Non-photochemical Fluorescence Quenching in Photosystem II Antenna Complexes by the Reaction Center Cation Radical
V. Z. Paschenko1*, V. V. Gorokhov1, N. P. Grishanova1, B. N. Korvatovskii1, M. V. Ivanov1, E. G. Maksimov1, and M. D. Mamedov2
1Lomonosov Moscow State University, Faculty of Biology, 119991 Moscow, Russia; fax: +7 (495) 939-1115; E-mail: vz.paschenko@gmail.com2Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received November 24, 2015; Revision received February 3, 2016
In direct experiments, rate constants of photochemical (kP) and non-photochemical (kP+) fluorescence quenching were determined in membrane fragments of photosystem II (PSII), in oxygen-evolving PSII core particles, as well as in core particles deprived of the oxygen-evolving complex. For this purpose, a new approach to the pulse fluorometry method was implemented. In the “dark” reaction center (RC) state, antenna fluorescence decay kinetics were measured under low-intensity excitation (532 nm, pulse repetition rate 1 Hz), and the emission was registered by a streak camera. To create a “closed” [P680+QA–] RC state, a high-intensity pre-excitation pulse (pump pulse, 532 nm) of the sample was used. The time advance of the pump pulse against the measuring pulse was 8 ns. In this experimental configuration, under the pump pulse, the [P680+QA–] state was formed in RC, whereupon antenna fluorescence kinetics was measured using a weak testing picosecond pulsed excitation light applied to the sample 8 ns after the pump pulse. The data were fitted by a two-exponential approximation. Efficiency of antenna fluorescence quenching by the photoactive RC pigment in its oxidized (P680+) state was found to be ~1.5 times higher than that of the neutral (P680) RC state. To verify the data obtained with a streak camera, control measurements of PSII complex fluorescence decay kinetics by the single-photon counting technique were carried out. The results support the conclusions drawn from the measurements registered with the streak camera. In this case, the fitting of fluorescence kinetics was performed in three-exponential approximation, using the value of τ1 obtained by analyzing data registered by the streak camera. An additional third component obtained by modeling the data of single photon counting describes the P680+Pheo– charge recombination. Thus, for the first time the ratio of kP+/kP = 1.5 was determined in a direct experiment. The mechanisms of higher efficiency for non-photochemical antenna fluorescence quenching by RC cation radical in comparison to that of photochemical quenching are discussed.
KEY WORDS: photosystem II, non-photochemical quenching, photochemical reactions, light-harvesting antenna, reaction centerDOI: 10.1134/S0006297916060043
High (~100%) photosynthetic quantum yield is realized at relatively moderate light intensities, when all energy of the light absorbed by a photosynthetic organism’s pigment apparatus is utilized in photochemical reactions. At high light intensities, not all energy of antenna-excited states is utilized by reaction centers for product formation. As a result, excess energy can cause destructive changes in the photosynthetic apparatus [1, 2]. To prevent this, photosynthetic organisms have developed various mechanisms for quenching excessive absorbed energy, notably non-photochemical quenching (NPQ) in photosystem II (PSII) antenna complexes, as well as antenna excitation quenching by reaction center (RC) cation radicals. The first of these mechanisms, NPQ in PSII antenna complexes, has been studied quite well. Its essence is the fact that under intense illumination, the pH of thylakoid lumen decreases, and the process of violaxanthin deepoxidation is triggered, transforming violaxanthin into zeaxanthin [1, 3, 4]. Compared to violaxanthin, zeaxanthin has a greater number of π-bonds and a lower S1 level, and therefore it is a more efficient acceptor for energy of chlorophyll (Chl) excited states. For NPQ to occur, the PsbS protein, sensitive to low pH levels, is also essential. This protein binds zeaxanthins in its protonated state and is a NPQ site. The second NPQ mechanism, energy dissipation by the PSII RC cation radical (the oxidized photoactive pigment P680+) is mentioned in a large number of studies, notably [5-11]. However, in reports, both those mentioned and others related to the subject of the current study, quantitative estimates for the efficiency of NPQ by the photoactive pigment P680+ oxidized state were not given. Only in [7] a theoretical calculation is given, according to which non-photochemical quenching of antenna fluorescence by oxidized PSII RC (P680+) is three times lower than that of photochemical quenching (P680).
From a functional point of view, PSII can be described in terms of three domains: the central photochemical domain, the plastoquinone-reducing domain, and the water-oxidizing domain. It should be noted that the water oxidation complex, located on the donor side, is the most labile PSII site and is susceptible to oxidative damage [12]. Figure 1 shows the location of cofactors in the protein matrix.
