CELSR1 Is a Positive Regulator of Endothelial Cell Migration and Angiogenesis
Yi-Hong Zhan1,2#, Qi-Cong Luo3#, Xiao-Rong Zhang2, Nai-An Xiao1, Cong-Xia Lu1, Cen Yue1, Ning Wang4*, and Qi-Lin Ma1*
1The First Affiliated Hospital of Xiamen University, Department of Neurology, 361003 Xiamen, China; E-mail: qilinma@yeah.net2Fujian Medical University, Center of Neuroscience, The First Affiliated Hospital, The First Clinical Medical College, 350005 Fuzhou, China
3The First Affiliated Hospital of Xiamen University, Center for Laboratory, 361003 Xiamen, China
4Fujian Medical University, Center of Neuroscience, The First Affiliated Hospital, Department of Neurology and Institute of Neurology, 350005 Fuzhou, China; E-mail: ningwang@mail.fjmu.edu.cn
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received January 28, 2016
Cadherin is an epidermal growth factor and laminin-G seven-pass G-type receptor 1 (CELSR1) is a key component of the noncanonical Wnt/planar cell polarity (PCP) pathway that critically regulates endothelial cell proliferation and angiogenesis. In this study, we examined the biological significance of CELSR1 in endothelial cell migration and angiogenesis. For this, we applied both gain-of-function and loss-of-function approaches. To increase the endogenous expression of CELSR1, we used the transcription activator-like effector (TALE) technology and constructed an artificial TALE-VP64 activator. To knock down the expression of CELSR1, we generated lentivirus containing short hairpin RNA sequences targeting different regions of CELSR1 mRNA. Following up- or down-regulation of CELSR1 in human aortic endothelial cells (HAEC), we assessed in vitro cell proliferation by MTT assay, migration by scratch and transwell migration assays, and angiogenesis by tube formation analysis. We found that CELSR1 was endogenously expressed in human umbilical vein endothelial cells (HUVEC) and HAEC. When focusing on HAEC, we found that upregulating CELSR1 expression significantly promoted cell growth, while knocking down CELSR1 inhibited the growth (p < 0.05). Using both scratch and transwell migration assays, we observed a positive correlation between CELSR1 expression and cell migratory capability. In addition, CELSR1 upregulation led to higher levels of tube formation in HAEC, while downregulating CELSR1 expression decreased tube formation (p < 0.05). Mechanistically, CELSR1-regulated migration and tube formation was mediated through disheveled segment polarity protein 3 (Dvl3). In conclusion, CELSR1 plays an important role in regulating multiple phenotypes of endothelial cells, including proliferation, migration, and formation of capillary-like structures.
KEY WORDS: CELSR1, endothelial cells, migration, angiogenesisDOI: 10.1134/S0006297916060055
Cadherin epidermal growth factor and laminin-G seven-pass G-type receptor 1 (CELSR1) is a large 35-exon gene on human chromosome 22q13.31. It encodes a cadherin protein with an atypical combination of a seven-transmembrane domain coupled to a large, extracellular, modular region containing epidermal growth factor-like and laminin-G like domains crowned by nine protocadherin repeats [1]. The structure of CELSR1 enables the molecule to self-recognize across neighboring cells in addition to traditional ligand–receptor interactions [1]. CELSR1 is a key component of the noncanonical Wnt/planar cell polarity (PCP) pathway, the latter critically regulating development, morphogenesis, and polarity of various tissues [2, 3]. Recent evidence suggests that the noncanonical Wnt/PCP pathway also regulates endothelial cell proliferation and angiogenesis [4-7], yet no studies have been carried out on the significance of CELSR1 in angiogenesis. The initial link suggesting the potential involvement of CELSR1 in vascular biology was first revealed by a genome-wide association study demonstrating CELSR1 as a susceptibility gene for ischemic stroke (IS) in Japanese individuals [8], which was later confirmed in Portuguese IS patients [9] and recently by our group among the Chinese Han population [10]. Besides being expressed in hair, inner ear, and central nervous system [11, 12], CELSR1 expression has also been identified in lymphatic endothelial cells in a recent study [13]. However, it is still not clear whether CELSR1 is also expressed in other vascular endothelial cells including arterial and venous endothelial cells and if CELSR1 directly regulates endothelial behaviors such as proliferation or migration and functionally modulates angiogenesis. To address these questions, we set up in vitro analyses to assess the role of CELSR1 in proliferation, migration, and tubulogenesis of vascular endothelial cells.
