mice prior to generation of PHF [83].
In our studies [84], we used an in vitro model characterized by: (i) increased expression of the natural form of full-size human Tau-protein by producer cells 3T3-4R-Tau; (ii) functionality of the expressed protein: its interaction with murine tubulin, accompanied by a change in the morphology of producer cell membranes; (iii) generation of fibrillar forms of Tau-protein within cells actively synthesizing this Tau-protein in response to increased amounts of toxic polymers of Aβ: high and low molecular weight forms of Aβ peptide or Tau-protein itself; (iv) existence of a similar receptor-mediated “infectiousness” of such toxic polymers in producer cells of Tau and in neurons of cell culture of the hippocampus of healthy mice. It was shown that enhanced expression of the 4R-Tau isoform by 3T3 cells has a cytotoxic effect on murine neurons. This was observed upon cocultivation of primary culture of the hippocampus and 3T3-4R-Tau cells secreting expression products (4R-isoforms of Tau-protein) [85]. Consequently, these results support the published data on toxicity of polymer Tau forms moving from the extracellular space into target neurons [86, 87].
About 30 years after Stanley B. Prusiner described for the first time the cause of bovine spongiform encephalopathy (“mad cow disease”) and its human equivalent, Creutzfeldt–Jakob disease, the term prion was coined and referred to a previously undescribed form of infection due to protein misfolding [88]. Prusiner proposed that this infectious process might take place in AD [85], and there appeared a great amount of data confirming a surprising similarity in biology and biochemistry of such disorders as AD and “classical” prion diseases. Contrary to PrP-associated diseases such as mad cow disease, scabies in sheep and chicken, most likely AD is not communicated from one organism to another. However, the ever-increasing data show that misfolded toxic oligomers of Aβ and Tau-protein propagate in brain tissues from the target affected neuron to adjacent neurons, this reminding greatly a similar process with involvement of PrP [89-94]. It was demonstrated both for Aβ [71, 95] and for Tau-protein [96-100] that in addition to the fact that their misfolding forms can be captured by neurons containing normal forms of this peptide and protein, this also results in latter pathological conformation, become toxic and propagating to other neurons.
Besides histopathological data [95, 97, 98, 100], during recent years some research teams have demonstrated mechanisms of prion-like propagation of Aβ and Tau-protein [101-105] and described a number of proteins in which an increase in the number of β-folds is the basis for well-known neurodegenerative diseases [90, 92, 94, 106]. In light of this, the most significant is the evidence that Aβ and Tau-protein interact with each other both directly upon molecule contact [107, 108] and indirectly via cell signaling [70, 72, 74, 102, 109-114]. Thus, AD can be considered as a disorder with manifestation of prion-like properties of two molecules.
PRION PROPERTIES OF BETA-AMYLOID
The described progressive aggregation of Aβ at AD suggested an assumption on the presence of prion-like mechanisms of its misfolding. At present, the data obtained in vivo and in vitro are direct evidence of prion-like properties of Aβ and have allowed determination of their specific biochemical and biophysical mechanisms. The most significant results obtained in numerous studies in vivo demonstrated that introduction of incorrectly folded Aβ from Aβ-loaded brain of APP23 transgenic mice or an extract of brain homogenate after autopsy of AD patients, to the hippocampus or the right-brain of young male APP23 mice accelerates generation of aggregated recombinant Aβ molecules in the brain [105, 115-117]. While these deposits of seed-induced Aβ are initially found in tissues only near the site of injection, their sequential propagation occurs gradually along axons to different brain regions bound to the hippocampus. This gives grounds to suggest the involvement of both the axonal transport and extracellular pathways of Aβ propagation over brain tissues.
In in vivo studies, several approaches were used that provided direct evidence for specific mechanisms of propagation of misfolded Aβ. Researchers studying AD long ago noticed the paradox that in many cases purified Aβ behaves unpredictably as concerns the induction of a neurotoxic effect, thereby suggesting the existence of different forms of its aggregates. These assumptions were recently corroborated experimentally when aliquots of monomeric Aβ of one sample were subjected to independent aggregation, which resulted in the formation of a variety of structurally and immunologically different Aβ oligomers specific for each aliquot [118]. These experiments also demonstrated that the interaction of different Aβ oligomers formed specifically with monomeric Aβ stimulates aggregation of the latter with generation of oligomer forms with the same dimensions and immune reactivity. Direct interpretation of these data suggests a model where specific oligomers formed earlier during aggregation propagate, “reproducing” themselves by increasing the probability of realization of such folding schemes inherent to them in newly formed fibrils.
A considerable part of studies devoted to Aβ is conducted with the use of synthetic peptide oligomers Aβ1-40 and Aβ1-42. However, Aβ preparations obtained from biological specimens, especially from brain tissues of AD patients, show much higher biological activity in different studies [119-121]. This may be associated, at least in part, with the possibility that Aβ from biological specimens contains a great variety of peptides including Aβ1-40 and Aβ1-42, distinguished by their biological activity, length of N- and C-termini, and posttranslational modifications of the main chain.
In fact, the recent study of Aβ from liquor specimens showed more than twenty different types of peptide molecules [122]. At least one of the Aβ types present in biological material is concurrently and exclusively toxic and has prion-like properties. Therefore, it is possible that represented in small volumes, but displaying considerable activity infectious Aβ forms, isolated from brain tissues or neural cultures, can explain the increased biological activity of natural Aβ as compared to its synthetic analog.
PRION PROPERTIES OF TAU-PROTEIN
During recent years, several lines of evidence have developed demonstrating that polymer filaments of Tau-protein reveal infectious features in respect to normal Tau-protein, which upon interaction with pathological Tau-protein acquires its properties. Such “self-replication” may take place due to protein aggregation. Monomeric Tau-protein is a soluble natively unfolded protein [123] with low capacity to form filaments in vitro, if this is not facilitated by the presence of proteins with misfolding or by the presence of strong anion reagents such as arachidonic acid [124]. Small oligomers of Tau-protein (especially dimers) exist as intermediates in the process of filament assembly [108, 125]. Upon polymerization of filaments, they can be fragmented by ultrasound, leading to generation of shorter and numerous structures that can serve as “seeds” causing aggregation of other Tau-protein monomers [126].
Preliminarily aggregated Tau-protein, representing a mixture of filaments and oligomer forms, can penetrate into cultured cells and induce misfolding and aggregation of intracellular Tau-protein [127, 128]. This general principle was demonstrated in an in vivo experiment when intracerebral introduction of mutant P301S Tau-protein, predisposed to aggregation, caused propagation of NFT generation over the murine cerebral cortex, expressing wild-type human Tau-protein which does not form filaments (NFT) spontaneously [129]. With account of the small amount of initially introduced material, it can be stated that wild-type human Tau-protein becomes able to have toxic effect inherent to aggregated mutant Tau-protein, and this facilitates propagation of pathological Tau-protein in brain tissues. The above-mentioned studies of P301L Tau-protein, expressed exclusively in the entorhinal cortex of transgenic mice but simultaneously inducing pathology in hippocampus structures [130-132], provide additional evidence for prion-like behavior of misfolded Tau-protein.
It is probable that Tau oligomers are responsible for propagation of pathology. The use of immunological methods made it possible to find such oligomers in brain tissues of AD patients, first of all in neurons in which NFT had not yet accumulated [125, 129]. Moreover, intracerebral introduction of Tau oligomers, rather than its monomer and fibrillar forms, causes neurotoxicity and also synaptic and mitochondrial dysfunction as well as memory impairment.
Are Tau-proteins formed under the action of Aβ? Several research teams have reported that the toxic effect of Aβ peptide is mediated by Tau-protein, thereby having confirmed that Aβ is involved in AD pathogenesis prior to Tau-protein [70, 72, 74, 109, 110-113, 131, 132]. Pathological phosphorylation of Tau-protein can be caused by activation of protein kinases dependent on Aβ, in particular GSK3 [74, 113, 114]. Data have been obtained demonstrating also direct interaction of Aβ and Tau-protein. In in vitro experiments, Aβ can bind to Tau-protein in the absence of other proteins and peptides [107], inducing oligomerization of its monomers [108]. These results demonstrate explicitly that Aβ oligomers provoke in vivo generation of Tau-protein oligomers, which later are able to propagate independently already in the absence of Aβ. If this process occurs in vivo, it can represent a “trigger mechanism” of AD pathogenesis. In addition, this process can explain why in clinical trials massive efforts on searching for AD treatment by effecting Aβ have been unsuccessful. The reason may evidently be that all patients, who took part in the clinical trials, already have had AD with corresponding clinical manifestations, which have advanced far beyond the stage of generation of Aβ oligomers when Tau-protein pathology may maintain itself independently.
Thus, Aβ can form toxic oligomers and induce oligomerization of Tau-protein, which is capable of propagating by the prion-like mechanism with matrix-mediated misfolding of normal proteins. Aβ oligomers can activate kinases that catalyze pathological phosphorylation of Tau-protein (pTau), and they themselves can be seeds inducing Tau-protein oligomerization. Tau oligomers can propagate by the prion-like mechanism and together with pathologically phosphorylated Tau-protein can provoke degeneration and death of neurons responsible for memory and cognitive functions. Temporary and causal connection between pathological phosphorylation of Tau-protein and its aggregation should still be determined [68].
APPROACHES FOR TREATMENT OF ALZHEIMER’S DISEASE
Studies in molecular genetics, molecular biology, and design of cell and animal models described above make it possible to show confidently the existence of several metabolic and signaling pathways leading to Aβ accumulation, hyperphosphorylation, and aggregation of Tau-protein inside neurons and activation of the immune system as well as generation of inflammatory processes. As molecular mechanisms of functioning of “node points” of these pathways are elucidated, it becomes more probable that systems biology approaches will reveal such pathways, which will open therapeutic perspectives [133-136]. As a matter of fact, in studies performed using simulation (in vitro, cell, and animal models), a variety of such regions of metabolic pathways – targets of possible therapeutic intervention – have been described [133, 137].
Aβ-directed therapy of Alzheimer’s disease. Below is a short description of several therapeutic targets referring both to mechanisms of Aβ replication and to the process of destroying it [138].
One of the most promising studied strategies for prevention and/or treatment of AD consists in decreasing the generation of Aβ either by increasing the activity of α-secretase, by decreasing the activity of BACE, or by inhibiting the activity of γ-secretase. An increase in the activity of α-secretase can be achieved experimentally by activation of 5-hydroxytryptamine-4 (5-HT4) receptors [139] and by enhancing the expression of matrix metalloproteinase 9 [140] or melatonin synthesis [141]. Attempts to inhibit the activity of BACE were unsuccessful initially due to the inaccessible catalytic center in BACE and the necessity to use compounds with high permeability through the blood–brain barrier. Unfortunately, anxiety is induced due to a possible effect of partial inhibitors of BACE on its other substrates such as neuregulin [142] or subunits of potential-regulated sodium channels [143]. However at least one highly effective inhibitor of BACE causing a considerable and prolonged inhibition of Aβ generation in human brain is now passing clinical trials [144, 145]. Its ability to significantly decrease the Aβ level in liquor in patients with moderate and severe AD has been demonstrated in randomized, double blind, placebo-controlled studies of the effectiveness of the new drug – the β-secretase (BACE1) inhibitor MK-8931. The described effect was dose-dependent; when the highest dose was applied, the Aβ level decreased more than by 80% as compared to the initial level (according to the data of the Alzheimer’s Association International Conference (AAIC 2013) in Boston).