Fig. 1. Three-dimensional structure of PSII core complex (view parallel to the membrane plane). P680 is the Chl a dimer in the RC; ChlD1 and ChlD2, chlorophyll a molecules in D1 and D2 subunits, respectively; PheoD1 and PheoD2, pheophytin molecules in D1 and D2 units, respectively; QA and QB, primary and secondary quinone acceptors; YZ and YD, redox-active tyrosine residues (adapted from [12]).
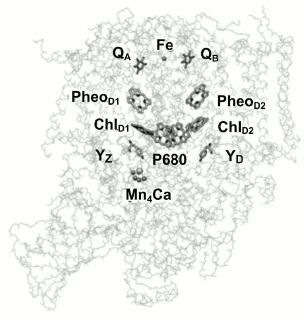
The aim of this study was comparative experimental analysis of antenna Chl fluorescence quenching efficiency in different pigment–protein PSII complexes – membrane fragments, oxygen-evolving core complexes, and PSII core complexes deprived of the Mn4 cluster by neutral [P680QA] and oxidized [P680+QA–] reaction centers. Some terminological explanations should be given here. First, it should be noted that the term “P680” originated from spectroscopic data, in which a bleaching band is observed at 680 nm in differential spectra (light-minus-dark). The band is often identified with the PD1PD2 special pair, by analogy with the RC of purple bacteria. Indeed, in the RC of purple bacteria the P870 special pair is an electron donor, and the hole is also located on it, forming the oxidized P870+ state. In the case of PSII, however, such identification is no longer correct. In fact, the excited state of the reaction center, 1P680*, from which the primary electron transport reaction starts, and the oxidized state of reaction center, P680+, on which a hole is stabilized, differ in their molecular nature. As follows from [13], for example, at room temperature excitation is delocalized on four reaction center Chl molecules. According to other data [14], all four Chl molecules and two Pheo molecules contribute to the 1P680* state, with maximum excitation density concentrated on ChlD1 (~30%), and the contributions from other pigments are: PD1 (~15%), PD2 (~11%), ChlD2 (~9%), PheoD1 (~15%), PheoD2 (~20%). Based on the fact that PSII RC excitation is distributed among six pigments, it cannot be definitely said which one is the primary electron donor at room temperature. On the other hand, it was shown [15] that at physiological temperatures, the hole is found on PD1 after an extremely short time. Consequently, the oxidized PSII RC state is P680+, meaning that the hole is on the PD1 pigment, which in turn is the RC cofactor closest to the Yz tyrosine (Fig. 1) and is capable of its efficient oxidation at physiological temperatures.
The only study known to us that has experimentally evaluated the ratio of LHC II fluorescence quenching efficiency by oxidized (kp+) and open (kp) reaction centers is the work of Renger et al. [16]. To determine the kp+/kp ratio, they measured the curve of fluorescence yield changes for PSII membrane fragments treated with hydroxylamine. After such treatment, electron transfer from Yz to P680+ is inhibited, and therefore the P680+ reduction rate is slowed to 150-200 µs. This fact allowed studying P680+ quenching properties in the nanosecond to microsecond time range. The authors [16] used a 10 ns laser flash with λex = 532 nm as the actinic light. They found that immediately after the actinic laser flash, an instantaneous decrease in antenna fluorescence yield is observed, followed by a two-component increase with times of 4.7 and 160 µs. The fast kinetics were attributed to the disappearance of the 3Car quencher [17], and the slow kinetics to recombination between P680+ and QA– [18]. This time (160 µs) coincides with the P680+ lifetime measured by a decrease in absorption in the 830 nm band. To describe the induction curve, they [16] proposed a three-quencher model for ΦII(t) antenna fluorescence quenching as follows:
In this equation, kf is the Chl fluorescence rate constant, kΣ the total rate constant for all non-radiative processes, and the remaining three members of the denominator are multiplications of rate constants by the concentrations of the respective states that describe photochemical (kp[QA(t)]) and non-photochemical (kcar[3Car] and kp+[P680+(t)]) fluorescence quenching. This equation is valid for describing an induction curve after a short pulse PSII complex excitation, when NPQ processes in antenna complexes do not have time to develop.
With the three-quencher model, Renger et al. [16] obtained the ratio of kp+/kp ≈ 1.7 for membrane fragments, and for intact Arabidopsis thaliana leaves a satisfactory computational modeling of the induction curve was achieved on the assumption of the ratio kp+/kp ≈ 2. Evidently, they [16] obtained their results in an indirect experiment – they introduced the kp+/kp parameter for modeling the induction curve most accurately.
