Upregulation of p72 Enhances Malignant Migration and Invasion of Glioma Cells by Repressing Beclin1 Expression
Zhenxing Zhang1, He Tian2, Ye Miao1, Xu Feng1, Yang Li1, Honglei Wang1, and Xiaofeng Song2*
1Liaoning Medical University, Department of Neurosurgery, 121001 Jinzhou, China2Liaoning Medical University, Department of Histology and Embryology, 121001 Jinzhou, China; E-mail: Songzhenxing151121@163.com
* To whom correspondence should be addressed.
Received November 21, 2015; Revision received February 21, 2016
p72 is the member of the DEAD-box RNA helicase family, which can unwind double-stranded RNA and is efficient for microRNA (miRNA, miR) processing. However, its specific role in glioma has not been elucidated. First, the expression of p72 in glioma cell lines and tissues was explored using Western blot. To explore the role of p72 on glioma progression, adenovirus inhibiting p72 was transfected into A172 and T98G cells. Cell autophagy was determined using GFP-LC3 dots, and cell apoptosis was determined using flow cytometry. The effect of Beclin1 was explored using GFP-LC3 dots, flow cytometry, and colony formation. The possible miRNAs that target the 3′-untranslated region (3′-UTR) of Beclin1 were predicted using TargetScan. Dual luciferase reporter assay was applied to determine whether these miRNAs bind to the 3′-UTR of Beclin1. The expression of p72 was significantly increased in glioma cell lines and tissues. Autophagy-related protein Beclin1 was found to be significantly enhanced when p72 was inhibited. The accumulation of GFP-LC3 dots was significant in cells transfected with ad-sh-p72 compared with ad-con. Colony formation capacity and cell apoptosis were also found to be significantly decreased with p72 inhibition. Furthermore, upregulation of Beclin1 contributes to A172 cell autophagy, invasion, and apoptosis. Overexpression of p72 induces increased miR-34-5p and miR-5195-3p expression in A172 and T98G cells. Beclin1 was the target gene of miR-34-5p and miR-5195-3p. In conclusion, we found for the first time that overexpression of p72 decreased Beclin1 expression partially by increasing miR-34-5p and miR-5195-3p expression in A172 and T98G cells.
KEY WORDS: glioma, p72, miR-34-5p, miR-5195-3p, Beclin1DOI: 10.1134/S0006297916060031
Malignant glioma is the most common and deadly primary brain tumor in adults [1, 2]. Despite aggressive treatment methods, they are characterized by high invasion, rapid cell growth, and poor survival [3]. It is reported that most glioma patients survive less than 1 year after accurate diagnosis. Even with the standard treatment of radiotherapy and chemotherapy, most patients die within 2 years [4, 5]. Thus, it is important to further explore the underlying mechanism and develop novel therapy strategies.
Recent studies have reported that autophagy is activated in glioma cells by chemotherapeutic agents and radiation, which may serve as a potential therapeutic target for cancer therapy [6, 7]. As a conserved catabolic process, autophagy was found to prompt the degradation of unnecessary cellular contents and damaged organelles by lysosomal enzymes [8, 9]. The process of autophagy includes the formation of autophagosomes, which then engulf the unwanted cell components and transfer them to lysosomes [10]. As an important cellular self-digestive process, autophagy plays a key role in cell survival, differentiation, homeostasis, and development [11]. Beclin1 is the first identified autophagy-related protein that tightly correlates with cancer [12]. At the early stage of autophagy, Beclin1 is involved in autophagosome nucleation [13]. More importantly, through interaction with positive and negative regulators, Beclin1 is involved in autophagic activity and tumorigenesis [14].
MicroRNAs (miRNAs or miRs) are small noncoding RNAs that widely regulate gene expression, and several miRs are found to bind the 3′-untranslated region (3′-UTR) of Beclin1, such as miR-Let7A miR-17-5p and miR-30d [15-17]. Primary microRNAs (pri-miRNAs) are processed by the nuclear RNase III Drosha, thereby forming hairpin-shaped precursor miRNAs (pre-miRNAs). In humans, such process is facilitated by the Asp-Glu-Ala-Asp (D-E-A-D) box helicases p68 (DDX5) and p72 (DDX17) [18]. RNA helicases participates in various cellular signalings mainly by regulating RNA splicing, transcription, translation, and miRNA biogenesis [19]. As the largest helicase family, the D-E-A-D box proteins share 12 conserved motifs [20]. p72 RNA helicases are found to be involved in RNA splicing and miRNA biogenesis [21]. For instance, they are reported to bind estrogen receptor α (ERα) and enhance downstream gene expression through transactivation [22]. Furthermore, DEAD-box RNA helicases are suggested to be necessary for primary miRNA processing [23]. This prompted us to carefully explore the expression and role of p72 in glioma cancer and reveal their importance for cancer progression.