Much attention was also paid to development of inhibitors/modulators of γ-secretase. Such compounds were quickly obtained, but quite often they were toxic, this being associated directly with the mechanism of action – the toxicity emerged because of inhibition of other signaling pathways depending on γ-secretase-cleaved receptors differing from APP (including Notch receptor) [146, 147]. As a result, the attention of researchers has been focused on an inhomogeneous class of substances called γ-secretase modulators (GSM) [148-151]. These compounds evidently have their effect due to modification of the dominating site of cleavage with γ-secretase that provokes a decrease in generation of a more amyloidogenic Aβ42 upon increase in generation of Aβ38 (less inclined to amyloidogenesis). In all probability, substances of this group do not affect the ε-cleavage. Molecular mechanisms of their action have not been finally determined, but they may include allosteric interactions of long-range order mediated by the noncatalytic binding site of the complex or the substrate itself. It was demonstrated recently that compounds of this group (GSM) cause conformational changes in the catalytic center by binding to the presenilin loop in the γ-secretase complex [152]. However, studies should be continued that would make it possible to clarify whether the given hypothetical mechanism is universal for representatives of the GSM group. An alternative approach to decreasing the generation of Aβ is such a change in APP processing that would allow breaking the APP transportation to later endosomes for its splitting first by β- and after that γ-secretases. Another alternative therapeutic approach, currently passing preclinical trials on cell models, is the use of small molecular chaperones for enhancing the stability of retromers [153].
Data showing that oligomers of Aβ are the main toxic forms have stimulated attempts to inhibit peptide aggregation with the use of a number of small molecules. One of these compounds, scyllo-inositol, can prevent Aβ oligomerization and lead to alleviation of both cognitive and neuropathological disturbances, which was shown in experiments on an animal model [154]. Nevertheless, the second phase of clinical trials of scyllo-inositol was terminated because of its toxicity [155]. Therefore, the therapeutic value of this approach remains undetermined. In our investigations, it has been established that such activity is inherent to recombinant protein YB1 present in human and animal organisms [156]. Also, several therapeutic targets have been selected in metabolic pathways implementing Aβ removal [157]. It is proposed to enhance the activity of the enzymes that presumably execute Aβ degradation (insulin-degrading enzyme, angiotensin-converting enzyme, and neprilysin), though it is necessary to take into account that these enzymes can affect many processes, in particular, stimulate vasoconstriction and induce side effects [158].
Modulation of Aβ transcytosis by the action of LRP1 or the receptor for advanced glycation end products (RAGE) can also be used for controlling Aβ level. According to published data, RAGE belongs to the so-called motif-recognizing receptors (MRR) that can bind the class of ligands having a common motif [159]. It is believed that in AD the interaction of Aβ and RAGE oligomers results in an increase of the amount of this receptor and stimulates transportation of amyloid peptides from the blood through the blood–brain barrier (BBB) and then into neurons, also due to RAGE. This increases the concentration and accumulation of oligomer Aβ in the brain parenchyma and in nerve cells per se, provoking their degeneration. The mechanism of RAGE-dependent effect on cell biology remains unclear. The accumulation of late products of glycosylation is associated with aging. The number of RAGE increases in cerebral vessels in AD. RAGE is involved in Aβ transcytosis and mediates its flow to brain tissues [160]. Therefore, it is presumed that RAGE inhibitors can be used for restriction of Aβ capture from the blood. In our studies, we found immunization by a short fragment of the extracellular domain of this receptor is effective in olfactory bulbectomized animals with Alzheimer’s type neurodegeneration [161]. A low molecular weight inhibitor of RAGE (TTP488) is now passing phases II and III of clinical trials [157]. The lipoprotein receptor LRP1 is vital for Aβ removal from brain tissues by transcytosis through the BBB. Aβ can bind to LRP1 directly or through APOE [162]. Though the interactions of APOE and Aβ have attracted considerable attention of researchers, a number of research teams demonstrated that APOE could compete with Aβ for LRP rather than play the role of a carrier [163, 164]. APOE expression is regulated by liver receptor X (LXR) in the structure of a heterodimer with other nuclear receptors, including retinoid receptors X (RXR) and peroxisome proliferator-activated receptor γ (PPARγ) [165]. In 2012, great attention was drawn to a report describing effective removal of Aβ deposits in a murine AD model after bexarotene (RXR agonist used for cancer therapy) injections [166]. Unfortunately, a number of subsequent attempts to reproduce the published results failed [167-169]. A possibly promising approach is being developed in our laboratory together with other Russian scientists, based on immunological blockade of Aβ binding sites with such receptors as the α-7 nicotine acetylcholine receptor, prion receptor, or neurotrophin p75 receptor [170-172].
The best-established therapeutic approach to impact Aβ accumulation appears to be the use of active or passive immunization. Initial studies, when transgenic mice expressing human mutant APP were used as a model, demonstrated a considerable capacity of anti-Aβ antibodies to stimulate disintegration of Aβ plaques and improve cognitive functions [173, 174]. Early experiments on active immunization involving AN-172 compounds (Elan Pharmaceuticals, USA) were interrupted subsequently to the development of encephalitis in some patients with AD [175]. In later studies, it was found that the Aβ level decreased in brain tissues of the immunized group of patients as compared to the control untreated group, but no effect on cognitive functions evaluated by simple tests was demonstrated [176, 177]. Further investigations with passive immunization by recombinant anti-Aβ antibodies did not reveal substantial effectiveness (at least under conditions of this experiment), but some of them (for example, Gantenerumab (Roche)) were effective based on the results of phase II tests [178]. Solanezumab, which was not effective overall, provoked though small but statistically significant improvements of cognitive functions in patients from a subgroup of moderate-stage AD [179], and, as a preparation with proved safety, it was chosen for further investigations.
The reason for almost complete absence of effectiveness of anti-Aβ immunotherapy of AD has not yet been determined. Several putative explanations have been suggested [136, 170, 180]. In particular, it is probable that the low capacity of these antibodies to penetrate into the brain and/or their comparatively low affinity (for some of these antibodies) could impede their positive effect. It is also probable that the antibodies could not reach the “target” oligomer neurotoxic Aβ forms. Finally, most likely therapy with the use of antibodies could have begun too late as concerns the developed pathology, and therefore could not have a considerable effect. Numerous data of neurovisualization studies as well as prolonged observations of asymptomatic carriers of mutations in presenilin and APP showed that amyloid pathology begins more than 10 years prior to the manifestation of the first symptoms, and that it is later followed by pathological changes in Tau-protein and development of inflammatory processes [181, 182].
Contemporary notions have stimulated attempts to find new sensitive biomarkers of Aβ accumulation and activation of subsequent biochemical changes that can be used for preclinical diagnostics and determination of the risk group, for which in future it is possible to perform additional studies to develop measures for AD prevention. At present, the most sensitive criterion of the risk of AD development is increased ratios of the Aβ42/Aβ40 in cerebrospinal fluid.
Tau-protein as a therapeutic target. The action on Tau-protein may also be promising. A number of approaches for correction of Tau pathology in AD have been proposed. They are summoned to slow neuronal degeneration caused by NFT generation, including the following: inhibition of one or several protein kinases phosphorylating Tau-protein (glycogen synthase kinase 3-β and cyclin-dependent protein kinase 5); activation of the basic phosphatase enzyme of Tau-protein – the so-called phosphatase-2A; amplification of Tau-protein modification by β-N-acetylglucosamine either using inhibition of β-N-acetylglucosaminidase or stimulation of glucose capture by brain tissues; removal of pathologically hyperphosphorylated Tau-protein by amplification of autophagy or through the ubiquitin/proteasome system [183].
CONCLUSION
The clarification of the mechanisms of AD pathogenesis on the cellular and molecular levels is complicated by simultaneous involvement in it of at least two pathological factors having toxic properties — Aβ and Tau-protein. Their combined involvement in the development of this pathology was known long ago, but the importance of Aβ and Tau-protein in this process and the chronology of their involvement in pathogenesis are still being discussed. Controversy was also expressed relative to the forms of these toxic substances most significant for the pathology, which are known to be characterized by polymorphism. For example, in brain tissues Aβ is represented by polypeptide chains of different lengths, the most numerous of them being Aβ1-40 and Aβ1-42, the most important role in AD pathogenesis being given to Aβ1-42. Separate Aβ molecules can join in different-sized nonfibrillar aggregates and fibrils, characterized by a correct periodical structure, and retain their low-molecular weight form. In its turn, Tau-protein is represented as a microtubule-associated hydrophilic low molecular weight form and a relatively insoluble filamentous form. Of importance also are posttranslational modifications of this protein: its phosphorylation on different epitopes can proceed in the cell as well as glycosylation, glycation, ubiquitination, and racemization.
Also, no less complexity is represented by the establishment of mechanisms of interaction of two toxic proteins that can take place due to direct intermolecular interaction, be mediated by cell receptors, or have a complex multicomponent nature. Evidence for the presence of such interaction can also be nontrivial, because there are assumptions on the possibility of parallel development of Aβ and Tau-protein pathology [66, 67] in different brain regions.
It should be noted that such exclusive complexity of pathogenic processes could be a reason that for more than 100 years no effective methods of AD treatment have been developed, in spite of great efforts to find them.
Therefore, one of the most promising directions of AD studies is investigation of interactions of the two mentioned toxic Aβ and Tau-protein taking into account which of their forms have key value for this interaction and manifestation of toxicity. In this case especially promising is the use of cell models when cocultivation is performed in common culturing medium of nerve cells (first of all, neuronal culture) and producer cells of toxic Aβ and Tau-protein forms obtained by transgenesis with introduction of genes responsible for the synthesis of the toxic agents.
In accord with the regulations of the dominating “amyloid” AD hypothesis, for several decades attempts have been made to develop treatment aimed at Aβ metabolism. As mentioned, these efforts proved ineffective. Relatively recently, researchers have paid their attention also to pathology of Tau-protein in AD, the validity of this being determined, in particular, by the presence of a direct correlation between the amount of fibrillar Tau-protein (PHF and NFT) and the degree of decrease in cognitive functions in AD against the background of the absence of such a correlation for amyloid plaques. This makes the process of their formation another promising target for therapeutic intervention in AD [184]. However, the presence of interaction of Tau-protein and Aβ should be taken into account. We have disclosed potentiation of the toxic effect of Tau-protein in the presence of Aβ on neurons in the primary culture of the hippocampus [85].
A new working hypothesis has been proposed quite recently, according to which the interaction of oligomer Aβ with RAGE leads to a considerable increase in expression of RAGE on the surface of the target cell, which in turn disturbs the capacity to recover from the damages on the cell membrane and make cells particularly vulnerable to the pathological effect of membranotropic oligomer Aβ and/or Tau-protein. In this case, the “infected” cells lose their ability for normal functioning and accumulate a greater amount of oligomers forms of Aβ and Tau-protein, which finally results in their degeneration [185, 186]. This process can lead to selective and directed propagation of the “infection” by these pathological forms of proteins between target cells (neurons or vascular wall cells), which makes the basis for vector propagation of the pathology characteristic of AD.
Thus, it is still a vital task to study the mechanism of interaction of Aβ and Tau-protein that would correlate the volume of available research data and could be consistently and unambiguously demonstrated on different AD models in vitro and in vivo. The clarification of this interconnection with corresponding pathological features (synaptic dysfunction, neurodegeneration, brain atrophy, inflammation) is necessary for developing a therapeutic strategy for prevention or treatment of AD.
This work was financially supported by the Russian Science Foundation within the project No. 14-14-00879 “Study of mechanisms of the YB-1 protein protective effect in neurodegeneration of Alzheimer’s type” and by the Russian Foundation for Basic Research within the project No. 13-04-00633 “Study of the role of the receptor for advanced glycation end products (RAGE) in mechanisms of β-amyloid neurotoxicity in the model of sporadic Alzheimer’s disease”.