Thus, there is a clear understanding of the fact that the PSII RC cation radical, P680+, participates in LHC II fluorescence NPQ processes, but there is no reliable data on the efficiency of this process. The reason is the very high recovery rate of P680+ from the redox-active Yz tyrosine residue (~40 ns), which greatly complicates LHC II fluorescence measurements under conditions where the photoactive RC pigment is oxidized. Therefore, for measuring PSII antenna fluorescence decay kinetics with the oxidized photoactive reaction center pigment, P680+, an experimental approach needs to be developed that will allow antenna fluorescence measurements after a very short time (≤10 ns) following the actinic light. For this purpose, we used the technique of fluorescent pump-probe analysis (picosecond flash photolysis) that allowed measuring PSII complex fluorescence decay kinetics under conditions where the RC photoactive pigment is in the neutral P680 state or in the oxidized P680+ state.
MATERIALS AND METHODS
PSII core complexes and membrane fragments from spinach were isolated as described by Haag et al. [19] and Schiller and Dau [20], respectively.
For obtaining PSII samples lacking the manganese cluster, core PSII complexes (0.5 mg of Chl a in 1 ml) were incubated in medium containing 0.9 M Tris-HCl, pH 9.0, for 30 min at 23°C and then washed twice in 25 mM Hepes, pH 7.5, containing 20 mM NaCl and 300 mM sucrose [21]. The samples were concentrated by ultrafiltration to 1.5-2.0 mg of Chl a in 1 ml, frozen in liquid nitrogen, and stored at –80°C. All measurements on samples were carried out at room temperature.
Light-induced absorption changes in the near infrared spectral region (820 nm) were detected by a single-beam differential spectrophotometer constructed in the Institute of Physico-Chemical Biology, Moscow State University. Probing light from a KGM-98 lamp passed a Jobin Yvon HL-1 monochromator (France), a sample cell, a SZS-21 glass light filter, a second UM-2 monochromator, and then impinged on a PMT-128 photomultiplier. The signal then went through an operational amplifier to a GAGE-8012 transient recorder connected to a computer. As an excitation light source, a Quantel laser was used (wavelength 532 nm, pulse half-width 12 ns, energy 50 mJ).
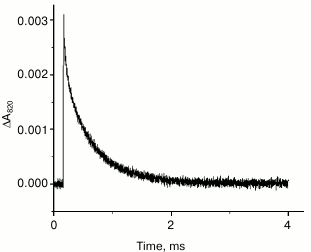
Figure 2 shows the kinetics of A820 changes in PSII complexes lacking the Mn4 cluster to be monoexponential, and its duration is ~150 µs. This time is very close to the result obtained in [16] for samples treated with hydroxylamine.
Fig. 2. Kinetics of absorption changes at 820 nm in PSII core complexes lacking the Mn4 cluster in 25 mM MES-NaOH, pH 6.5, containing 15 mM NaCl, 10 mM CaCl2, and 0.03% dodecylmaltoside. Chlorophyll concentration, ~10 µg/ml; excitation at 532 nm; pulse energy 50 mJ. The kinetic trace is a result of averaging 16 measurements.
For measuring PSII antenna complex fluorescence decay kinetics under the conditions where the photoactive RC pigment is in the neutral P680 or oxidized P680+ state, a modified version of a picosecond impulse fluorometer was used that was constructed at the Department of Biophysics, Biological Faculty of Moscow State University (Fig. 3) [22].
Fig. 3. Optical scheme of the experimental setup: 1) beam-splitting plate; 2) shutter; 3) mirror; 4) FEL-600 cutoff filter.
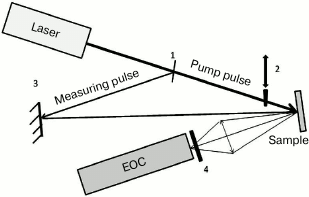
A sample was put in a cell with 2-mm pathlength and excited by light pulses (λex = 532 nm, duration ≈20 ps) obtained by the second harmonic of a PICAR-1 Nd:YAG laser (MSU, Russia) [23]. The laser pulse repetition frequency was 1 Hz, pulse energy ~2 µJ, stability of laser pulse generation with regard to energy and duration was over 95%, diameter of illuminated surface ~3 mm, and the energy absorbed by the sample did not exceed 4·1013 photons/cm2 per pulse. Sample optical density was 0.1, the sample was continuously stirred, and the cell was placed in a volume closed from external light. Under such conditions, the PSII sample was already back in a dark state in the time for the arrival of a next exciting light pulse. Fluorescence decay kinetics were recorded by an Agat SF3 electron-optical converter (EOC) (All-Russian Research Institute for Optical and Physical Measurements, Russia) coupled with a DAS C7041 multichannel array detector (Hamamatsu, Japan) cooled to –10°C. Through a C7557 controller (Hamamatsu), the detected signals were entered into the computer. To isolate the fluorescence signal, a FEL600 cutoff filter (Torlabs, USA) was placed between the EOC and the sample, permitting passage of light >600 nm. To improve the signal/noise ratio, 50 kinetics were accumulated and averaged. For measuring fluorescence decay kinetics of PSII samples with oxidized (P680+) RC, we used intense pulse sample pre-illumination. For this, a single light pulse (pump pulse) with duration of 20 ps, wavelength of 532 nm, and energy of 200 µJ, 8 ns ahead of the measuring pulse, was also applied to the sample cell. That impulse caused ~100% oxidation of P680. Then, fluorescence decay kinetics were measured with a weak measuring pulse. A controller-operated shutter was placed in the path of the pump pulse. The measurement procedure was similar to that used for analyzing transitional pump-probe absorption characteristics. Both light pulses (the pump pulse and the measuring pulse) passed the same sample area, with the pump pulse diameter being much larger than that of the measuring pulse.