In this study, we identified for the first time that p72 is upregulated in glioma cell lines and tissues. Through enhancing miR-34-5p and miR-5195-3p biogenesis in A172 and T98G cells, upregulation of p72 contributes to the malignancies of glioblastoma cells mainly by repressing Beclin1 expression.
MATERIALS AND METHODS
Cell lines and culture. Normal human astrocytes (NHA) were obtained from ScienCell Research Laboratories (Canada) and cultured according to the supplier’s instructions. Human U87MG, U251MG, U373, A172, U118, T98G, and SHU-44 glioblastoma cell lines were obtained from the American Type Culture Collection (ATCC). All cells were cultured in DMEM (HyClone, USA) supplemented with 10% fetal bovine serum (HyClone) and incubated in a humidified atmosphere containing 5% CO2 at 37°C without antibiotics.
Glioma specimens. Tumor specimens from patients with glioma (GBM) and non-neoplastic brain (NNB) tissues from patients without glioma were collected in the Department of Neurosurgery of the First Affiliated Hospital of Liaoning Medical University after accurate pathological confirmation. The tissues were immediately snap-frozen in liquid nitrogen and stored at –80°C for further study. All the experiments were approved by the Ethical Committee of the First Affiliated Hospital of Liaoning Medical University and informed consent was given by all patients.
Western blot. Cells were first rinsed with PBS three times and then lysed in RIPA buffer (1% Triton X-100, 150 mM NaCl, 5 mM EDTA, and 10 mM Tris-HCl, pH 7.0) (Solarbio, China). The cell lysates were collected after centrifugation at 12,000g for 15 min. Protein concentration was determined using a BCA protein assay kit (Merck Millipore, Germany). The cell lysates were separated by 10% SDS-PAGE and then transferred onto a PVDF membrane. The membranes were blocked with 5% nonfat dried milk in TBST for 1 h at room temperature. Then, the specific primary antibodies were added for incubation overnight at 4°C. Subsequently, the membranes were washed with TBST three times and incubated with the appropriate HRP-conjugated anti-rabbit IgG secondary antibodies (Zhongshanjinqiao, China) at room temperature for 1 h. The blots were detected using the ECL plus detection system (Millipore), and digitized data were quantified by Image J software.
Reverse transcription PCR. RNA was extracted with RNAvizol reagent (Vigorous, China) and reverse-transcribed with EasyScript One-Step gDNA Removal and cDNA Synthesis SuperMix (AE311-03; Transgene, China). The PCR program was as follows: 48°C for 45 min; 96°C for 2 min; 25 repeats of 94°C for 30 s, 55°C for 45 s, and 68°C for 80 s; followed by a final extension at 65°C for 6 min. In addition, TaqMan miRNA assays (Applied Biosystems, USA) were applied to detect the expression level of mature miRNAs using the stem-loop method. All PCRs were run in triplicate, and gene expression was calculated by the comparative ΔCt method relative to U6 small nuclear RNA (RNU6).
Colony formation assay. A172 cells were exposed to miR mimetics/inhibitors or negative control (NC) for 48 h. After transfection, 500 cells were seeded in the well of a 6-well plate and grown for 2 weeks. Then, the cells were fixed with methanol–acetic acid (3 : 1 v/v) and stained with Crystal violet.
RNA oligoribonucleotides. All RNA oligoribonucleotides were purchased from Genepharma (China). The small interfering RNAs (siRNAs) targeting the mRNA of human p72 were designed as si-p72.
siRNA transfection. Human glioblastoma cells were seeded at the concentration of 2·105 cells/well. Twenty-four hours later, a nonspecific control siRNA (NC) or a specific siRNA for Beclin1 was transfected into the wells at final concentration 50 nM using Hiperfect transfection reagent according to the manufacturer’s instructions (Qiagen, Germany).