MATERIALS AND METHODS
Cells and reagents. Human umbilical vein endothelial cells (HUVEC) and human aortic endothelial cells (HAEC) were purchased from Cascade Biologics (USA) and cultured in M200 medium supplemented with 2% Low Serum Growth Supplement (LSGS). The cells were maintained as monolayer cultures at 37°C in a humidified atmosphere of 5% CO2. Human breast carcinoma MDA-MB231 cells and human glioma U251 cells were provided by Dr. Li Boan (Xiamen University, Xiamen, China) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS).
Plasmids. Upregulation of CELSR1 expression was achieved using the artificial transcription activator-like effector (TALE) technology [14], where a construct (TALE-CELSR1) containing TALE repeats fused to a nuclease (TALEN) domain that specifically targets the sequence TTCTGAAAGCTCTAAGGT within human CELSR1 promoter, a heterologous transcriptional activation domain VP64, and a puromycin-resistant gene, was custom-designed by and obtained from Viewsolid Biotech (China). To stably enhance the endogenous expression of CELSR1, TALE-CELSR1 was transfected into HAEC using Lipofectamine 2000 (Invitrogen, USA) following the manufacturer’s instructions. Transfection was followed by selection using puromycin (1 µg/ml) for two to seven days. Cells transfected with the blank vector were used as controls.
Two short hairpin RNA molecules (shRNA) specifically targeting human CELSR1 (shRNA1: CCGGAGGAGGTGCAACCTGTATATACTCGAGTATATACAGGTTGCACCTCC TTTTTTG, shRNA2: CCGGACAACGGCATCCCGCAGAAATCTCGAGATTTCTGCGGGATGCCGTTGTTTTTTG) and a LacZ-specific shRNA molecule (control, CCGGCGTACGCGGAATACTTCGACTCGAGTCGAAGTATTCCGCGTACGTTTTTG) were subcloned into the pLKO.1 lentiviral RNA interference (RNAi) vector.
The disheveled segment polarity protein 3 (Dvl3)-expressing vector was kindly provided by Dr. Lin Li, and the lentiviral vector expressing Dvl3 shRNA was purchased from GeneChem (China).
Lentivirus production and infection of HAEC. The lentiviral vector containing either CELSR1-specific (LV-CELSR1 shRNA) or LacZ-specific (LV-LacZ shRNA) shRNA was cotransfected with packaging vectors pHR and pVSV-G into 293T using Lipofectamine 2000. At 48 h after cotransfection, the virus-containing supernatant was collected and added together with 10 µg/ml Polybrene onto HAECs at 50 to 70% confluence for 24 h. The medium was then replaced with fresh HAEC growth medium for another 48 h before puromycin was added at 1 µg/ml for seven days.
Quantitative reverse transcription PCR (RT-qPCR). Total RNA was extracted from HAEC using Trizol reagent (Invitrogen) and reverse transcribed into cDNAs using a ReverTra Ace qPCR RT kit (Toyobo, Japan). Real-time PCR was performed using the following primers: CELSR1 forward, 5′-CGCTTCCACTTCACCATCTCCCT-3′; CELSR1 reverse, 5′- GCCACGGTCGTTGTTGTCTCG-3′; 18S RNA forward, 5′-GCGGCTTAATTTGACTCAACAC-3′; and 18S RNA reverse, 5′-GGCCTCACTAAACCATCCAATC-3′. The expression levels of CELSR1 mRNA were normalized against 18S RNA levels, and the results were calculated using the 2–ΔΔCT method as previously described [15].