The optical scheme of the described fluorometer (Fig. 3) allows working in two modes: 1) the pump pulse may be blocked by the shutter, and the required number of measuring light fluorescence kinetics traces can be measured. In this optical configuration, antenna fluorescence kinetics is obtained under conditions where RCs are in the open [P680QA] state; 2) when the shutter is open, the sample is excited by two pulses – a powerful pump pulse and, after 8 ns, a measuring pulse whose signal is detected as fluorescence kinetics. With this experimental setup, LHC II fluorescence decay kinetics is registered when the light-oxidized RC pigment is in the oxidized [P680+QA–] state. In the experiment, we successively measured fluorescence kinetics with a pump pulse and without it; therefore, having recorded 100 kinetics, we obtained 50 traces for the situation where RCs were in the open state and 50 traces for oxidized RCs. The experimental kinetic traces were approximated by a biexponential theoretical curve I(t) = A1exp(–t/τ1) + A2exp(–t/τ2) convoluted with the pulse fluorometer instrument function. In the given formula, τ1, τ2 and A1, A2 are the durations and normalized amplitudes for fluorescence decay kinetic components, A1 + A2 = 1. The accuracy of the fast component duration measurement was ~5 ps.
The main limitation of the Agat SF3 EOC compared to, for example, the single photon counting method, is low dynamic range ≤100. Therefore, at high EOC scanning speeds ~1 ns/cm only the two fastest fluorescence kinetic components are registered with sufficient accuracy. On the other hand, the calculation of the parameters of the fastest components, obtained by single photon counting, is also based on a routine deconvolution procedure and depends on the accuracy of the registration of the instrumental function of the device. For that reason, we measured the full kinetics of fluorescence decay by the single photon counting method in testing experiments and used the τ1 parameter obtained from operating the EOC for model reconstruction.
For single photon counting, a Simple Tau 140 (Becker&Hickl, Germany) picosecond measuring complex was used, equipped with a 16 channel multi-anode PMT PML-16 (Becker&Hickl) with a diffraction grating (1200 lines/mm, a spectral resolution of 6.25 nm/channel). Fluorescence was excited with a LDN-405 pulsed LED laser (InTop, Russia) with wavelength of 405 nm, pulse duration 25 ps, pulse rate 25 MHz, and single pulse energy 13 pJ.
To determine the value of the kp+/kp ratio, the known relation of quantum yield value φ and fluorescence duration τ (φ = kf·τ) was used, as well as Eq. (1). PSII antenna complex fluorescence duration was measured for two extreme cases, when all RCs were either in the open [P680QA] or in the oxidized [P680+QA–] state. Therefore, the measured fluorescence duration τ1 = 1/k1, determined by the denominator value in Eq. (1), equals 1/(kf + kΣ + kcar + kp) for open RCs and 1/(kf + kΣ + kcar + kp+) for oxidized RCs. From this, expressions for determining the required values are obtained: kp = 1/τ1 – (kf + kΣ + kcar); kp+ = 1/τ1+ – (kf + kΣ + kcar). Here, τ1 and τ1+ are the fast fluorescence component durations measured under the conditions where all RCs are either in the open or in the oxidized state, respectively. The values of rate constants given in the parentheses were taken from [16, 17], according to which kf = (15 ns)–1, kΣ = (6 ns)–1, and kcar = (0.6 ns)–1. The error in determining the α = kp+/kp ratio can be estimated from the accuracy, Δτ1, of calculating τ1: Δα = Δτ1·kp+(1 + kp+/kp). With the τ1 measuring accuracy of 5 ps (95% confidence interval), the value of α = kp+/kp ≈ 1.50 ± 0.08.
RESULTS AND DISCUSSION
Typical fluorescence decay kinetics of native PSII core complexes containing the Mn4 cluster and capable of O2 release, taken by the Agat SF3 EOC, are given in Fig. 4.
Fig. 4. Typical fluorescence decay kinetics registered by the EOC. Theoretical curves are obtained by a two-exponential approximation I(t) = a1exp(–t/τ1) + a2exp(–t/τ2). The a1, a2, τ1, and τ2 values are given in Table 1. The incubation medium contained 50 mM MES, pH 6.5, 15 mM NaCl, and 300 mM sucrose.