Luciferase reporter assay. To test whether miR-34-5p or miR-5195-3p targets the 3′-UTR of Beclin1, the sequence containing the predicted binding sites was included into pmirGLO plasmids (Promega, USA). For luciferase reporter assay, A172 cells were seeded into 96-well plates at 10,000 cells per well on the day before transfection. Then, a mixture of 100 ng pmirGLO-Beclin1-3′-UTR and 50 nM miRNA mimetic or negative control were transfected into A172 cells using Lipofectamine 2000 transfection reagent (Invitrogen Life Technologies, USA). After 48 h, firefly and Renilla luciferase activity was determined using the Dual-luciferase Reporter Assay System (Promega). The firefly luciferase activities were used as an internal control for transfection efficiency.
Autophagy measurement using GFP-LC3. A72 cells were transfected with a green fluorescent protein–microtubule-associated protein 1 light chain 3 (GFP-LC3) expression plasmid (Invitrogen Life Technologies). After 24 h, cells were treated with ad-p72, and the fluorescence of GFP-LC3 was observed under a fluorescence microscope. Then, the LC3 punctate spots were counted 2 days later. The percentage of cells undergoing autophagy was calculated from the ratio of autophagic cells to normal cells bearing GFP-LC3 fluorescence.
Statistical analyses. Data are presented as mean ± S.E. from three independent experiments. The figures show representative results of at least three independent experiments. The significance of differences between different test conditions was determined using one-way ANOVA and Student’s t-test; p < 0.05 was considered statistically significant.
RESULTS
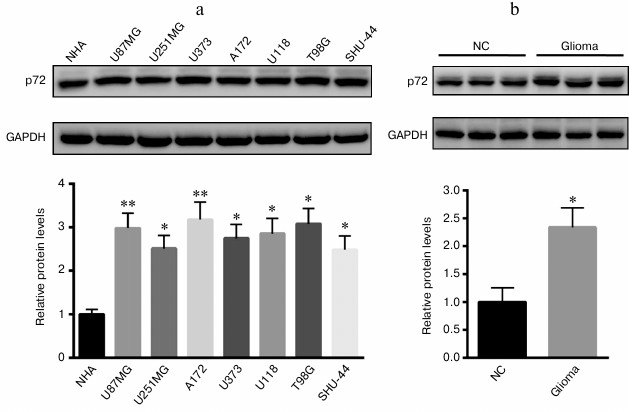
Increased p72 expression in glioma cell lines and tissues. First, we explored the expression of p72 in glioma cell lines and tissues; we found that p72 was significantly increased in human U87MG, U251MG, U373, A172, U118, T98G, and SHU-44 glioblastoma cell lines in comparison with NHA cells (Fig. 1a). Besides, the protein level of p72 was much higher than that of the tumor specimens from glioma patients (Fig. 1b). Since p72 was most significantly increased in A172 and T98G cell lines, both were selected for further study.
Fig. 1. Expression of p72 was significantly increased in glioma cell lines and tissues. a) Protein level of p72 was explored in human U87MG, U251MG, U373, A172, U118, T98G, and SHU-44 glioblastoma cell lines compared with NHA cells. b) Protein level of p72 was much higher than that of the tumor specimens from patients with glioma compared with normal control (NC). Data are presented as mean ± S.E. from three independent experiments; * p < 0.05, ** p < 0.01.
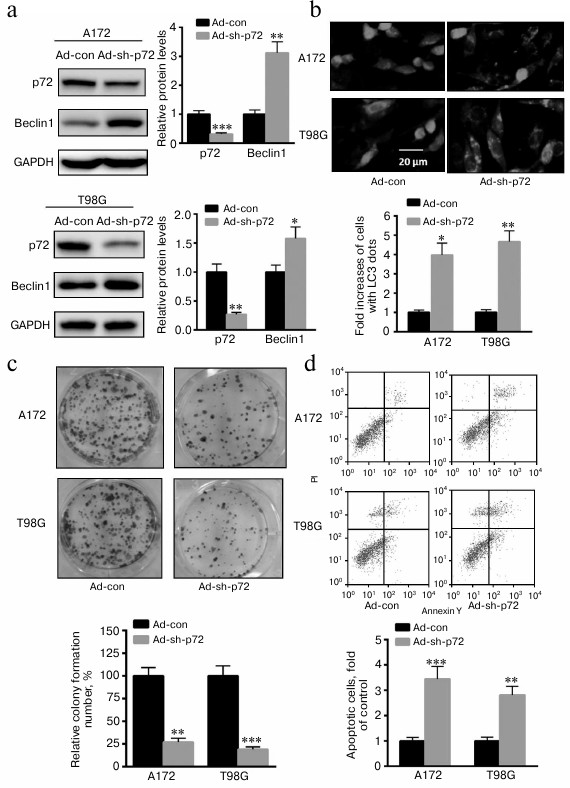
Knockdown of p72 enhances A172 and T98G cell autophagy, invasion, and apoptosis. To explore the role of p72 on glioma progression, adenovirus inhibiting p72 was transfected into A172 and T98G cells. As shown in Fig. 2a, autophagy-related protein Beclin1 was significantly enhanced when p72 was inhibited in both cell types. Meanwhile, accumulation of GFP-LC3 dots was significant in A172 and T98G cells transfected with ad-sh-p72 compared with ad-con (Fig. 2b). Moreover, colony formation capacity was also significantly decreased with p72 inhibition in A172 and T98G cells (Fig. 2c). In addition, cell apoptosis was also enhanced in A172 and T98G cells transfected with ad-sh-p72 (Fig. 2d). These data suggest an oncogenic role of p72 in glioma cells.