REFERENCES
1.Duthey, B. (2013) Background paper.
Alzheimer’s disease and other dementias, in A Public
Health Approach to Innovation, Update on 2004 Background Paper, pp.
1-74.
2.Lee, V., Goedert, M., and Trojanowski, J. (2001)
Neurodegenerative tauopathies, Annu. Rev. Neurosci., 24,
1121-1159.
3.Jayadev, S., Nochlin, D., Poorkaj, P., Steinbart,
E., Mastrianni, J., Montine, T., Ghetti, B., Schellenberg, G., Bird,
T., and Leverenz, J. (2011) Familial prion disease with
Alzheimer’s disease-like tau pathology and clinical phenotype,
Ann. Neurol., 69, 712-720.
4.Hardy, J., and Selkoe, D. (2002) The amyloid
hypothesis of Alzheimer’s disease: progress and problems on the
road to therapeutics, Science, 297, 353-356.
5.Ryazantseva, M. A., Mozhaeva, G. N., and
Kaznacheeva, E. V. (2014) The pathogenesis of Alzheimer’s disease
and potassium homeostasis, in Neurodegenerative Diseases: from
Genome to the Whole Organism (Ugrumov, M. V., ed.) Vol. 2, Nauka,
Moscow, pp. 163-181.
6.Le, M., Kim, W., Lee, S., McKee, A., and Hall, G.
(2012) Multiple mechanisms of extracellular tau spreading in a
non-transgenic tauopathy model, Am. J. Neurodegener. Dis.,
1, 316-333.
7.Avila, J., Lucas, J., Perez, M., and Hernandez, F.
(2004) Role of tau protein in both physiological and pathological
conditions, Physiol. Rev., 84, 361-384.
8.Schliebs, R., and Arendt, T. (2006) The
significance of the cholinergic system in the brain during aging and in
Alzheimer’s disease, J. Neural Transm., 113,
1625-1644.
9.Crow, T., Cross, A., Cooper, S., Deakin, J.,
Ferrier, I., Johnson, J., Joseph, M., Owen, F., Poulter, M., Lofthouse,
R., Corsellis, J., Chambers, D., Blessed, G., Perry, E., Perry, R., and
Tomlinson, B. (1984) Neurotransmitter receptors and monoamine
metabolites in the brains of patients with Alzheimer-type dementia and
depression, and suicides, Neuropharmacology, 23, 1561-1569.
10.Brouwers, N., Sleegers, K., and Van Broeckhoven,
C. (2008) Molecular genetics of Alzheimer’s disease: an update,
Ann. Med., 40, 562-583.
11.Barger, S., DeWall, K., Liu, L., Mrak, R., and
Griffin, W. (2008) Relationships between expression of apolipoprotein E
and beta-amyloid precursor protein are altered in proximity to
Alzheimer’s beta-amyloid plaques: potential explanations from
cell culture studies, J. Neuropathol. Exp. Neurol., 67,
773-783.
12.Holtzman, D., Herz, J., and Bu, G. (2012)
Apolipoprotein E and apolipoprotein E receptors: normal biology and
roles in Alzheimer’s disease, Cold Spring Harb. Perspect.
Med., 2, a006312.
13.Bertram, L., and Tanzi, R. (2008) Thirty years of
Alzheimer’s disease genetics: the implications of systematic
meta-analyses, Nat. Rev. Neurosci., 9, 768-778.
14.Weller, R., Yow, H., Preston, S., Mazanti, I.,
and Nicoll, J. (2002) Cerebrovascular disease is a major factor in the
failure of elimination of amyloid beta from the aging human brain,
Ann. N. Y. Acad. Sci., 977, 162-168.
15.Carson, J., and Turner, A. (2002) Beta-amyloid
catabolism: role for neprilysin (NEP) and other metallopeptidases?
J. Neurochem., 81, 1-8.
16.Nalivaeva, N., Fisk, L., Belyaev, N., and Turner,
A. (2008) Amyloid-degrading enzymes as therapeutic targets in
Alzheimer’s disease, Curr. Alzheimer Res., 5,
212-224.
17.Fisk, L., Nalivaeva, N., Boyle, J., Peers, C.,
and Turner, A. (2007) Effects of hypoxia and oxidative stress on
expression of neprilysin in human neuroblastoma cells and rat cortical
neurons and astrocytes, Neurochem. Res., 32,
1741-1748.
18.Dubrovskaya, N., Nalivaeva, N., Plesneva, S.,
Feponova, A., Turner, A., and Zhuravin, I. (2010) Changes in the
activity of amyloid-degrading metallopeptidases leads to disruption of
memory in rats, Neurosci. Behav. Physiol., 40,
975-980.
19.Bohm, C., Chen, F., Sevalle, J., Qamar, S., Dodd,
R., Li, Y., and St. George-Hyslop, P. H. (2015) Current and future
implications of basic and translational research on amyloid-β
peptide production and removal pathways, Mol. Cell. Neurosci.,
66, 3-11.
20.Ray, B., Long, J., Sokol, D., and Lahiri, D.
(2011) Increased secreted amyloid precursor protein-α
(sAPPα) in severe autism: proposal of a specific, anabolic
pathway and putative biomarker, PLoS One, 6, e20405.
21.Asai, M., Hattori, C., Szabo, B., Sasagawa, N.,
Maruyama, K., Tanuma, S., and Ishiura, S. (2003) Putative function of
ADAM9, ADAM10, and ADAM17 as APP alpha-secretase, Biochem. Biophys.
Res. Commun., 301, 231-235.
22.Fahrenholz, F., Gilbert, S., Kojro, E., Lammich,
S., and Postina, R. (2000) Alpha-secretase activity of the disintegrin
metalloprotease ADAM 10. Influences of domain structure, Ann. N. Y.
Acad. Sci., 920, 215-222.
23.Fuwa, H., Takahashi, Y., Konno, Y., Watanabe, N.,
Miyashita, H., Sasaki, M., Natsugari, H., Kan, T., Fukuyama, T.,
Tomita, T., and Iwatsubo, T. (2007) Divergent synthesis of
multifunctional molecular probes to elucidate the enzyme specificity of
dipeptidic gamma-secretase inhibitors, ACS Chem. Biol.,
2, 408-418.
24.Suh, J., Choi, S., Romano, D., Gannon, M.,
Lesinski, A., Kim, D., and Tanzi, R. (2013) ADAM10 missense mutations
potentiate β-amyloid accumulation by impairing prodomain chaperone
function, Neuron, 80, 385-401.
25.Vassar, R., Kuhn, P., Haass, C., Kennedy, M.,
Rajendran, L., Wong, P., and Lichtenthaler, S. (2014) Function,
therapeutic potential and cell biology of BACE proteases: current
status and future prospects, J. Neurochem., 130,
4-28.
26.Rogaeva, E., Meng, Y., Lee, J., Gu, Y., Kawarai,
T., Zou, F., Katayama, T., Baldwin, C., Cheng, R., Hasegawa, H., Chen,
F., Shibata, N., Lunetta, K., Pardossi-Piquard, R., Bohm, C., Wakutani,
Y., Cupples, A., Cuenco, K., Green, R., Pinessi, L., Rainero, I.,
Sorbi, S., Bruni, A., Duara, R., Friedland, R., Inzelberg, R., Hampe,
W., Bujo, H., Song, Y., Andersen, O., Willnow, T., Graff-Radford, N.,
Petersen, R., Dickson, D., Der, S., Fraser, P., Schmitt-Ulms, G.,
Younkin, S., Mayeux, R., Farrer, L., and St. George-Hyslop, P. (2007)
The neuronal sortilin-related receptor SORL1 is genetically associated
with Alzheimer’s disease, Nat. Genet., 39,
168-177.
27.Bhalla, A., Vetanovetz, C., Morel, E., Chamoun,
Z., Di Paolo, G., and Small, S. (2012) The location and trafficking
routes of the neuronal retromer and its role in amyloid precursor
protein transport, Neurobiol. Dis., 47, 126-134.
28.Seaman, M. (2012) The retromer complex –
endosomal protein recycling and beyond, J. Cell Sci.,
125, 4693-4702.
29.Vardarajan, B., Bruesegem, S., Harbour, M.,
Inzelberg, R., Friedland, R., St. George-Hyslop, P., Seaman, M., and
Farrer, L. (2012) Identification of Alzheimer disease-associated
variants in genes that regulate retromer function, Neurobiol.
Aging, 33, e15-2231.
30.Edbauer, D., Winkler, E., Regula, J., Pesold, B.,
Steiner, H., and Haass, C. (2003) Reconstitution of gamma-secretase
activity, Nat. Cell Biol., 5, 486-488.
31.Sobhanifar, S., Schneider, B., Lohr, F.,
Gottstein, D., Ikeya, T., Mlynarczyk, K., Pulawski, W., Ghoshdastider,
U., Kolinski, M., Filipek, S., Guntert, P., Bernhard, F., and Dotsch,
V. (2010) Structural investigation of the C-terminal catalytic
fragment of presenilin 1, Proc. Natl. Acad. Sci. USA, 5,
9644-9649.
32.De Strooper, B. (2003) Aph-1, Pen-2, and
Nicastrin with Presenilin generate an active gamma-secretase complex,
Neuron, 10, 9-12.
33.Glenner, G., and Wong, C. (1984)
Alzheimer’s disease: initial report of the purification and
characterization of a novel cerebrovascular amyloid protein,
Biochem. Biophys. Res. Commun., 120, 885-890.
34.Bekris, L., Yu. C., Bird, T., and Tsuang, D.
(2010) Genetics of Alzheimer’s disease, J. Geriatr. Psychiatr.
Neurol., 23, 213-227.
35.Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le
Meur, N., Laquerriere, A., Vital, A., Dumanchin, C., Feuillette, S.,
Brice, A., Vercelletto, M., Dubas, F., Frebourg, T., and Campion, D.
(2006) APP locus duplication causes autosomal dominant early-onset
Alzheimer’s disease with cerebral amyloid angiopathy, Nat.
Genet., 38, 24-26.
36.Bornemann, K., and Staufenbiel, M. (2000)
Transgenic mouse models of Alzheimer’s disease, Ann. N. Y.
Acad. Sci., 908, 260-266.
37.Nilsberth, C., Westlind-Danielsson, A., Eckman,
C., Condron, M., Axelman, K., Forsell, C., Stenh, C., Luthman, J.,
Teplow, D., Younkin, S., and Lannfelt, L. (2001) The
“Arctic” APP mutation (E693G) causes Alzheimer’s
disease by enhanced Abeta-protofibril formation, Nat. Neurosci.,
4, 887-893.