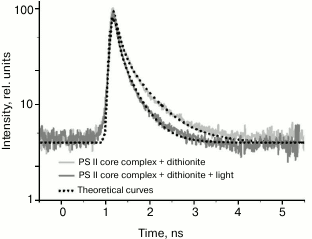
Table 1. Kinetic parameters for
approximation of experimental kinetics of PSII complex fluorescence
decay recorded by the EOC; a1, a2
and τ1, τ2 are values of amplitudes and
durations for the fast and slow components. The
k+p/kp value
characterizes the relative efficiency of excitation capture by PSII
reaction center in the P680+ and P680 states
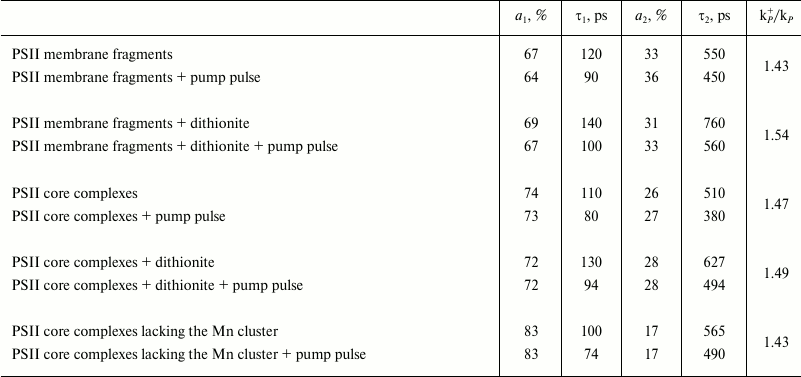
Modeling these kinetics in a biexponential approximation yielded the following results. In the case of dark-adapted samples (the [P680QA] state), the durations of components were τ1 = 110 ps, τ2 = 510 ps, and the contributions of the A1 and A2 components were 74 and 26%, respectively. These durations are in good agreement with the results obtained earlier, according to which for PSII core complexes with open RCs, τ1 = 40-100 ps, τ2 = 200-500 ps [24, 25]. A small difference in τ1 and τ2 durations towards longer lifetimes obtained in our study compared to the quoted papers may be easily attributed to the difference in the analyzed core complexes and instrumental approaches. During the oxidation of the photoactive pigment by an early pump pulse, a significant shortening of τ1 and τ2 was observed, down to 80 and 380 ps, respectively. To modify the RC and test the trend of changing τ1 and τ2, dithionine-treated PSII core complexes were studied. As it is known, dithionite reduces the quinone acceptor QA in the dark. Therefore, by using only the measuring light, we detected PSII antenna fluorescence decay kinetics, when the RC was in the [P680QA–] state. In the second variation of measurements, where we used an early pump pulse, the RC was in the [P680+QA–] state. Apparently, we compared the efficiency of photochemical (P680) and non-photochemical (P680+) quenching of antenna Chl fluorescence by neutral and oxidized RC in the presence of dithionite as well. The results of the measurements are given in Table 1. It appears that in dark-adapted samples in the presence of dithionite, a certain increase in τ1 and τ2 is observed, up to 130 and 627 ps, respectively. An increase in lifetimes was to be expected, as electron transfer from P680* to Pheo in charge separation reaction occurs against the gradient of the electric field generated by the excess electron on QA, which appears as a result of dark reduction of the primary quinone by dithionite. At the same time, upon pre-illuminating a sample with a pump pulse both in the presence and in the absence of dithionite, τ1 and τ2 are reduced almost in equal parts, down to 94 and 494 ps. Apparently, in this case as well, oxidized P680 is an effective non-photochemical quencher of excited states of antenna Chl molecules. It was shown earlier [26, 27] that under reducing conditions (QA–), after charge separation in PSII RC, the [P680+Pheo–QA–] state is formed, whose transitions into the original [P680PheoQA–] state occurs in 2-4 ns, emitting a quantum of recombination fluorescence (P680+Pheo– → P680*Pheo → P680Pheo + fluorescence quantum). Apparently, if charge recombination occurs with 100% probability, then virtually all RCs should be back in the original [P680PheoQA–] state by the arrival of the measuring pulse. Therefore, no shortening of τ1 and τ2 should have occurred. The shortening of τ1 and τ2 that we found indicates the charge recombination to be a minor channel of [P680+Pheo–] state decay. Apparently, electron transfer from Pheo– to QA– is more effective, causing a two-electron reduction of the quinone acceptor. In this case, the photoactive RC pigment is in the oxidized P680+ state by the arrival of the measuring light pulse, and it carries out non-photochemical fluorescence quenching, causing a decrease in its duration.