Western blot. Cells were lysed in RIPA buffer containing the protease inhibitor phenylmethylsulfonyl fluoride (PMSF) at a final concentration of 1 mM. Total protein concentration was measured using the BCA assay (Thermo Scientific, USA). A 30-µg total protein sample was run through SDS-PAGE gel and subsequently transferred onto PVDF membrane. After blocking with 5% milk in phosphate buffered saline with Tween (PBST), membranes were incubated with anti-CELSR1 antibody (Santa Cruz, USA) or anti-β-actin (internal control) (Sigma Aldrich, USA) at 4°C overnight, followed by a 1 h incubation with HRP-conjugated goat-anti-mouse IgG at room temperature (Pierce Biotechnology, USA). ECL substrate reagents were used for signal development.
MTT assay. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed as previously described [16]. Briefly, cells in exponential growth phase were seeded in triplicate into 96-well plates at 5000 cells/well. Cells were cultured in complete growth media for one to four days. To measure the number of living cells, 20 µl of MTT solution (5 mg/ml) was added to each well and incubated in the dark at 37°C for 4 h. The supernatant was then removed, and dimethyl sulfoxide (DMSO) (150 µl/well) was added to dissolve formazan crystals. The optical density (OD) in each well was then measured at 490 nm. The growth inhibition (%) was calculated as (1 – ODtreated/ODcontrol) × 100%. Three independent experiments were performed for each condition.
Scratch assay. For endothelial migration experiments, 5·105 cells were seeded in a 35-mm cell culture dish with lines marked at the plate bottom. When cells became confluent after overnight culture, a scratch was made in the confluent cell monolayer using pipet tips (0.2-10 µl) followed by PBS wash. Cells were cultured in media at 37°C without serum, and cell migration into the scratched area was documented after 10 h. Images were taken at indicated time points on the scratched positions, and three images from each plate were analyzed. Three independent experiments were performed for each condition.
Migration assay. The transwell migration assay was performed as previously described [17]. Briefly, 2·105 cells were loaded on top of the filter membrane of the transwell insert. Growth medium supplemented with 2% LSGS was added to the lower well. After a 7-h incubation at 37°C with 5% CO2, the transwell insert was removed from the plate. A cotton-tipped applicator was used to carefully remove the medium and non-migrating cells from the top of the membrane without damaging it. The transwell insert with migrated cells was placed into methanol for 10 min to fix the cells. After methanol evaporation at room temperature, the transwell insert was placed into 0.1% Crystal violet for 30 min. After removing excessive Crystal violet, the migrated cells were imaged under an inverted microscope (100×), and the number of cells from five different fields (upper, lower, right, left, and center) of view was quantified to get an average sum of cells that migrated through the membrane. Three independent experiments were performed for each condition.
Tube formation assay. The Matrigel tube formation assay was performed following the instructions provided by BD Biosciences (USA). Briefly, 0.1 ml growth factor-reduced Matrigel (BD Biosciences) was seeded into 96-well plate and solidified at 37°C for 1 h. Cells from different groups were then seeded at 20,000 cells/well on top of the Matrigel in complete HAEC growth medium. After 18 h of incubation at 37°C, images of each well were taken at 4× magnification using a LEICA DMIL LED microscope with a Leica DFC295 camera (Leica Microsystems, USA). Each condition was assessed in triplicate. Tube formation was quantified using Wimasis Image Analysis (http://www.wimasis.com), which measured the total tube length (pixels, px), and the total number of tubes, loops, and branching points.
RESULTS
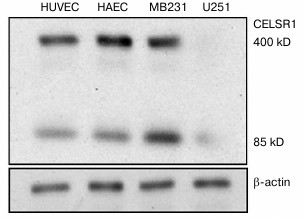
CELSR1 expression is detectable in vascular endothelial cells. The endogenous expression of CELSR1 has been detected in different human cell types including lymphatic endothelial cells, but not yet in other vascular endothelial cells [11, 13]. In this study, we first examined the expression of CELSR1 in HAEC and HUVEC. Western blot analysis indicated that CELSR1 is expressed in HAEC and HUVEC at a level comparable to that in the positive control MDA-MB231 cells and significantly higher than in the negative control U251 cells (Fig. 1). In addition to the full-length CELSR1 protein at 400 kDa, there was an approximately 85-kDa protein recognized by the anti-CELSR1 antibody, which is consistent with a previous report that this 85-kDa fragment was generated by an unknown proteolytic event of the full-length CELSR1 on the cell surface [1].