38.Cruchaga, C., Karch, C., Jin, S., Benitez, B.,
Cai, Y., Guerreiro, R., Harari, O., Norton, J., Budde, J., Bertelsen,
S., Jeng, A., Cooper, B., Skorupa, T., Carrell, D., Levitch, D., Hsu,
S., Choi, J., Ryten, M., Hardy, J., Ryten, M., Trabzuni, D., Weale, M.,
Ramasamy, A., Smith, C., Sassi, C., Bras, J., Gibbs, J., Hernandez, D.,
Lupton, M., Powell, J., Forabosco, P., Ridge, P., Corcoran, C.,
Tschanz, J., Norton, M., Munger, R., Schmutz, C., Leary, M., Demirci,
F., Bamne, M., Wang, X., Lopez, O., Ganguli, M., Medway, C., Turton,
J., Lord, J., Braae, A., Barber, I., Brown, K., Passmore, P., Craig,
D., Johnston, J., McGuinness, B., Todd, S., Heun, R., Kolsch, H.,
Kehoe, P., Hooper, N., Vardy, E., Mann, D., Pickering-Brown, S., Brown,
K., Kalsheker, N., Lowe, J., Morgan, K., David, S., Wilcock, G.,
Warden, D., Holmes, C., Pastor, P., Lorenzo-Betancor, O., Brkanac, Z.,
Scott, E., Topol, E., Morgan, K., Rogaeva, E., Singleton, A., Hardy,
J., Kamboh, M., St. George-Hyslop, P., Cairns, N., Morris, J., Kauwe,
J., and Goate, A. (2014) Rare coding variants in the phospholipase D3
gene confer risk for Alzheimer’s disease, Nature,
505, 550-554.
39.Guerreiro, R., Wojtas, A., Bras, J.,
Carrasquillo, M., Rogaeva, E., Majounie, E., Cruchaga, C., Sassi, C.,
Kauwe, J., Younkin, S., Hazrati, L., Collinge, J., Pocock, J., Lashley,
T., Williams, J., Lambert, J., Amouyel, P., Goate, A., Rademakers, R.,
Morgan, K., Powell, J., St. George-Hyslop, P., Singleton, A., Hardy,
J., and Alzheimer’s Genetic Analysis Group (2013) TREM2 variants
in Alzheimer’s disease, N. Engl. J. Med., 368,
117-127.
40.Harold, D., Abraham, R., Hollingworth, P., Sims,
R., Gerrish, A., Hamshere, M., Pahwa, J., Moskvina, V., Dowzell, K.,
Williams, A., Jones, N., Thomas, C., Stretton, A., Morgan, A.,
Lovestone, S., Powell, J., Proitsi, P., Lupton, M., Brayne, C.,
Rubinsztein, D., Gill, M., Lawlor, B., Lynch, A., Morgan, K., Brown,
K., Passmore, P., Craig, D., McGuinness, B., Todd, S., Holmes, C.,
Mann, D., Smith, A., Love, S., Kehoe, P., Hardy, J., Mead, S., Fox, N.,
Rossor, M., Collinge, J., Maier, W., Jessen, F., Schurmann, B., Heun,
R., Bussche, H., Heuser, I., Kornhuber, J., Wiltfang, J., Dichgans, M.,
Frolich, L., Hampel, H., Hull, M., Rujescu, D., Goate, A., Kauwe, J.,
Cruchaga, C., Nowotny, P., Morris, J., Mayo, K., Sleegers, K., Bettens,
K., Engelborghs, S., De Deyn, P., Broeckhoven, C., Livingston, G.,
Bass, N., Gurling, H., McQuillin, A., Gwilliam, R., Deloukas, P.,
Al-Chalabi, A., Shaw, C., Tsolaki, M., Singleton, A., Guerreiro, R.,
Muhleisen, T., Nothen, M., Moebus, S., Jockel, K., Klopp, N., Wichmann,
H., Carrasquillo, M., Pankratz, V., Younkin, S., Holmans, P.,
O’Donovan, M., Owen, M., and Williams, J. (2009) Genome-wide
association study identifies variants at CLU and PICALM associated with
Alzheimer’s disease, Nat. Genet., 4, 1088-1093.
41.Hoglinger, G., Melhem, N., Dickson, D., Sleiman,
P., Wang, L., Klei, L., Rademakers, R., de Silva, R., Litvan, I.,
Riley, D., Swieten, J., Heutink, P., Wszolek, Z., Uitti, R.,
Vandrovcova, J., Hurtig, H., Gross, R., Maetzler, W., Goldwurm, S.,
Tolosa, E., Borroni, B., Pastor, P., Cantwell, L., Han, M., Dillman,
A., Brug, M., Gibbs, J., Cookson, M., Hernandez, D., Singleton, A.,
Farrer, M., Yu, C., Golbe, L., Revesz, T., Hardy, J., Lees, A., Devlin,
B., Hakonarson, H., Muller, U., and Schellenberg, G. (2011)
Identification of common variants influencing risk of the tauopathy
progressive supranuclear palsy, Nat. Genet., 43,
699-705.
42.Lambert, J., Ibrahim-Verbaas, C., Harold, D.,
Naj, A., Sims, R., Bellenguez, C., Jun, G., Destefano, A., Bis, J.,
Beecham, G., Grenier-Boley, B., Russo, G., Thorton-Wells, T., Jones,
N., Smith, A., Chouraki, V., Thomas, C., Ikram, M., Zelenika, D.,
Vardarajan, B., Kamatani, Y., Lin, C., Gerrish, A., Schmidt, H.,
Kunkle, B., Dunstan, M., Ruiz, A., Bihoreau, M., Choi, S., Reitz, C.,
Pasquier, F., Cruchaga, C., Craig, D., Amin, N., Berr, C., Lopez, O.,
De Jager, P., Deramecourt, V., Johnston, J., Evans, D., Lovestone, S.,
Letenneur, L., Moron, F., Rubinsztein, D., Eiriksdottir, G., Sleegers,
K., Goate, A., Fievet, N., Huentelman, M., Gill, M., Brown, K., Kamboh,
M., Keller, L., Barberger-Gateau, P., McGuiness, B., Larson, E., Green,
R., Myers, A., Dufouil, C., Todd, S., Wallon, D., Love, S., Rogaeva,
E., Gallacher, J., St. George-Hyslop, P., Clarimon, J., Lleo, A.,
Bayer, A., Tsuang, D., Yu, L., Tsolaki, M., Bossu, P., Spalletta, G.,
Proitsi, P., Collinge, J., Sorbi, S., Sanchez-Garcia, F., Fox, N.,
Hardy, J., Deniz Naranjo M., Bosco, P., Clarke, R., Brayne, C.,
Galimberti, D., Mancuso, M., Matthews, F., European Alzheimer’s
Disease Initiative (EADI), Genetic and Environmental Risk in
Alzheimer’s Disease, Alzheimer’s Disease Genetic
Consortium, and Cohorts for Heart and Aging Research in Genomic
Epidemiology (2013) Meta-analysis of 74,046 individuals identifies 11
new susceptibility loci for Alzheimer’s disease, Nat.
Genet., 45, 1452-1458.
43.Naj, A., Jun, G., Beecham, G., Wang, L.,
Vardarajan, B., Buros, J., Gallins, P., Buxbaum, J., Jarvik, G., Crane,
P., Larson, E., Bird, T., Boeve, B., Graff-Radford, N., De Jager, P.,
Evans, D., Schneider, J., Carrasquillo, M., Ertekin-Taner, N., Younkin,
S., Cruchaga, C., Kauwe, J., Nowotny, P., Kramer, P., Hardy, J.,
Huentelman, M., Myers, A., Barmada, M., Demirci, F., Baldwin, C.,
Green, R., Rogaeva, E., St. George-Hyslop, P., Arnold, S., Barber, R.,
Beach, T., Bigio, E., Bowen, J., Boxer, A., Burke, J., Cairns, N.,
Carlson, C., Carney, R., Carroll, S., Chui, H., Clark, D., Corneveaux,
J., Cotman, C., Cummings, J., DeCarli, C., DeKosky, S., Diaz-Arrastia,
R., Dick, M., Dickson, D., Ellis, W., Faber, K., Fallon, K., Farlow,
M., Ferris, S., Frosch, M., Galasko, D., Ganguli, M., Gearing, M.,
Geschwind, D., Ghetti, B., Gilbert, J., Gilman, S., Giordani, B.,
Glass, J., Growdon, J., Hamilton, R., Harrell, L., Head, E., Honig, L.,
Hulette, C., Hyman, B., Jicha, G., Jin, L., Johnson, N., Karlawish, J.,
Karydas, A., Kaye, J., Kim, R., Koo, E., Kowall, N., Lah, J., Levey,
A., Lieberman, A., Lopez, O., Mack, W., Marson, D., Martiniuk, F.,
Mash, D., Masliah, E., McCormick, W., McCurry, S., McDavid, A., McKee,
A., Mesulam, M., Miller, B., Miller, C., Miller, J., Parisi, J., Perl,
D., Peskind, E., Petersen, R., Poon, W., Quinn, J., Rajbhandary, R.,
Raskind, M., Reisberg, B., Ringman, J., Roberson, E., Rosenberg, R.,
Sano, M., Schneider, L., Seeley, W., Shelanski, M., Slifer, M., Smith,
C., Sonnen, J., Spina, S., Stern, R., Tanzi, R., Trojanowski, J.,
Troncoso, J., Van Deerlin, V., Vinters, H., Vonsattel, J., Weintraub,
S., Welsh-Bohmer, K., Williamson, J., Woltjer, R., Cantwell, L.,
Dombroski, B., Beekly, D., Lunetta, K., Martin, E., Kamboh, M., Saykin,
A., Reiman, E., Bennett, D., Morris, J., Montine, T., Goate, A.,
Blacker, D., Tsuang, D., Hakonarson, H., Kukull, W., Foroud, T.,
Haines, J., Mayeux, R., Pericak-Vance, M., Farrer, L., and
Schellenberg, G. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33
and EPHA1 are associated with late-onset Alzheimer’s disease,
Nat. Genet., 43, 436-441.
44.Vardarajan, B., Zhang, Y., Lee, J., Cheng, R.,
Bohm, C., Ghani, M., Reitz, C., Reyes-Dumeyer, D., Shen, Y., Rogaeva,
E., St. George-Hyslop, P., and Mayeux, R. (2015) Coding mutations in
SORL1 and Alzheimer’s disease, Ann. Neurol., 77,
215-27.
45.Bettens, K., Brouwers, N., Engelborghs, S.,
Lambert, J., Rogaeva. E., Vandenberghe, R., Le Bastard, N., Pasquier,
F., Vermeulen, S., Van Dongen, J., Mattheijssens, M., Peeters, K.,
Mayeux, R., St. George-Hyslop, P., Amouyel, P., De Deyn, P., Sleegers,
K., and Broeckhoven, C. (2012) Both common variations and rare
non-synonymous substitutions and small insertion/deletions in CLU are
associated with increased Alzheimer’s risk, Mol.
Neurodegener., 7, 3.
46.Narayan, P., Orte, A., Clarke, R., Bolognesi, B.,
Hook, S., Ganzinger, K., Meehan, S., Wilson, M., Dobson, C., and
Klenerman, D. (2012) The extracellular chaperone clusterin sequesters
oligomeric forms of the amyloid-β(1-40) peptide, Nat. Struct.
Mol. Biol., 19, 79-83.
47.Hazrati, L., Van Cauwenberghe, C., Brooks, P.,
Brouwers, N., Ghani, M., Sato, C., Cruts, M., Sleegers, K., St.
George-Hyslop, P., Van Broeckhoven, C., and Rogaeva, E. (2012) Genetic
association of CR1 with Alzheimer’s disease: a tentative disease
mechanism, Neurobiol. Aging, 33, 2949.
48.Jun, G., Naj, A. C., Beecham, G. W., Wang, L.,
Buros, J., Gallins, P., Buxbaum, J., Ertekin-Taner, N., Fallin, M.,
Friedland, R., Inzelberg, R., Kramer, P., Rogaeva, E., St.
George-Hyslop, P., and Alzheimer’s Disease Genetics Consortium
(2010) Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer’s
disease risk loci and reveals interactions with APOE genotypes,
Arch. Neurol., 67, 1473-1484.