Similar results were obtained in experiments with other PSII complexes – membrane fragments and core particles lacking the Mn complex (see Table 1). It can be seen that for different kinds of measurements under the conditions when PSII RCs are in the oxidized P680+ state, the duration of fluorescence from antenna Chl shortens. However, for the three studied PSII complexes, the kp+/kp ratio varies little, remaining in the 1.43-1.54 range.
We noted that the dynamic range of EOC registration system is relatively low, not exceeding 100. Therefore, fluorescence decay kinetics were modeled by a biexponential approximation. To verify the data from the EOC, test measurements were taken for PSII complex fluorescence decay kinetics by single-photon counting. The results are given in Fig. 5 and Table 2. It should be noted that in this case, the kinetics were modeled by a three-exponential approximation, using the τ1 value from Table 1 obtained from analyzing the data registered by the EOC. The additional third component, obtained by modeling data from single-photon counting, describes P680+Pheo– charge recombination, as shown earlier [26, 28].
Fig. 5. Fluorescence decay kinetics of PSII dark-adapted core complexes (shown in black) and the same complexes in the presence of dithionite (shown in gray) registered by the single photon counting system. Fluorescence was excited by a laser pulse at 405 nm with duration of 25 ps and pulse rate of 25 MHz; dithionite concentration was 10 mM.
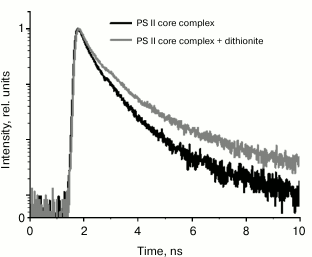
Table 2. Kinetic parameters for the
approximation of experimental kinetics of PSII complex fluorescence
decay obtained by the single-photon counting technique. The
τ1 parameter value was taken from Table 1, the data
were approximated with three components. a1,
a2, a3 and τ1,
τ2, τ3 are the values of amplitudes and
durations of the kinetic components, respectively. χ2 is
the standard deviation of the model curve from the experimental
data
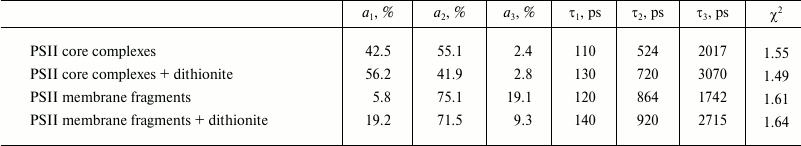
According to [28], the fluorescence decay kinetics are supposed to contain at least three components, reflecting electron excitation energy migration from antenna Chl to RC, charge separation and electron transfer to QA, as well as charge recombination in the primary ion-radical pair [P680+Pheo–]. Since these three PSII states are linked by reverse processes, each of the components will have mixed contributions from all three. To the fullest extent, this mixing affects the duration of the second component, while due to significant difference in lifetimes the first and third kinetic components remain virtually “pure”. In addition, the fastest component reflects electron excitation energy migration from antenna to RC, and the slowest reflects the charge recombination time in the P680+Pheo– pair. Therefore, to compare excitation quenching efficiency in PSII antenna, we analyzed changes in the fast component duration when the RCs were in the [P680QA] and [P680+QA–] states. In this case, we viewed P680 and P680+ as acceptors for excitation energy originating from antenna complexes (P680 is a cluster of four Chl molecules and two Pheo molecules, as described in the introduction). The results of the comparison are given in Table 1 as the kp+/kp ratio. The table shows that the fast component duration after PSII pumping by a powerful light pulse is significantly decreased. We suppose that this shortening occurs due to more efficient fluorescence quenching. It should be noted that in different PSII complexes the kp+/kp ratio is approximately the same, about 1.5. At first glance, it is surprising that the process of non-photochemical quenching by the P680+ cation radical is more effective than photochemical quenching. Indeed, during the oxidation of the special pair PD1PD2, a band at 830 nm appears in the stationary PSII absorption spectrum, which was the one attributed to P680+ absorption. The excitation migration time from antenna Chl to P680+ (~100 ps) implies that this process occurs via the Förster mechanism. Critical parameters for its realization are the distance between donor and acceptor R, orientation factor k2, and overlap integral for donor fluorescence and acceptor absorption spectra. The distance R can be assumed to not undergo significant changes during RC oxidation, while the k2 parameter is supposed to change. Unfortunately, changes in this parameter have not been reported by anyone to date. Nevertheless, since P680+ quenches antenna fluorescence [5-11, 16], the k2 parameter is not zero. For a long time, P680+ absorption was associated with a single band at 830 nm. Evidently, overlap of the antenna Chl fluorescence spectrum (λmax ~ 680 nm) with this band at 830 nm will be insignificant. However, detailed study of RC spectral properties in PSII RC core complexes [13] revealed bands at 675-684 nm in differential P680+Pheo– – P680Pheo and P680+QA– – P680QA spectra, as well as in T – S spectra, which effectively overlap with the core complex fluorescence spectrum. Apparently, the combination of the k2 parameter and the overlap integral provides high efficiency of antenna fluorescence quenching by the oxidized P680+ state of the PSII reaction center.