Fig. 1. CELSR1 is expressed in vascular endothelial cells HAEC and HUVEC. The expression of CELSR1 in HAEC and HUVEC was detected by Western blot, and MDA-MB231 and U251 cells were used as positive and negative control for CELSR1 expression, respectively. The expression of β-actin was used as internal control.
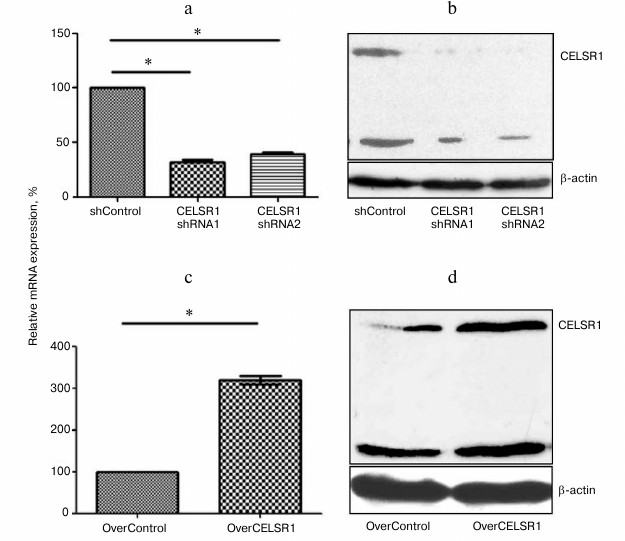
CELSR1 is successfully upregulated by TALE-CELSR1 and inhibited by shRNA. To evaluate the biological significance of CELSR1 in angiogenesis, we focused on HAEC cells and adopted both loss-of-function and gain-of-function approaches. For loss-of-function, we generated lentivirus expressing shRNA specifically targeting the CELSR1 gene. For the gain-of-function, we upregulated the endogenous expression of CELSR1 using TALE technology [14]. RT-qPCR analysis of HAEC stably infected with lentivirus expressing CELSR1 shRNA1 and CELSR1 shRNA2 demonstrated a significant decrease in the steady-state mRNA level of CELSR1 (CELSR1 shRNA1, (0.32 ± 0.03)-fold; CELSR1 shRNA2, (0.40 ± 0.03)-fold) compared to shControl (p < 0.05) (Fig. 2a). Western blot analysis of CELSR1 shRNA1 and CELSR1 shRNA2 from stably transfected HAEC also confirmed a significant decrease in CELSR1 protein level (Fig. 2b). In contrast, compared to the HAEC transfected with control vector (OverControl), TALE-CELSR1 significantly increased CELSR1 expression at both the mRNA level ((3.19 ± 0.10)-fold; p < 0.05) compared to OverControl, and the protein level (p < 0.05) (Figs. 2c and 2d, respectively).
Fig. 2. Upregulation and downregulation of endogenous CELSR1 level was successfully achieved in HAEC. The expression of CELSR1 in HAEC infected with lentivirus expressing LacZshRNA (shControl) or CELSR1 shRNA (CELSR1 shRNA1 and shRNA2) (a, b) or transfected with OverControl or TALE-CELSR1 (c, d) was measured by real-time PCR (a, c) and Western blot (b, d). The expression of β-actin was used as internal control. Three independent experiments were performed; * p < 0.05, compared to the corresponding control cells.
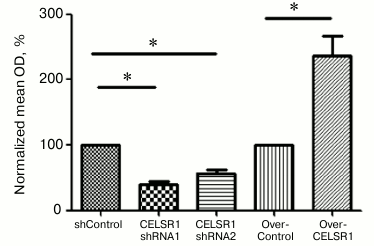
CELSR1 is an essential regulator of HAEC proliferation. Endothelial proliferation and migration are integral processes for angiogenesis. Therefore, we focused on these two processes to assess the significance of CELSR1 for angiogenesis. Using the MTT assay, we found that CELSR1 is necessary and sufficient to drive HAEC proliferation (Fig. 3). When the CELSR1 level was reduced by shRNA1 and shRNA2, HAEC proliferation decreased (0.40 ± 0.05 and 0.57 ± 0.06, respectively; p < 0.05), relative to shControl cells. In contrast, upregulating CELSR1 promoted proliferation ((2.37 ± 0.29)-fold; p < 0.05, compared to OverControl cells).