Fig. 2. Knockdown of p72 enhances A172 and T98G cell autophagy, invasion, and apoptosis. a) Autophagy related protein Beclin1 was significantly enhanced when p72 was inhibited (ad-sh-p72) compared with ad-con. b) Accumulation of GFP-LC3 dots was significant in A172 and T98G cells transfected with ad-sh-p72 compared with ad-con. c) Colony formation capacity was also significantly decreased with p72 inhibition. d) Cell apoptosis was also enhanced in A172 and T98G cells transfected with ad-sh-p72. Data are presented as mean ± S.E. from three independent experiments; * p < 0.05, ** p < 0.01, *** p < 0.001.
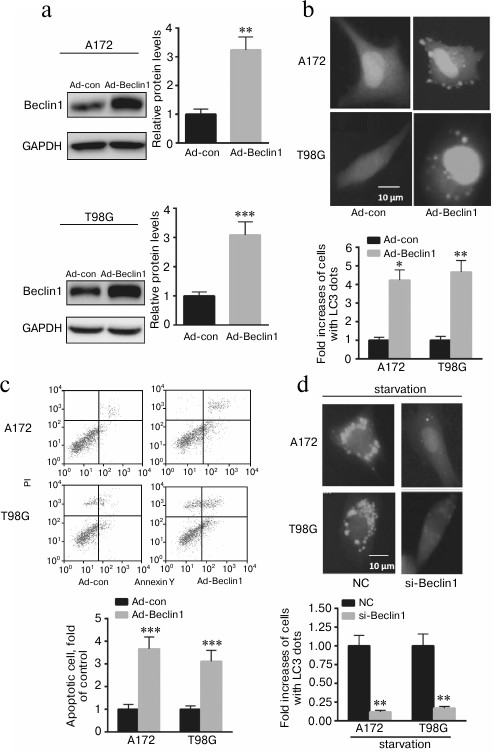
Upregulation of Beclin1 contributes to A172 and T98G cell autophagy, invasion, and apoptosis. To explore whether p72 exerts its function mainly through Beclin1, adenovirus overexpressing Beclin1 was transfected into A172 and T98G cells. As shown in Fig. 3a, overexpression of Beclin1 significantly enhanced the ratio of GFP-LC3 dots in A172 and T98G cells. In addition, overexpression of Beclin1 enhanced A172 and T98G cell invasion and apoptosis (Fig. 3, b and c). Furthermore, we found that knockdown of Beclin1 significantly decreased GFP-LC3 dots in A172 and T98G cells after starvation for 6 h (Fig. 3d). These data indicate that p72 prompted A172 cell autophagy, invasion, and apoptosis partially through positive regulation of Beclin1 expression.
Fig. 3. Beclin1 prompted A172 and T98G cell autophagy, invasion, and apoptosis partially through regulation of Beclin1 expression. a) Transfection of ad-Beclin1 enhanced the protein level of Beclin1 in A172 and T98G cells. b) Overexpressing Beclin1 significantly enhanced the ratio of GFP-LC3 dots in A172 and T98G cells. Cells showing six or more GFP-LC3 dots were considered undergoing autophagy and quantified as percentages of autophagic cells. c) Overexpression of Beclin1 enhanced A172 and T98G cell apoptosis. d) Knockdown of Beclin1 significantly decreased GFP-LC3 dots compared with A172 and T98G cells transfected with a nonspecific control siRNA (NC). Data are presented as mean ± S.E. from three independent experiments; * p < 0.05, ** p < 0.01, *** p < 0.001.