49.Wyss-Coray, T., Yan, F., Lin, A., Lambris, J.,
Alexander, J., Quigg, R., and Masliah, E. (2002) Prominent
neurodegeneration and increased plaque formation in
complement-inhibited Alzheimer’s mice, Proc. Natl. Acad. Sci.
USA, 99, 10837-10842.
50.Biffi, A., Shulman, J., Jagiella, J., Cortellini,
L., Ayres, A., Schwab, K., Brown, D., Silliman, S., Selim, M., Worrall,
B., Meschia, J., Slowik, A., De Jager, P., Greenberg, S., Schneider,
J., Bennett, D., and Rosand, J. (2012) Genetic variation at CR1
increases risk of cerebral amyloid angiopathy, Neurology,
78, 334-341.
51.Neher, M., Rich, M., Keene, C., Weckbach, S.,
Bolden, A., Losacco, J., Patane, J., Flierl, M., Kulik, L., Holers, V.,
and Stahel, P. (2014) Deficiency of complement receptors CR2/CR1 in
Cr2−/− mice reduces the extent of secondary
brain damage after closed head injury, J. Neuroinflamm.,
11, 95.
52.Thambisetty, M., An, Y., Nalls, M., Sojkova, J.,
Swaminathan, S., Zhou, Y., Singleton, A., Wong, D., Ferrucci, L.,
Saykin, A., Resnick, S., Baltimore Longitudinal Study of Aging, and the
Alzheimer’s Disease Neuroimaging Initiative (2013) The effect of
CR1 on brain amyloid burden during aging and its modification by APOE
genotype, Biol. Psychiatry, 73, 422-428.
53.Hollingworth, P., Harold, D., Sims, R., Gerrish,
A., Lambert, J., Carrasquillo, M., Abraham, R., Hamshere, M., Pahwa,
J., Moskvina, V., Dowzell, K., Jones, N., Stretton, A., Thomas, C.,
Richards, A., Ivanov, D., Widdowson, C., Chapman, J., Lovestone, S.,
Powell, J., Proitsi, P., Lupton, M., Brayne, C., Rubinsztein, D., Gill,
M., Lawlor, B., Lynch, A., Brown, K., Passmore, P., Craig, D.,
McGuinness, B., Todd, S., Holmes, C., Mann, D., Smith, A., Beaumont,
H., Warden, D., Wilcock, G., Love, S., Kehoe, P., Hooper, N., Vardy,
E., Hardy, J., Mead, S., Fox, N., Rossor, M., Collinge, J., Maier, W.,
Jessen, F., Ruther, E., Schurmann, B., Heun, R., Kolsch, H., Bussche,
H., Heuser, I., Kornhuber, J., Wiltfang, J., Dichgans, M., Frolich, L.,
Hampel, H., Gallacher, J., Hull, M., Rujescu, D., Giegling, I., Goate,
A., Kauwe, J., Cruchaga, C., Nowotny, P., Morris, J., Mayo, K.,
Sleegers, K., Bettens, K., Engelborghs, S., De Deyn, P., Broeckhoven,
C., Livingston, G., Bass, N., Gurling, H., McQuillin, A., Gwilliam, R.,
Deloukas, P., Al-Chalabi, A., Shaw, C., Tsolaki, M., Singleton, A.,
Guerreiro, R., Muhleisen, T., Nothen, M., Moebus, S., Jockel, K.,
Klopp, N., Wichmann, H., Pankratz, V., Sando, S., Aasly, J.,
Barcikowska, M., Wszolek, Z., Dickson, D., Graff-Radford, N., Petersen,
R., Alzheimer’s Disease Neuroimaging Initiative, and CHARGE
consortium (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33
and CD2AP are associated with Alzheimer’s disease, Nat.
Genet., 43, 429-435.
54.Jonsson, T., Stefansson, H., Steinberg, S.,
Jonsdottir, I., Jonsson, P., Snaedal, J., Bjornsson, S., Huttenlocher,
J., Levey, A., Lah, J., Rujescu, D., Hampel, H., Giegling, I.,
Andreassen, O., Engedal, K., Ulstein, I., Djurovic, S.,
Ibrahim-Verbaas, C., Hofman, A., Ikram, M., Duijn, C.,
Thorsteinsdottir, U., Kong, A., and Stefansson, K. (2013) Variant of
TREM2 associated with the risk of Alzheimer’s disease, N.
Engl. J. Med., 368, 107-116.
55.Benitez, B., Jin, S., Guerreiro, R., Graham, R.,
Lord, J., Harold, D., Sims, R., Lambert, J., Gibbs, J., Bras, J.,
Sassi, C., Harari, O., Bertelsen, S., Lupton, M., Powell, J.,
Bellenguez, C., Brown, K., Medway, C., Haddick, P., Brug, M., Bhangale,
T., Ortmann, W., Behrens, T., Mayeux, R., Pericak-Vance, M., Farrer,
L., Schellenberg, G., Haines, J., Turton, J., Braae, A., Barber, I.,
Fagan, A., Holtzman, D., Morris, J., 3C Study Group, EADI consortium,
Alzheimer’s Disease Genetic Consortium (ADGC), Alzheimer’s
Disease Neuroimaging Initiative (ADNI), and GERAD consortium (2014)
Missense variant in TREML2 protects against Alzheimer’s disease,
Neurobiol. Aging, 35, 19-26.
56.Griciuc, A., Serrano-Pozo, A., Parrado, A.,
Lesinski, A., Asselin, C., Mullin, K., Hooli, B., Choi, S., Hyman, B.,
and Tanzi, R. (2013) Alzheimer’s disease risk gene CD33 inhibits
microglial uptake of amyloid beta, Neuron, 78,
631-643.
57.Kleinberger, G., Yamanishi, Y., Suarez-Calvet,
M., Czirr, E., Lohmann, E., Cuyvers, E., Struyfs, H., Pettkus, N.,
Wenninger-Weinzierl, A., Mazaheri, F., Tahirovic, S., Lleo, A.,
Alcolea, D., Fortea, J., Willem, M., Lammich, S., Molinuevo J.,
Sanchez-Valle, R., Antonell, A., Ramirez, A., Heneka, M., Sleegers, K.,
Zee, J., Martin, J., Engelborghs, S., Demirtas-Tatlidede, A.,
Zetterberg, H., Broeckhoven, C., Gurvit, H., Wyss-Coray, T., Hardy, J.,
Colonna, M., and Haass, C. (2014) TREM2 mutations implicated in
neurodegeneration impair cell surface transport and phagocytosis,
Sci. Transl. Med., 6, 243.
58.Goedert, M. (2005) Tau gene mutations and their
effects, Mov. Disord., 20, 45-52.
59.Nelson, P., Braak, H., and Markesbery, W. (2009)
Neuropathology and cognitive impairment in Alzheimer’s disease: a
complex but coherent relationship, J. Neuropathol. Exp. Neurol.,
68, 1-14.
60.Serrano-Pozo, A., Frosch, M., Masliah, E., and
Hyman, B. (2011) Neuropathological alterations in Alzheimer’s
disease, Cold Spring Harb. Perspect. Med., 1,
a006189.
61.Jack, C., and Holtzman, D. (2013) Biomarker
modeling of Alzheimer’s disease, Neuron, 80,
1347-1358.
62.Lleo, A., Cavedo, E., Parnetti, L.,
Vanderstichele, H., Herukka, S., Andreasen, N., Ghidoni, R., Lewczuk,
P., Jeromin, A., Winblad, B., Tsolaki, M., Mroczko, B., Visser, P.,
Santana, I., Svenningsson, P., Blennow, K., Aarsland, D., Molinuevo,
J., Zetterberg, H., and Mollenhauer, B. (2015) Cerebrospinal fluid
biomarkers in trials for Alzheimer’s and Parkinson’s
diseases, Nat. Rev. Neurol., 11, 41-55.
63.Risacher, S., and Saykin, A. (2013) Neuroimaging
biomarkers of neurodegenerative diseases and dementia, Semin.
Neurol., 33, 386-416.
64.Thal, D., Attems, J., and Ewers, M. (2014)
Spreading of amyloid, tau, and microvascular pathology in
Alzheimer’s disease: findings from neuropathological and
neuroimaging studies, J. Alzheimer’s Dis., 42
(Suppl. 4), 421-429.
65.Wicklund, M., and Petersen, R. (2013) Emerging
biomarkers in cognition, Clin. Geriatr. Med., 29,
809-828.
66.Crary, J., Trojanowski, J., Schneider, J.,
Abisambra, J., Abner, E., Alafuzoff, I., Arnold, S., Attems, J., Beach,
T., Bigio, E., Cairns, N., Dickson, D., Gearing, M., Grinberg, L., Hof,
P., Hyman, B., Jellinger, K., Jicha, G., Kovacs, G., Knopman, D.,
Kofler, J., Kukull, W., Mackenzie, I., Masliah, E., McKee, A., Montine,
T., Murray, M., Neltner, J., Santa-Maria, I., Seeley, W., Serrano-Pozo,
A., Shelanski, M., Stein, T., Takao, M., Thal, D., Toledo, J.,
Troncoso, J., Vonsattel, J., White, C., 3rd, Wisniewski, T., Woltjer,
R., Yamada, M., and Nelson, P. (2014) Primary age-related tauopathy
(PART): a common pathology associated with human aging, Acta
Neuropathol., 128, 755-766.
67.Jack, C., Wiste, H., Knopman, D., Vemuri, P.,
Mielke, M., Weigand, S., Senjem, M., Gunter, J., Lowe, V., Gregg, B.,
Pankratz, V., and Petersen, R. (2014) Rates of beta-amyloid
accumulation are independent of hippocampal neurodegeneration,
Neurology, 82, 1605-1612.
68.Nussbaum, J., Seward, M., and Bloom, G. (2013)
Alzheimer’s disease: a tale of two prions, Prion,
7, 14-19.
69.Selkoe, D. (2001) Alzheimer’s disease:
genes, proteins, and therapy, Physiol. Rev., 81,
741-766.
70.King, M., Kan, H., Baas, P., Erisir, A., Glabe,
C., and Bloom, G. (2006) Tau-dependent microtubule disassembly
initiated by prefibrillar β-amyloid, J. Cell Biol.,
175, 541-546.
71.Nath, S., Agholme, L., Kurudenkandy, F.,
Granseth, B., Marcusson, J., and Hallbeck, M. (2012) Spreading of
neuro-degenerative pathology via neuron-to-neuron transmission of
β-amyloid, J. Neurosci., 32, 8767-8777.
72.Nussbaum, J., Schilling, S., Cynis, H., Silva,
A., Swanson, E., Wangsanut, T., Tayler, K., Wiltgen, B., Hatami, A.,
Ronicke, R., Reymann, K., Hutter-Paier, B., Alexandru, A., Jagla, W.,
Graubner, S., Glabe, C., Demuth, H., and Bloom, G. (2012) Prion-like
behavior and tau-dependent cytotoxicity of pyroglutamylated
amyloid-β, Nature, 485, 651-655.
73.Picone, P., Carrotta, R., Montana, G., Nobile,
M., San Biagio, P., and Di Carlo, M. (2009) Abeta oligomers and
fibrillar aggregates induce different apoptotic pathways in LAN5
neuroblastoma cell cultures, Biophys. J., 96,
4200-4211.
74.Seward, M., Swanson, E., Roberson, E., and Bloom,
G. (2012) Amyloid-β signals through tau to drive neuronal cell
cycle re-entry in Alzheimer’s disease, J. Cell Sci.,
126, 1278-1286.