Thus, the efficiency of non-photochemical Chl fluorescence quenching by PSII RC radical cation was shown to be ~1.5 times higher than that of photochemical quenching. We have demonstrated a new approach for pulse fluorometry for direct measurement of photochemical and non-photochemical fluorescence quenching efficiency. By analogy with the method of picosecond pump–probe differential absorption spectroscopy, we used high-intensity pulse pre-illumination (pump pulse) of a sample, which preceded the measuring light pulse by 8 ns. This approach allows registering fluorescence decay kinetics for PSII samples with oxidized (P680+) reaction centers with picosecond time resolution. The analysis was carried out on different pigment–protein PSII complexes – membrane fragments, oxygen-evolving core complexes, and PSII core complexes lacking the Mn4 cluster. For all these three PSII complexes, the kp+/kp ratio value changed little, remaining in the 1.43-1.54 range (see Table 1). Given the error in determining the kp+/kp value, about 0.08 (see “Materials and Methods”), it can be assumed that kp+ ≈ 1.5kp. This value differs significantly from the data reported in [16], where kp+/kp ≈ 1.7 for PSII membrane fragments, and for whole Arabidopsis thaliana leaves [16] the reported value was kp+/kp ≈ 2. From our point of view, such a discrepancy is explained by the fact that induction curves in [16] were taken in the time range from 100 ns to 10 s, where fluorescence signal from PSII antenna Chl, excited by 10 ns actinic light pulse, is no longer detected (antenna Chl fluorescence kinetics is multicomponent, its duration does not exceed a few nanoseconds; see Table 2). The kp+/kp parameter was introduced in [16] for computational modeling of an induction curve and is not an experimentally determined value. We suppose that the efficiency ratio of photochemical and non-photochemical PSII fluorescence quenching efficiencies by open and closed RC states, measured in our direct experiment, is more realistic than that calculated in [16].
We are grateful to the Russian Foundation for Basic Research (V. Z. Paschenko, V. V. Gorokhov, B. N. Korvatovskii, E. G. Maksimov – projects Nos. 14-04-01536 and 15-29-01167; M. D. Mamedov – project No. 14-04-00519) for partial financial support of this study.
REFERENCES
1.Karapetyan, N. V. (2007) Non-photochemical
quenching of fluorescence in cyanobacteria, Biochemistry
(Moscow), 72, 1127-1135
2.Barber, J., and Andersson, B. (1992) Too much of a
good thing: light can be bad for photosynthesis, Trends Biochem.
Sci., 17, 61-66.
3.Demmig-Adams, B. (1990) Carotenoids and
photoprotection in plants: a role for xanthophyll zeaxanthin,
Biochim. Biophys. Acta, 1020, 1-24.
4.Horton, P., and Ruban, A. V. (2005) Molecular
design of the photosystem II light-harvesting antenna: photosynthesis
and photoprotection, J. Exp. Bot., 56, 365-373.
5.Butler, W. (1972) On the primary nature of
fluorescence yield changes associated with photosynthesis, Proc.
Natl. Acad. Sci. USA, 89, 3420-3422.
6.Allakhverdiev, S. I., Shafiev, M. A., and Klimov,
V. V. (1986) Effect of reversible extraction of manganese on
photooxidation of chlorophyll P680 in photosystem II
preparations, Photobiochem. Photobiophys., 12, 61-65.
7.Shinkarev, V. P., and Govindjee (1993) Insight into
the relationship of chlorophyll a fluorescence yield to the
concentration of its natural quenchers in oxygenic photosynthesis,
Proc. Natl. Acad. Sci. USA, 90, 7466-7469.
8.Ruban, A. V., and Horton, P. (1995) Regulation of
non-photochemical quenching of chlorophyll fluorescence in plants,
Austr. J. Plant Physiol., 22, 221-230.
9.Anderson, J. M., Park, Y.-I., and Chow, W. S.
(1998) Unifying model for the photoinactivation of photosystem II in
vivo under steady-state photosynthesis, Photosynth. Res.,
56, 1-13.
10.Ivanov, B., and Edvards, G. (2000) Influence of
ascorbate and the Mehler peroxidase reaction on non-photochemical
quenching of chlorophyll fluorescence in maize mesophyll chloroplasts,
Planta, 210, 765-774.