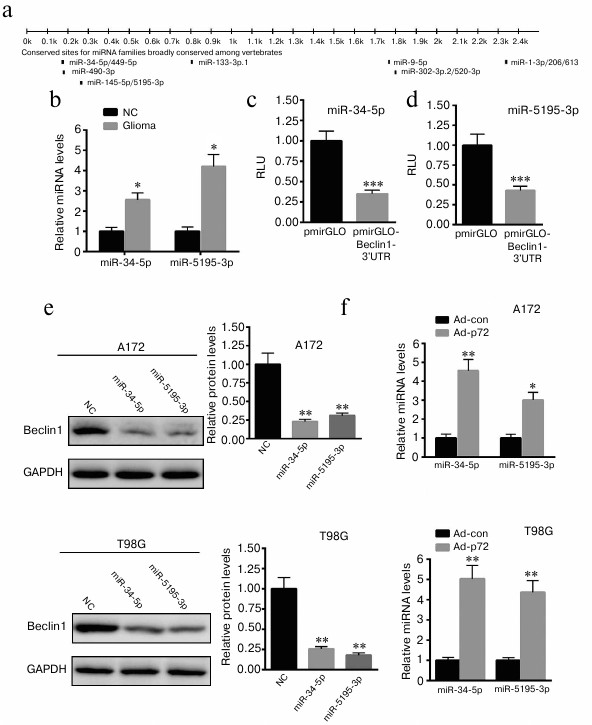
Overexpression of p72 induces increased miR-34-5p and miR-5195-3p expression in A172 and T98G cells. As an RNA helicase, p72 plays a key role in miRNA biogenesis. Thus, we explore possible miRNAs that regulate the expression of Beclin1. First, we predicted the possible miRNAs that bind the 3′-UTR of Beclin1 using targetScan (http://www.targetscan.org) (Fig. 4a). We explored the expression of 11 miRNAs. We found that only miR-34-5p and miR-5195-3p levels were significantly increased in glioma tissues (Fig. 4b). Then, the 3′-UTR of Beclin1 was cloned into pmirGLO plasmid. Dual luciferase reporter assay demonstrated that miR-34-5p and miR-5195-3p significantly decreased the luciferase activity of pmirGLO-Beclin1-3′-UTR (Fig. 4, c and d). Western blot analysis revealed that overexpression of miR-34-5p and miR-5195-3p significantly reduced the expression of Beclin1 in A172 and T98G cells (Fig. 4e). We also found that overexpression of p72 significantly enhanced the level of miR-34-5p and miR-5195-3p in A172 and T98G cells (Fig. 4f). These data indicate that overexpression of p72 induced decreased miR-34-5p and miR-5195-3p expression in A172 cells.
Fig. 4. Overexpression of p72 induces increased miR-34-5p and miR-5195-3p expression in A172 and T98G cells. a) Possible miRNAs that bind the 3′-UTR of Beclin1 were predicted using targetScan (http://www.targetscan.org). Only miR-34-5p and miR-5195-3p levels were significantly increased in glioma tissues compared with normal control samples (NC) (b). Dual luciferase reporter assay demonstrated that miR-34-5p (c) and miR-5195-3p (d) significantly decreased the luciferase activity of pmirGLO-Beclin1-3′-UTR. e) Western blot analysis revealed that overexpression of miR-34-5p and miR-5195-3p significantly reduced the expression of Beclin1 compared with A172 and T98G cells transfected with a nonspecific control miRNA (NC). f) Overexpression of p72 significantly enhanced the level of miR-34-5p and miR-5195-3p in A172 and T98G cells. Data are presented as mean ± S.E. from three independent experiments; * p < 0.05, ** p < 0.01, *** p < 0.001.
DISCUSSION
Glioma is a common primary malignant brain tumor among adults [24]. It was reported that the general median survival rate of patients with high-grade glioma is less than 12 months [25]. Thus, it is necessary to develop novel and effective treatment methods for glioma patients [26, 27]. RNA helicases are indicated to play key roles in tumorigenesis [28, 29]. They can not only unwind RNA, but also disturb the interaction between RNA and proteins [30, 31]. p72 helicase belongs to the DDX17 family with the typical sequence of DEAD, which is important for ATP hydrolysis [31]. In this study, we first demonstrated that p72 RNA helicase was decreased in glioma cell lines and tissues. Further study revealed that overexpression of p72 contributed to enhanced glioma cell autophagy, invasion, and apoptosis. Thus, p72 may serve as a therapeutic target for glioma.