75.Westerman, M., Cooper-Blacketer, D., Mariash, A.,
Kotilinek, L., Kawarabayashi, T., Younkin, L., and Ashe, K. (2002) The
relationship between Abeta and memory in the Tg2576 mouse model of
Alzheimer’s disease, J. Neurosci., 22,
1858-1867.
76.Weingarten, M., Lockwood, A., Hwo, S.-Y., and
Kirschner, M. (1975) A protein factor essential for microtubule
assembly, Proc. Natl. Acad. Sci. USA, 72, 1858-1862.
77.Grundke-Iqbal, I., Iqbal, K., Tung, Y., Quinlan,
M., Wisniewski, H., and Binder, L. (1986) Abnormal phosphorylation of
the microtubule-associated protein tau (tau) in Alzheimer’s
cytoskeletal pathology, Proc. Natl. Acad. Sci. USA, 83,
4913-4917.
78.Kondo, J., Honda, T., Mori, H., Hamada, Y.,
Miura, R., Ogawara, M., and Ihara, Y. (1988) The carboxyl third of tau
is tightly bound to paired helical filaments, Neuron, 1,
827-834.
79.Kosik, K., Orecchio, L., Binder, L., Trojanowski,
J., Lee, V., and Lee, G. (1988) Epitopes that span the tau molecule are
shared with paired helical filaments, Neuron, 1,
817-825.
80.Stamer, K., Vogel, R., Thies, E., Mandelkow, E.,
and Mandelkow, E. (2002) Tau blocks traffic of organelles,
neurofilaments, and APP vesicles in neurons and enhances oxidative
stress, J. Cell Biol., 156, 1051-1063.
81.Kolarova, M., Garcia-Sierra, F., Bartos, A.,
Ricny, J., and Ripova, D. (2012) Structure and pathology of tau protein
in Alzheimer disease, Int. J. Alzheimer’s Dis.,
2012, 731526.
82.Hanger, D., Anderton, B., and Noble, W. (2009)
Tau phosphorylation: the therapeutic challenge for neurodegenerative
disease, Trends Mol. Med., 15, 112-119.
83.Porzig, R., Singer, D., and Hoffmann, R. (2007)
Epitope mapping of mAbs AT8 and Tau5 directed against
hyperphosphorylated regions of the human tau protein, Biochem.
Biophys. Res. Commun., 358, 644-649.
84.Klenyaeva, A., Chuprov-Netochin, R., Marusich,
E., Tatarnikova, O., Orlov, M., and Bobkova, N. (2014) Development of
mouse fibroblast cell line expressing human tau protein and evaluation
of tau-dependent cytotoxity, Biochemistry (Moscow), Ser. A: Membr.
Cell Biol., 8, 232-239.
85.Tatarnikova, O., Klenyaeva, A., Orlov, M.,
Panchenko, M., Sergeev, A., and Bobkova, N. (2014) Tau-mediated
toxicity of beta-amyloid, Neurocomp. Dev. Appl., 4,
55-56.
86.Beekes, M., Thomzig, A., Schulz-Schaeffer, W. J.,
and Burger, R. (2014) Is there a risk of prion-like disease
transmission by Alzheimer- or Parkinson-associated protein particles?
Acta Neuropathol., 128, 463-476.
87.Brundin, P., Melki, R., and Kopito, R. (2010)
Prion-like transmission of protein aggregates in neurodegenerative
diseases, Nat. Rev. Mol. Cell Biol., 11, 301-307.
88.Prusiner, S. (1982) Novel proteinaceous
infectious particles cause scrapie, Science, 216,
136-144.
89.Prusiner, S. (1984) Some speculations about
prions, amyloid, and Alzheimer’s disease, N. Engl. J.
Med., 310, 661-663.
90.Frost, B., and Diamond, M. (2010) Prion-like
mechanisms in neurodegenerative diseases, Nat. Rev. Neurosci.,
11, 155-159.
91.Goedert, M., Clavaguera, F., and Tolnay, M.
(2010) The propagation of prion-like protein inclusions in
neurodegenerative diseases, Trends Neurosci., 33,
317-325.
92.Lee, S., Desplats, P., Sigurdson, C., Tsigelny,
I., and Masliah, E. (2010) Cell-to-cell transmission of nonprion
protein aggregates, Nat. Rev. Neurol., 6, 702-706.
93.Novak, P., Prcina, M., and Kontsekova, E. (2011)
Tauons and prions: infamous cousins? J. Alzheimer’s Dis.,
26, 413-430.
94.Prusiner, S. (2012) Cell biology. A unifying role
for prions in neurodegenerative diseases, Science, 336,
1511-1513.
95.Harris, J., Devidze, N., Verret, L., Ho, K.,
Halabisky, B., Thwin, M., Kim, D., Hamto, P., Lo, I., Yu, G., Palop,
J., Masliah, E., and Mucke, L. (2010) Transsynaptic progression of
amyloid-β-induced neuronal dysfunction within the
entorhinal-hippocampal network, Neuron, 68, 428-441.
96.Braak, H., and Del Tredici, K. (2011)
Alzheimer’s pathogenesis: is there neuron-to-neuron propagation?
Acta Neuropathol., 121, 589-595.
97.Clavaguera, F., Bolmont, T., Crowther, R.,
Abramowski, D., Frank, S., Probst, A., Fraser, G., Stalder, A., Beibel,
M., Staufenbiel, M., Jucker, M., Goedert, M., and Tolnay, M. (2009)
Transmission and spreading of tauopathy in transgenic mouse brain,
Nat. Cell Biol., 11, 909-913.
98.De Calignon, A., Polydoro, M., Suarez-Calvet, M.,
William, C., Adamowicz, D., Kopeikina, K., Pitstick, R., Sahara, N.,
Ashe, K., Carlson, G., Spires-Jones, T., and Hyman, B. (2012)
Propagation of tau pathology in a model of early Alzheimer’s
disease, Neuron, 73, 685-697.
99.Guo, J., and Lee, V. (2011) Seeding of normal Tau
by pathological Tau conformers drives pathogenesis of Alzheimer-like
tangles, J. Biol. Chem., 286, 15317-15331.
100.Liu, L., Drouet, V., Wu, J., Witter, M., Small,
S., Clelland, C., and Duff, K. (2012) Trans-synaptic spread of tau
pathology in vivo, PLoS One, 7, e31302.
101.Nussbaum, J., Schilling, S., Cynis, H., Silva,
A., Swanson, E., Wangsanut, T., Tayler, K., Wiltgen, B., Hatami, A.,
Ronicke, R., Reymann, K., Hutter-Paier, B., Alexandru, A., Jagla, W.,
Graubner, S., Glabe, C., Demuth, H., and Bloom, G. (2012) Prion-like
behavior and tau-dependent cytotoxicity of pyroglutamylated
amyloid-β, Nature, 485, 651-655.
102.Hurtado, D., Molina-Porcel, L., Iba, M.,
Aboagye, A., Paul, S., Trojanowski, J., and Lee, V. (2010) Abeta
accelerates the spatiotemporal progression of tau pathology and
augments tau amyloidosis in an Alzheimer’s mouse model, Am. J.
Pathol., 177, 1977-1988.
103.Miller, Y., Ma, B., and Nussinov, R. (2011)
Synergistic interactions between repeats in tau protein and Aβ
amyloids may be responsible for accelerated aggregation via polymorphic
states, Biochemistry, 50, 5172-5181.
104.Pauwels, K., Williams, T., Morris, K.,
Jonckheere, W., Vandersteen, A., Kelly, G., Schymkowitz, J., Rousseau,
F., Pastore, A., Serpell, L., and Broersen, K. (2012) Structural basis
for increased toxicity of pathological aβ42/aβ40 ratios in
Alzheimer’s disease, J. Biol. Chem., 287,
5650-5660.
105.Stohr, J., Watts, J., Mensinger, Z., Oehler,
A., Grillo, S., DeArmond, S., Prusiner S., and Giles, K. (2012)
Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions,
Proc. Natl. Acad. Sci. USA, 109, 11025-11030.
106.Jucker, M., and Walker, L. (2011) Pathogenic
protein seeding in Alzheimer’s disease and other
neurodegenerative disorders, Ann. Neurol., 70,
532-740.
107.Guo, J., Arai, T., Miklossy, J., and McGeer, P.
(2006) Abeta and tau form soluble complexes that may promote
self-aggregation of both into the insoluble forms observed in
Alzheimer’s disease, Proc. Natl. Acad. Sci. USA,
103, 1953-1958.
108.Lasagna-Reeves, C., Castillo-Carranza, D.,
Guerrero-Muoz, M., Jackson, G., and Kayed, R. (2010) Preparation and
characterization of neurotoxic tau oligomers, Biochemistry,
49, 10039-10041.
109.Gotz, J., Chen, F., Van Dorpe, J., and Nitsch,
R. (2001) Formation of neurofibrillary tangles in P301l tau transgenic
mice induced by Abeta 42 fibrils, Science, 293,
1491-1495.
110.Lewis, J., Dickson, D., Lin, W.-L., Chisholm,
L., Corral, A., Jones, G., Yen, S., Sahara, N., Skipper, L., Yager, D.,
Eckman, C., Hardy, J., Hutton, M., and McGowan, E. (2001) Enhanced
neurofibrillary degeneration in transgenic mice expressing mutant tau
and APP, Science, 293, 1487-1491.
111.Rapoport, M., Dawson, H., Binder, L., Vitek,
M., and Ferreira, A. (2002) Tau is essential to β-amyloid-induced
neurotoxicity, Proc. Natl. Acad. Sci. USA, 99,
6364-6369.
112.Roberson, E., Scearce-Levie, K., Palop, J.,
Yan, F., Cheng, I., Wu, T., Gerstein, H., Yu, G., and Mucke, L. (2007)
Reducing endogenous tau ameliorates amyloid beta-induced deficits in an
Alzheimer’s disease mouse model, Science, 316,
750-754.
113.Seino, Y., Kawarabayashi, T., Wakasaya, Y.,
Watanabe, M., Takamura, A., Yamamoto-Watanabe, Y., Kurata, T., Abe, K.,
Ikeda, M., Westaway, D., Murakami, T., Hyslop, P., Matsubara, E., and
Shoji, M. (2010) Amyloid β accelerates phosphorylation of tau and
neurofibrillary tangle formation in an amyloid precursor protein and
tau double-transgenic mouse model, J. Neurosci. Res., 88,
3547-3554.
114.Zempel, H., Thies, E., Mandelkow, E., and
Mandelkow, E. (2010) Abeta oligomers cause localized Ca(2+) elevation,
missorting of endogenous Tau into dendrites, Tau phosphorylation, and
destruction of microtubules and spines, J. Neurosci., 30,
11938-11950.
115.Eisele, Y., Obermuller, U., Heilbronner, G.,
Baumann, F., Kaeser, S., Wolburg, H., Walker, L., Staufenbiel, M.,
Heikenwalder, M., and Jucker, M. (2010) Peripherally applied
Abeta-containing inoculates induce cerebral beta-amyloidosis,
Science, 330, 980-982.
116.Meyer-Luehmann, M., Coomaraswamy, J., Bolmont,
T., Kaeser, S., Schaefer, C., Kilger, E., Neuenschwander, A.,
Abramowski, D., Frey, P., Jaton, A., Vigouret, J., Paganetti, P.,
Walsh, D., Mathews, P., Ghiso, J., Staufenbiel, M., Walker, L., and
Jucker, M. (2006) Exogenous induction of cerebral beta-amyloidogenesis
is governed by agent and host, Science, 313,
1781-1784.
117.Walker, L., Callahan, M., Bian, F., Durham, R.,
Roher, A., and Lipinski, W. (2002) Exogenous induction of cerebral
beta-amyloidosis in betaAPP-transgenic mice, Peptides,
23, 1241-1247.