11.Bukhov, N. G., Heber, U., Wiese, C., and
Shuvalov, V. A. (2001) Energy dissipation in photosynthesis: does the
quenching of chlorophyll fluorescence originate from antenna complexes
of photosystem II or from the reaction center? Planta,
212, 749-758.
12.Semenov, A. Yu., Kurashov, V. N., and Mamedov, M.
D. (2011) Transmembrane charge transfer in photosynthetic reaction
centers: some similarities and distinctions, J. Photochem.
Photobiol. B, 104, 326-332.
13.Mamedov, M., Govindjee, Nadtochenko, V., and
Semenov, A. (2015) Primary electron transfer processes in
photosynthetic reaction centers from oxygenic organisms, Photosynth.
Res., 125, 51-63.
14.Raszewski, G., Diner, B. A., Schlodder, E., and
Renger, T. (2008) Spectroscopic properties of reaction center pigments
in photosystem II core complexes: revision of the multimer model,
Biophys. J., 95, 105-110.
15.Renger, G., and Renger, T. (2008) Photosystem II:
the machinery of photosynthetic water splitting, Photosynth.
Res., 98, 53-80.
16.Steffen, R., Eckert, H.-J., Kelly, A. A.,
Dormann, P., and Renger, G. (2005) Investigation on the reaction
pattern of photosystem II in leaves from Arabidopsis thaliana by
time-resolved fluorometric analysis, Biochemistry, 44,
3123-3133.
17.Schodel, R., Irrgang, K.-D., Voigt, I., and
Renger, G. (1999) Quenching of chlorophyll fluorescence by triplets in
solubilized light-harvesting complex II (LHCII), Biophys. J.,
79, 2238-2248.
18.Eckert, H.-J., Geiken, B., Bernarding, J.,
Napiwotzki, A., Eichler, H.-J., and Renger, G. (1991) Two sites of
photoinhibition of the electron transfer in oxygen evolving and
Tris-treated PS II membrane fragments from spinach, Photosynth.
Res., 27, 97-108.
19.Haag, E., Irrgang, K. D., Boekema, E. J., and
Renger, G. (1990) Functional and structural analysis of photosystem II
core complexes from spinach with high oxygen evolution capacity,
Eur. J. Biochem., 189, 47-53.
20.Schiller, H., and Dau, H. (2000) Preparation
protocols for high-activity photosystem II membrane particles of green
algae and higher plants, pH dependence of oxygen evolution and
comparison of the S2-state multiline signal by X-band EPR spectroscopy,
J. Photochem. Photobiol. B, 55, 138-144.
21.Gopta, O. A., Tyunyatkina, A. A., Kurashov, V.
N., Semenov, A. Yu., and Mamedov, M. D. (2008) Effect of redox
mediators on the flash-induced membrane potential generation in
Mn-depleted photosystem II core particles, Eur. Biophys. J.,
37, 1045-1050.
22.Volgusheva, A. A., Zagidullin, V. E., Antal, T.
K., Korvatovskii, B. N., Krendeleva, T. E., Paschenko, V. Z., and
Rubin, A. B. (2007) Examination of chlorophyll fluorescence decay
kinetics in sulfur derived algae Chlamydomonas reinhardtii,
Biochim. Biophys. Acta, 1767, 559-564.
23.Gorbunkov, M. V., Shabalin, Y. V., Konyashkin, A.
V., Kostryukov, P. V., Olenin, A. N., Tunkin, V. G., Morozov, V. B.,
Rusov, V. A., Telegin, L. S., and Yakovlev, D. V. (2005)
Pulsed-diode-pumped, all-solid-state, electro-optically controlled
picosecond ND:YAG lasers, Quantum Electronics, 35,
2-6.
24.Vassiliev, S., Lee, C. I., Brudvig, G. W., and
Bruce, D. (2002) Structure-based kinetic modeling histidine-tagged
photosystem II core complexes from Synechocystis,
Biochemistry, 41, 12236-12243.
25.Tian, L., Farooq, S., and Amerongen, H. (2013)
Probing the picosecond kinetics of the photosystem II core complex
in vivo, Phys. Chem. Chem. Phys., 15,
3146-3154.
26.Klimov, V. V., Allakhverdiev, S. I., and
Paschenko, V. Z. (1978) Activation energy and lifetime changes of PSII
chlorophyll fluorescence, Proc. USSR AS, 242,
1204-1207.
27.Klimov, V. V., Klevanik, A. V., Shuvalov, V. A.,
and Krasnovsky, A. A. (1977) Reduction of pheophytin in the primary
reaction of photosystem II, FEBS Lett., 829, 183-186.
28.Schatz, G. H., Brock, H., and Holzwarth, A. R.
(1988) Kinetic and energetic model for the primary processes in
photosystem II, Biophys. J., 54, 397-405.