Autophagy has been indicated to be involved in tumor progression and suppression [32]. It is reported that autophagy can maintain cellular homeostasis, genomic stability, and metabolism by suppressing malignant transformation of normal cells [33]. It was reported that the actual function of autophagy on tumorigenesis is largely dependent on the microenvironment [34]. According to different tissue types and genetic context, autophagy exerts different functions on cell growth [35]. Beclin1 is suggested to interact with the class III phosphoinositide 3-kinase complex, which is necessary for autophagy initiation [36]. Our data are the first to demonstrate that overexpression of p72 significantly decreased the protein expression of Beclin1. Further study revealed that upregulation of Beclin1 contributes to enhanced A172 cell autophagy, apoptosis, and colony formation capacity.
Another function of p72 is related to the processing of miRNA. It is well known that primary miRNA transcript (pri-miRNA) can be processed in the nucleus through the Drosha complex. Researchers found that p72 is an important component of the Drosha complex [21, 37]. More importantly, knockdown of p72 significantly decreases the processing of pri-miRNAs, since it can unwind RNA and enhances the cleavage of pri-miRNA in the Drosha complex. In this study, we further explored whether p72 negatively regulated Beclin1 expression through affecting miRNA biogenesis. We found that increased p72 expression significantly enhances the level of miR-34-5p and miR-5195-3p. Luciferase analysis and Western blot revealed that Beclin1 is a target gene of miR-34-5p and miR-5195-3p. In summary, we are the first to show that overexpression of p72 decreases Beclin1 expression partially by decreasing miR-34-5p and miR-5195-3p expression in A172 cells.
In this study, we first determined the oncogenic role of p72 in glioma tissues and cell lines. Further study revealed that p72 contributes to glioblastoma cell malignancies mainly by enhancing miR-34-5p and miR-5195-3p biogenesis, thereby repressing Beclin1 expression.
This study was supported by the Program for Excellent Talents in Liaoning Province in China (No. LJQ2013088).
REFERENCES
1.Omuro, A., and DeAngelis, L. M. (2013) Glioblastoma
and other malignant gliomas: a clinical review, JAMA,
310, 1842-1850.
2.Koshkin, P. A., Chistiakov, D. A., and Chekhonin,
V. P. (2013) Role of microRNAs in mechanisms of glioblastoma resistance
to radio- and chemotherapy, Biochemistry (Moscow), 78,
325-334.
3.Stupp, R., and Weber, D. C. (2005) The role of
radio- and chemotherapy in glioblastoma, Onkologie, 28,
315-317.
4.Ryu, C. H., Yoon, W. S., Park, K. Y., Kim, S. M.,
Lim, J. Y., Woo, J. S., Jeong, C. H., Hou, Y., and Jeun, S. S. (2012)
Valproic acid downregulates the expression of MGMT and sensitizes
temozolomide-resistant glioma cells, J. Biomed. Biotechnol.,
987495.
5.Stupp, R., Hegi, M. E., Mason, W. P., Van den Bent,
M. J., Taphoorn, M. J., Janzer, R. C., Ludwin, S. K., Allgeier, A.,
Fisher, B., Belanger, K., Hau, P., Brandes, A. A., Gijtenbeek, J.,
Marosi, C., Vecht, C. J., Mokhtari, K., Wesseling, P., Villa, S.,
Eisenhauer, E., Gorlia, T., Weller, M., Lacombe, D., Cairncross, J. G.,
and Mirimanoff, R. O.; European Organisation for Research and Treatment
of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer
Institute of Canada Clinical Trials Group (2009) Effects of
radiotherapy with concomitant and adjuvant temozolomide versus
radiotherapy alone on survival in glioblastoma in a randomised phase
III study: 5-year analysis of the EORTC-NCIC trial, Lancet
Oncol., 10, 459-466.
6.Yan, Y., Xu, Z., Dai, S., Qian, L., Sun, L., and
Gong, Z. (2016) Targeting autophagy to sensitive glioma to temozolomide
treatment, J. Exp. Clin. Cancer Res., 35, 23.