118.Kayed, R., Canto, I., Breydo, L., Rasool, S.,
Lukacsovich, T., Wu, J., Albay, R., 3rd, Pensalfini, A., Yeung, S.,
Head, E., Marsh, J., and Glabe, C. (2010) Conformation dependent
monoclonal antibodies distinguish different replicating strains or
conformers of prefibrillar Aβ oligomers, Mol.
Neurodegener., 5, 57.
119.Davis, R., Marsden, I., Maloney, M., Minamide,
L., Podlisny, M., Selkoe, D., and Bamburg, J. (2011) Amyloid beta
dimers/trimers potently induce cofilin–actin rods that are
inhibited by maintaining cofilin-phosphorylation, Mol.
Neurodegener., 6, 10.
120.Shankar, G., Bloodgood, B., Townsend, M.,
Walsh, D., Selkoe, D., and Sabatini, B. (2007) Natural oligomers of the
Alzheimer amyloid-beta protein induce reversible synapse loss by
modulating an NMDA-type glutamate receptor-dependent signaling pathway,
J. Neurosci., 27, 2866-2875.
121.Shankar, G., Li, S., Mehta, T., Garcia-Munoz,
A., Shepardson, N., Smith, I., Brett, F., Farrell, M., Rowan, M.,
Lemere, C., Regan, C., Walsh, D., Sabatini, B., and Selkoe, D. (2008)
Amyloid-beta protein dimers isolated directly from Alzheimer’s
brains impair synaptic plasticity and memory, Nat. Med.,
14, 837-842.
122.Portelius, E., Westman-Brinkmalm, A.,
Zetterberg, H., and Blennow, K. (2006) Determination of beta-amyloid
peptide signatures in cerebrospinal fluid using
immunoprecipitation-mass spectrometry, J. Proteome. Res.,
5, 1010-1016.
123.Jeganathan, S., Von Bergen, M., Mandelkow,
E.-M., and Mandelkow, E. (2008) The natively unfolded character of tau
and its aggregation to Alzheimer-like paired helical filaments,
Biochemistry, 47, 10526-10539.
124.Gamblin, T., King, M., Dawson, H., Vitek, M.,
Kuret, J., Berry, R., and Binder, L. (2000) In vitro
polymerization of tau protein monitored by laser light scattering:
method and application to the study of FTDP-17 mutants,
Biochemistry, 39, 6136-6144.
125.Patterson, K., Remmers, C., Fu, Y., Brooker,
S., Kanaan, N., Vana, L., Ward, S., Reyes, J., Philibert, K.,
Glucksman, M., and Binder, L. (2011) Characterization of prefibrillar
Tau oligomers in vitro and in Alzheimer’s disease, J.
Biol. Chem., 286, 23063-23076.
126.Friedhoff, P., Von Bergen, M., Mandelkow, E.,
Davies, P., and Mandelkow, E. (1998) A nucleated assembly mechanism of
Alzheimer paired helical filaments, Proc. Natl. Acad. Sci. USA,
95, 15712-15717.
127.Frost, B., Jacks, R., and Diamond, M. (2009)
Propagation of tau misfolding from the outside to the inside of a cell,
J. Biol. Chem., 284, 12845-12852.
128.Nonaka, T., Watanabe, S., Iwatsubo, T., and
Hasegawa, M. (2010) Seeded aggregation and toxicity of alpha-synuclein
and tau: cellular models of neurodegenerative diseases, J. Biol.
Chem., 285, 34885-34898.
129.Lasagna-Reeves, C., Castillo-Carranza, D.,
Sengupta, U., Sarmiento, J., Troncoso, J., Jackson, G., and Kayed, R.
(2012) Identification of oligomers at early stages of tau aggregation
in Alzheimer’s disease, FASEB J., 26,
1946-1959.
130.Lasagna-Reeves, C., Castillo-Carranza, D.,
Sengupta, U., Clos, A., Jackson, G., and Kayed, R. (2011) Tau oligomers
impair memory and induce synaptic and mitochondrial dysfunction in
wild-type mice, Mol. Neurodegener., 6, 39.
131.Ittner, L., Ke, Y., Delerue, F., Bi, M.,
Gladbach, A., Van Eersel, J., Wolfing, H., Chieng, B. C., Christie, M.
J., Napier, I. A., Eckert, A., Staufenbiel, M., Hardeman, E., and Gotz,
J. (2010) Dendritic function of tau mediates amyloid-beta toxicity in
Alzheimer’s disease mouse models, Cell, 142,
387-397.
132.Vossel, K., Zhang, K., Brodbeck, J., Daub, A.,
Sharma, P., Finkbeiner, S., Cui, B., and Mucke, L. (2010) Tau reduction
prevents Abeta-induced defects in axonal transport, Science,
330, 198.
133.Rhinn, H., Fujita, R., Qiang, L., Cheng, R.,
Lee, J., and Abeliovich, A. (2013) Integrative genomics identifies APOE
epsilon 4 effectors in Alzheimer’s disease, Nature,
500, 45-50.
134.Santiago, J., and Potashkin, J. (2014) A
network approach to clinical intervention in neurodegenerative
diseases, Trends Mol. Med., 20, 694-703.
135.Zhang, B., Gaiteri, C., Bodea, L., Wang, Z.,
McElwee, J., Podtelezhnikov, A., Zhang, C., Xie, T., Tran, L., Dobrin,
R., Fluder, E., Clurman, B., Melquist, S., Narayanan, M., Suver, C.,
Shah, H., Mahajan, M., Gillis, T., Mysore, J., MacDonald, M. E., Lamb,
J. R., Bennett, D. A., Molony, C., Stone, D. J., Gudnason, V., Myers,
A. J., Schadt, E. E., Neumann, H., Zhu, J., and Emilsson, V. (2013)
Integrated systems approach identifies genetic nodes and networks in
late-onset Alzheimer’s disease, Cell, 153,
707-720.
136.Hampel, H., Schneider, L., Giacobini, E.,
Kivipelto, M., Sindi, S., Dubois, B., Broich, K., Nistico, R., Aisen,
P., and Lista, S. (2014) Advances in the therapy of Alzheimer’s
disease: targeting amyloid beta and tau and perspectives for the
future, Exp. Rev. Neurother., 15, 83-105.
137.Huang, Y., and Mucke, L. (2012) Alzheimer
mechanisms and therapeutic strategies, Cell, 148,
1204-1222.
138.Narayan, P., Ehsani, S., and Lindquist, S.
(2014) Combating neurodegenerative disease with chemical probes and
model systems, Nat. Chem. Biol., 10, 911-920.
139.Pimenova, A., Thathiah, A., De Strooper, B.,
and Tesseur, I. (2014) Regulation of amyloid precursor protein
processing by serotonin signaling, PLoS One, 9,
e87014.
140.Fragkouli, A., Tsilibary, E., and Tzinia, A.
(2014) Neuroprotective role of MMP-9 overexpression in the brain of
Alzheimer’s 5xFAD mice, Neurobiol. Dis., 70,
179-189.
141.Shukla, M., Htoo, H., Wintachai, P., Hernandez,
J., Dubois, C., Postina, R., Xu, H., Checler, F., Smith, D.,
Govitrapong, P., and Vincent, B. (2015) Melatonin stimulates the
nonamyloidogenic processing of betaAPP through the positive
transcriptional regulation of ADAM10 and ADAM17, J. Pineal Res.,
58, 151-165.
142.Willem, M., Garratt, A., Novak, B., Citron, M.,
Kaufmann, S., Rittger, A., DeStrooper, B., Saftig, P., Birchmeier, C.,
and Haass, C. (2006) Control of peripheral nerve myelination by the
β-secretase BACE1, Science, 314, 664-666.
143.Kim, D., Carey, B., Wang, H., Ingano, L.,
Binshtok, A., Wertz, M., Pettingell, W., He, Lee, V., Woolf, C., and
Kovacs, D. (2007) BACE1 regulates voltage-gated sodium channels and
neuronal activity, Nat. Cell Biol., 9, 755-764.
144.Butini, S., Brogi, S., Novellino, E., Campiani,
G., Ghosh, A., Brindisi, M., and Gemma, S. (2013) The structural
evolution of beta-secretase inhibitors: a focus on the development of
small-molecule inhibitors, Curr. Top. Med. Chem., 13,
1787-1807.
145.Vassar, R., Kuhn, P.-H., Haass, C., Kennedy,
M., Rajendran, L., Wong, P., and Lichtenthaler, S. (2014) Function,
therapeutic potential and cell biology of BACE proteases: current
status and future prospects, J. Neurochem., 130,
4-28.
146.Cummings, J. (2010) What can be inferred from
the interruption of the semagacestat trial for treatment of
Alzheimer’s disease? Biol. Psychiatry, 68,
876-878.
147.Doody, R., Raman, R., Farlow, M., Iwatsubo, T.,
Vellas, B., Joffe, S., Kieburtz, K., He, F., Sun, X., Thomas, R.,
Aisen, P., Siemers, E., Sethuraman, G., Mohs, R., and Semagacestat
Study Group (2013) A phase 3 trial of semagacestat for treatment of
Alzheimer’s disease, N. Engl. J. Med., 369,
341-350.
148.Golde, T., Koo, E., Felsenstein, K., Osborne,
B., and Miele, L. (2013) γ-Secretase inhibitors and modulators,
Biochim. Biophys. Acta, 1828, 2898-2907.
149.Hall, A., and Patel, T. R. (2014)
Gamma-secretase modulators: current status and future directions,
Prog. Med. Chem., 53, 101-145.
150.Pettersson, M., Stepan, A., Kauffman, G., and
Johnson, D. (2013) Novel gamma-secretase modulators for the treatment
of Alzheimer’s disease: a review focusing on patents from 2010 to
2012, Expert Opin. Ther. Pat., 23, 1349-1366.
151.Crump, C., Johnson, D., and Li, Y.-M. (2013)
Development and mechanism of γ-secretase modulators for
Alzheimer’s disease, Biochemistry, 52,
3197-3216.
152.Takeo, K., Tanimura, S., Shinoda, T., Osawa,
S., Zahariev, I., Takegami, N., Ishizuka-Katsura, Y., Shinya, N.,
Takagi-Niidome, S., Tominaga, A., Ohsawa, N., Kimura-Someya, T.,
Shirouzu, M., Yokoshima, S., Yokoyama, S., Fukuyama, T., Tomita, T.,
and Iwatsubo, T. (2014) Allosteric regulation of γ-secretase
activity by a phenylimidazole-type γ-secretase modulator,
Proc. Natl. Acad. Sci. USA, 111, 10544-10549.
153.Mecozzi, V., Berman, D., Simoes, S.,
Vetanovetz, C., Awal, M., Patel, V., Schneider, R., Petsko, G., Ringe,
D., and Small, S. (2014) Pharmacological chaperones stabilize retromer
to limit APP processing, Nat. Chem. Biol., 10,
443-449.
154.McLaurin, J., Kierstead, M., Brown, M., Hawkes,
C., Lambermon, M., Phinney, A., Darabie, A., Cousins, J., French, J.,
Lan, M., Chen, F., Wong, S., Mount, H., Fraser, P., Westaway, D., and
St. George-Hyslop, P. (2006) Cyclohexanehexol inhibitors of Aβ
aggregation prevent and reverse Alzheimer phenotype in a mouse model,
Nat. Med., 12, 801-808.