7.Wang, L., Long, L., Wang, W., and Liang, Z. (2015)
Resveratrol, a potential radiation sensitizer for glioma stem cells
both in vitro and in vivo, J. Pharmacol. Sci.,
129, 216-225.
8.Alers, S., Loffler, A. S., Wesselborg, S., and
Stork, B. (2012) Role of AMPK-mTOR-Ulk1/2 in the regulation of
autophagy: cross talk, shortcuts, and feedbacks, Mol. Cell.
Biol., 32, 2-11.
9.Levine, B., and Kroemer, G. (2008) Autophagy in the
pathogenesis of disease, Cell, 132, 27-42.
10.Araya, J., Hara, H., and Kuwano, K. (2013)
Autophagy in the pathogenesis of pulmonary disease, Intern.
Med., 52, 2295-2303.
11.Xavier, R. J., Huett, A., and Rioux, J. D. (2008)
Autophagy as an important process in gut homeostasis and Crohn’s
disease pathogenesis, Gut, 57, 717-720.
12.Liu, J. L., Chen, F. F., Chang, S. F., Chen, C.
N., Lung, J., Lo, C. H., Lee, F. H., Lu, Y. C., and Hung, C. H. (2015)
Expression of Beclin family proteins is associated with tumor
progression in oral cancer, PLoS One, 10,
e0141308.
13.Yue, Z., Jin, S., Yang, C., Levine, A. J., and
Heintz, N. (2003) Beclin 1, an autophagy gene essential for
early embryonic development, is a haploinsufficient tumor suppressor,
Proc. Natl. Acad. Sci. USA, 100, 15077-15082.
14.Qu, X., Yu, J., Bhagat, G., Furuya, N.,
Hibshoosh, H., Troxel, A., Rosen, J., Eskelinen, E. L., Mizushima, N.,
Ohsumi, Y., Cattoretti, G., and Levine, B. (2003) Promotion of
tumorigenesis by heterozygous disruption of the Beclin 1
autophagy gene, J. Clin. Invest., 112, 1809-1820.
15.Song, J., Oh, Y., and Lee, J. E. (2015) miR-Let7A
modulates autophagy induction in LPS-activated microglia, Exp.
Neurobiol., 24, 117-125.
16.Chatterjee, A., Chattopadhyay, D., and
Chakrabarti, G. (2014) miR-17-5p downregulation contributes to
paclitaxel resistance of lung cancer cells through altering Beclin1
expression, PLoS One, 9, e95716.
17.Zhang, Y., Yang, W. Q., Zhu, H., Qian, Y. Y.,
Zhou, L., Ren, Y. J., Ren, X. C., Zhang, L., Liu, X. P., Liu, C. G.,
Ming, Z. J., Li, B., Chen, B., Wang, J. R., Liu, Y. B., and Yang, J. M.
(2014) Regulation of autophagy by miR-30d impacts sensitivity of
anaplastic thyroid carcinoma to cisplatin, Biochem. Pharmacol.,
87, 562-570.
18.Chu, Y. D., Chen, H. K., Huang, T., and Chan, S.
P. (2016) A novel function for the DEAD-box RNA helicase DDX-23 in
primary microRNA processing in Caenorhabditis elegans, Devel.
Biol., 409, 459-472.
19.Linder, P., and Jankowsky, E. (2011) From
unwinding to clamping – the DEAD box RNA helicase family,
Nat. Rev. Mol. Cell Biol., 12, 505-516.
20.Jalal, C., Uhlmann-Schiffler, H., and Stahl, H.
(2007) Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82
(Ddx17) in ribosome biogenesis and cell proliferation, Nucleic Acids
Res., 35, 3590-3601.
21.Retraction: Fukuda, T., Yamagata, K., Fujiyama,
S., Matsumoto, T., Koshida, I., Yoshimura, K., Mihara, M., Naitou, M.,
Endoh, H., Nakamura, T., Akimoto, C., Yamamoto, Y., Katagiri, T.,
Foulds, C., Takezawa, S., Kitagawa, H., Takeyama, K., O’Malley,
B. W., and Kato, S. (2014) DEAD-box RNA helicase subunits of the Drosha
complex are required for processing of rRNA and a subset of microRNAs,
Nat. Cell Biol., 16, 1126.
22.Moy, R. H., Cole, B. S., Yasunaga, A., Gold, B.,
Shankarling, G., Varble, A., Molleston, J. M., tenOever, B. R., Lynch,
K. W., and Cherry, S. (2014) Stem-loop recognition by DDX17 facilitates
miRNA processing and antiviral defense, Cell, 158,
764-777.
23.Fukuda, T., Yamagata, K., Fujiyama, S.,
Matsumoto, T., Koshida, I., Yoshimura, K., Mihara, M., Naitou, M.,
Endoh, H., Nakamura, T., Akimoto, C., Yamamoto, Y., Katagiri, T.,
Foulds, C., Takezawa, S., Kitagawa, H., Takeyama, K., O’Malley,
B. W., and Kato, S. (2007) DEAD-box RNA helicase subunits of the Drosha
complex are required for processing of rRNA and a subset of microRNAs,
Nat. Cell Biol., 9, 604-611.
24.Buckner, J. C. (2003) Factors influencing
survival in high-grade gliomas, Semin. Oncol., 30,
10-14.
25.DeAngelis, L. M. (2001) Brain tumors, N. Engl.
J. Med., 344, 114-123.
26.Kakkar, A., Sharma, M. C., Suri, V., Kaushal, S.,
Chandra, S. P., Garg, A., and Sarkar, C. (2014) Angiocentric glioma: a
treatable cause of epilepsy: report of a rare case, Neurol.
India, 62, 677-679.
27.Kong, B. H., Moon, J. H., Huh, Y. M., Shim, J.
K., Lee, J. H., Kim, E. H., Chang, J. H., Kim, D. S., Hong, Y. K., Kim,
S. H., Lee, S. J., and Kang, S. G. (2014) Prognostic value of glioma
cancer stem cell isolation in survival of primary glioblastoma
patients, Stem Cells Int., 838950.
28.Russell, R. (2015) Unwinding the mechanisms of a
DEAD-box RNA helicase in cancer, J. Mol. Biol., 427,
1797-1800.
29.Epling, L. B., Grace, C. R., Lowe, B. R.,
Partridge, J. F., and Enemark, E. J. (2015) Cancer-associated mutants
of RNA helicase DDX3X are defective in RNA-stimulated ATP hydrolysis,
J. Mol. Biol., 427, 1779-1796.
30.Rocak, S., and Linder, P. (2004) DEAD-box
proteins: the driving forces behind RNA metabolism, Nat. Rev. Mol.
Cell Biol., 5, 232-241.
31.Caretti, G., Schiltz, R. L., Dilworth, F. J., Di
Padova, M., Zhao, P., Ogryzko, V., Fuller-Pace, F. V., Hoffman, E. P.,
Tapscott, S. J., and Sartorelli, V. (2006) The RNA helicases p68/p72
and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle
differentiation, Dev. Cell, 11, 547-560.
32.Galluzzi, L., Pietrocola, F., Bravo-San Pedro, J.
M., Amaravadi, R. K., Baehrecke, E. H., Cecconi, F., Codogno, P.,
Debnath, J., Gewirtz, D. A., Karantza, V., Kimmelman, A., Kumar, S.,
Levine, B., Maiuri, M. C., Martin, S. J., Penninger, J., Piacentini,
M., Rubinsztein, D. C., Simon, H. U., Simonsen, A., Thorburn, A. M.,
Velasco, G., Ryan, K. M., and Kroemer, G. (2015) Autophagy in malignant
transformation and cancer progression, EMBO J., 34,
856-880.
33.Guo, J. Y., Xia, B., and White, E. (2013)
Autophagy-mediated tumor promotion, Cell, 155,
1216-1219.
34.Kenific, C. M., and Debnath, J. (2015) Cellular
and metabolic functions for autophagy in cancer cells, Trends Cell
Biol., 25, 37-45.
35.Perez, E., Das, G., Bergmann, A., and Baehrecke,
E. H. (2015) Autophagy regulates tissue overgrowth in a
context-dependent manner, Oncogene, 34, 3369-3376.
36.Kihara, A., Kabeya, Y., Ohsumi, Y., and
Yoshimori, T. (2001) Beclin–phosphatidylinositol 3-kinase complex
functions at the trans-Golgi network, EMBO Rep.,
2, 330-335.
37.Janknecht, R. (2010) Multi-talented DEAD-box
proteins and potential tumor promoters: p68 RNA helicase (DDX5) and its
paralog, p72 RNA helicase (DDX17), Am. J. Transl. Res.,
2, 223-234.