155.Salloway, S., Sperling, R., Keren, R.,
Porsteinsson, A., Van Dyck, C., Tariot, P., Gilman, S., Arnold, D.,
Abushakra, S., Hernandez, C., Crans, G., Liang, E., Quinn, G., Bairu,
M., Pastrak, A., Cedarbaum, J., and ELND005-AD201 Investigators (2011)
A phase 2 randomized trial of ELND005, scylloinositol, in mild to
moderate Alzheimer’s disease, Neurology, 77,
1253-1262.
156.Bobkova, N., Lyabin, D., Medvinskaya, N.,
Samokhin, A., Nekrasov, P., Nesterova, I., Aleksandrova, I.,
Tatarnikova, O., Bobylev, A., Vikhlyantsev, I., Kukharsky, M.,
Ustyugov, A., Polyakov, D., Eliseeva, I., Kretov, D., Guryanov, S., and
Ovchinnikov, L. (2015) The Y-box binding protein 1 suppresses
Alzheimer’s disease progression in two animal models, PLoS
One, 10, e0138867.
157.Saito, S., and Ihara, M. (2014) New therapeutic
approaches for Alzheimer’s disease and cerebral amyloid
angiopathy, Front. Aging Neurosci., 6, 290.
158.Miners, J., Palmer, J., Tayler, H., Palmer, L.,
Ashby, E., Kehoe, P., and Love, S. (2014) Abeta degradation or cerebral
perfusion? Divergent effects of multifunctional enzymes, Front.
Aging Neurosci., 6, 238.
159.Ibrahim, Z., Armour, C., Phipps, S., and
Sukkar, M. (2013) RAGE and TLRs: relatives, friends or neighbors?
Mol. Immunol., 56, 739-744.
160.Deane, R., Du Yan, S., Submamaryan, R., LaRue,
B., Jovanovic, S., Hogg, E., Welch, D., Manness, L., Lin, C., Yu, J.,
Zhu, H., Ghiso, J., Frangione, B., Stern, A., Schmidt, A., Armstrong,
D., Arnold, B., Liliensiek, B., Nawroth, P., Hofman, F., Kindy, M.,
Stern, D., and Zlokovic, B. (2003) RAGE mediates amyloid-beta peptide
transport across the blood-brain barrier and accumulation in brain,
Nat. Med., 9, 907-913.
161.Volpina, O., Koroev, D., Volkova, T., Kamynina,
A., Filatova, A., Zaporozhskaya, Y., Samokhin, A., and Bobkova, N.
(2015) Fragment of the receptor for advanced glycation end products
recovery of spatial memory of animals in Alzheimer’s disease
model, Bioorg. Chem., in press.
162.Holtzman, D., Herz, J., and Bu, G. (2012)
Apolipoprotein E and apolipoprotein E receptors: normal biology and
roles in Alzheimer’s disease, Cold Spring Harb. Perspect.
Med., 2, a006312.
163.Sagare, A., Bell, R., Srivastava, A., Sengillo,
J., Singh, I., Nishida, Y., Chow, N., and Zlokovic, B. (2013) A
lipoprotein receptor cluster IV mutant preferentially binds
amyloid-β and regulates its clearance from the mouse brain, J.
Biol. Chem., 288, 15154-15166.
164.Verghese, P., Castellano, J., Garai, K., Wang,
Y., Jiang, H., Shah, A., Bu, G., Frieden, C., and Holtzman, D. (2013)
ApoE influences amyloid-β (Aβ) clearance despite minimal
apoE/Aβ association in physiological conditions, Proc. Natl.
Acad. Sci. USA, 110, 1807-1816.
165.Laffitte, B., Repa, J., Joseph, S., Wilpitz,
D., Kast, H., Mangelsdorf, D., and Tontonoz, P. (2001) LXRs control
lipid-inducible expression of the apolipoprotein E gene in macrophages
and adipocytes, Proc. Natl. Acad. Sci. USA, 98,
507-512.
166.Cramer, P., Cirrito, J., Wesson, D., Lee, C.,
Karlo, J., Zinn, A., Casali, B., Restivo, J., Goebel, W., James, M.,
Brunden, K., Wilson, D., and Landreth, G. (2012) ApoE-directed
therapeutics rapidly clear β-amyloid and reverse deficits in AD
mouse models, Science, 335, 1503-1506.
167.Price, A., Xu, G., Siemienski, Z., Smithson,
L., Borchelt, D., Golde, T., and Felsenstein, K. (2013) Comment on
“ApoE-directed therapeutics rapidly clear β-amyloid and
reverse deficits in AD mouse models”, Science, 340,
924-924.
168.Tesseur, I., Lo, A., Roberfroid, A., Dietvorst,
S., Broeck, B., Borgers, M., Gijsen, H., Moechars, D., Mercken, M.,
Kemp, J., D’Hooge, R., and De Strooper, B. (2013) Comment on
“ApoE-directed therapeutics rapidly clear β-amyloid and
reverse deficits in AD mouse models”, Science, 340,
924-924.
169.Veeraraghavalu, K., Zhang, C., Miller, S.,
Hefendehl, J., Rajapaksha, T., Ulrich, J., Jucker, M., Holtzman, D.,
Tanzi, R., Vassar, R., and Sisodia, S. (2013) Comment on
“ApoE-directed therapeutics rapidly clear β-amyloid and
reverse deficits in AD mouse models”, Science, 340,
924-924.
170.Kamynina, A., Volpina, O., Medvinskaya, N.,
Aleksandrova, I., Volkova, T., Koroev, D., Samokhin, A., Nesterova, I.,
Shelukhina, I., Kryukova, E., Tsetlin, V., Ivanov, V., and Bobkova, N.
(2010) Vaccination with peptide 173-193 of acetylcholine receptor
α7-subunit prevents memory loss in olfactory bulbectomized mice,
J. Alzheimer’s Dis., 21, 249-261.
171.Bobkova, N., Medvinskaya, N., Kamynina, A.,
Aleksandrova, I., Nesterova, I., Samokhin, A., Koroev, D., Filatova,
M., Nekrasov, P., Abramov, A., Leonov, S., and Volpina, O. (2014)
Immunization with either prion protein fragment 95-123 or the
fragment-specific antibodies rescue memory loss and neurodegenerative
phenotype of neurons in olfactory bulbectomized mice, Neurobiol.
Learn. Mem., 107, 50-64.
172.Volpina, O., Medvinskava, N., Kamynina, A.,
Zaporozhskaia, Ia., Aleksandrova, I., Koroev, D., Samokhin, A.,
Volkova, T., Arsenev, A., and Bobkova, N. (2014) Immunization with a
synthetic fragment 155-164 of neurotrophin receptor p75 prevents memory
loss and decreases beta-amyloid level in mice with experimentally
induced Alzheimer’s disease, Bioorg. Khim., 40,
451-457.
173.Janus, C., Pearson, J., McLaurin, J., Mathews,
P., Jiang, Y., Schmidt, S., Chishti, M., Horne, P., Heslin, D., French,
J., Mount, H., Nixon, R., Mercken, M., Bergeron, C., Fraser, P., St.
George-Hyslop, P., and Westaway, D. (2000) Abeta peptide immunization
reduces behavioral impairment and plaques in a model of
Alzheimer’s disease, Nature, 408, 979-982.
174.Schenk, D., Barbour, R., Dunn, W., Gordon, G.,
Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K.,
Kholodenko, D., Lee, M., Liao, Z., Lieberburg, I., Motter, R., Mutter,
L., Soriano, F., Shopp, G., Vasquez, N., Vandevert, C., Walker, S.,
Wogulis, M., Yednock, T., Games, D., and Seubert, P. (1999)
Immunization with amyloid-beta attenuates
Alzheimer’s-disease-like pathology in the PDAPP mouse,
Nature, 400, 173-177.
175.Orgogozo, J., Gilman, S., Dartigues, J.,
Laurent, B., Puel, M., Kirby, L., Jouanny, P., Dubois, B., Eisner, L.,
Flitman, S., Michel, B., Boada, M., Frank, A., and Hock, C. (2003)
Subacute meningoencephalitis in a subset of patients with AD after
Abeta42 immunization, Neurology, 61, 46-54.
176.Holmes, C., Boche, D., Wilkinson, D.,
Yadegarfar, G., Hopkins, V., Bayer, A., Jones, R., Bullock, R., Love,
S., Neal, J., Zotova, E., and Nicoll, J. (2008) Long-term effects of
Abeta42 immunization in Alzheimer’s disease: follow-up of a
randomized, placebo-controlled phase I trial, Lancet,
372, 216-223.
177.St. George-Hyslop, P., and Morris, J. (2008)
Will anti-amyloid therapies work for Alzheimer’s disease?
Lancet, 372, 180-182.
178.Ostrowitzki, S., Deptula, D., Thurfjell, L.,
Barkhof, F., Bohrmann, B., Brooks, D. J., Klunk, W. E., Ashford, E.,
Yoo, K., Xu, Z.-X., Loetscher, H., and Santarelli, L. (2012) Mechanism
of amyloid removal in patients with Alzheimer’s disease treated
with gantenerumab, Arch. Neurol., 69, 198-207.
179.Doody, R. S., Thomas, R. G., Farlow, M.,
Iwatsubo, T., Vellas, B., Joffe, S., Kieburtz, K., Raman, R., Sun, X.,
Aisen, P. S., Siemers, E., Liu-Seifert, H., Mohs, R., Alzheimer’s
Disease Cooperative Study Steering Committee, and Solanezumab Study
Group (2014) Phase 3 trials of solanezumab for mild-to-moderate
Alzheimer’s disease, N. Engl. J. Med., 370,
311-321.
180.Spencer, B., and Masliah, E. (2014)
Immunotherapy for Alzheimer’s disease: past, present and future,
Front. Aging Neurosci., 6, 114.
181.Bateman, R., Xiong, C., Benzinger, T., Fagan,
A., Goate, A., Fox, N., Marcus, D., Cairns, N., Xie, X., Blazey, T.,
Holtzman, D., Santacruz, A., Buckles, V., Oliver, A., Moulder, K.,
Aisen, P., Ghetti, B., Klunk, W., McDade, E., Martins, R., Masters, C.,
Mayeux, R., Ringman, J., Rossor, M., Schofield, P., Sperling, R.,
Salloway, S., Morris, J., and Dominantly Inherited Alzheimer’s
Network (2012) Clinical and biomarker changes in dominantly inherited
Alzheimer’s disease, N. Engl. J. Med., 367,
795-804.
182.Lippa, C., Nee, L., Mori, H., and St.
George-Hyslop, P. (1998) Abeta-42 deposition precedes other changes in
PS-1 Alzheimer’s disease, Lancet, 352,
1117-1118.
183.Gong, C., Grundke-Iqbal, I., and Iqbal, K.
(2010) Targeting tau protein in Alzheimer’s disease, Drugs
Aging, 27, 351-365.
184.Navarrete, L., Perez, P., Morales, I., and
Maccioni, R. (2011) Novel drugs affecting tau behavior in the treatment
of Alzheimer’s disease and tauopathies, Curr.
Alzheimer’s Res., 8, 678-685.
185.Taniguchi, T., Kawamata, T., Mukai, H.,
Hasegawa, H., Isagawa, T., Yasuda, M., Hashimoto, T., Terashima, A.,
Nakai, M., Mori, H., Ono, Y., and Tanaka, C. (2001) Phosphorylation of
tau is regulated by PKN, J. Biol. Chem., 276,
10025-10031.
186.Bandyopadhyay, B., Li, G., Yin, H., and Kuret,
J. (2007) Tau aggregation and toxicity in a cell culture model of
tauopathy, J. Biol. Chem., 282, 16454-16464.