Fig. 3. CELSR1 is necessary and sufficient for HAEC proliferation. The proliferation of HAEC with altered CELSR1 expression was examined by MTT assay. Three independent experiments were performed; * p < 0.05, compared to the corresponding control cells.
CELSR1 is a critical regulator for HAEC migration. To determine the effects of altering CELSR1 on endothelial cell migration, we applied both scratch assay and transwell migration analyses. The scratch assay showed that reducing CELSR1 level in HAEC significantly slowed migration, while overexpressing CELSR1 dramatically enhanced migration of the endothelial cells compared to the corresponding control cells (p < 0.001; Fig. 4). Similar results were obtained using the transwell migration analysis (Fig. 5). These data suggests that CELSR1 is a critical regulator of HAEC migration.
Fig. 4. CELSR1 is an essential regulator of HAEC migration as detected by scratch assay analysis. The migratory behavior of HAEC with downregulated CELSR1 level (a, b) and upregulated CELSR1 level (c, d) was examined by the scratch assay. Representative images of the cells from each group at 0 and 48 h after the scratch are shown on the left (a, c), and quantification of the scratch closure from three independent experiments is shown on the right (b, d). ** p < 0.001, compared to the corresponding control cells.
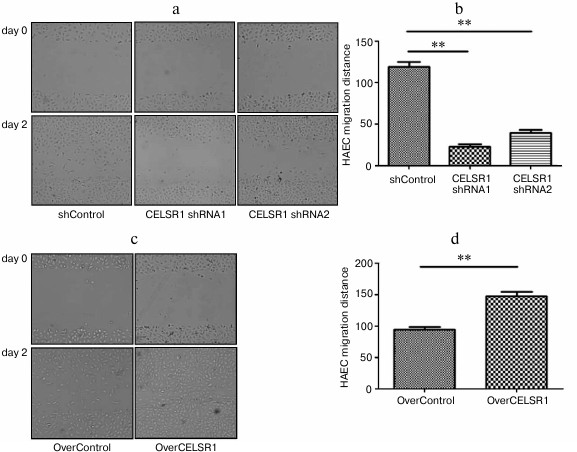
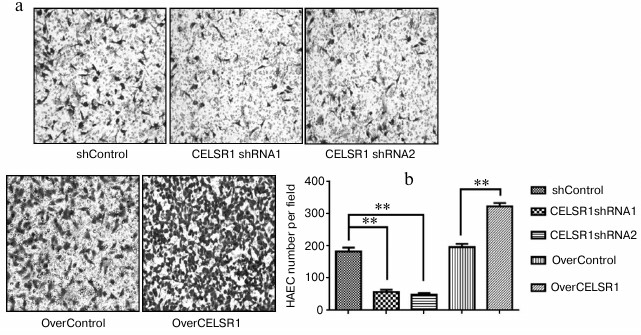
Fig. 5. CELSR1 is an essential regulator of HAEC migration as detected by the transwell migration assay. The migratory behavior of HAEC with downregulated or upregulated CELSR1 level was examined by the transwell migration assay. Representative images of the transwell insert from each group after migration are shown on the left (a), and the quantification of the number of migrated cells per field from three independent experiments is shown on the right (b). ** p < 0.001, compared to the corresponding control cells.
CELSR1 promotes endothelial tube formation. We investigated HAEC tubulogenesis on the extracellular matrix as a functional consequence of endothelial proliferation and migration. The shControl and OverControl cells showed similar levels of tube formation as represented by total tube length in pixels and total number of branching points, loops, and tubes after 18 h growth on Matrigel. This suggests that infection with lentivirus or transfection with TALE vector did not affect tube formation in the endothelial cells. The knockdown of CELSR1 in HAEC, however, significantly reduced tubulogenesis compared to shControl cells, while upregulating CELSR1 expression in HAEC dramatically stimulated tubulogenesis compared to the OverControl cells (p < 0.05; Fig. 6, a and b).
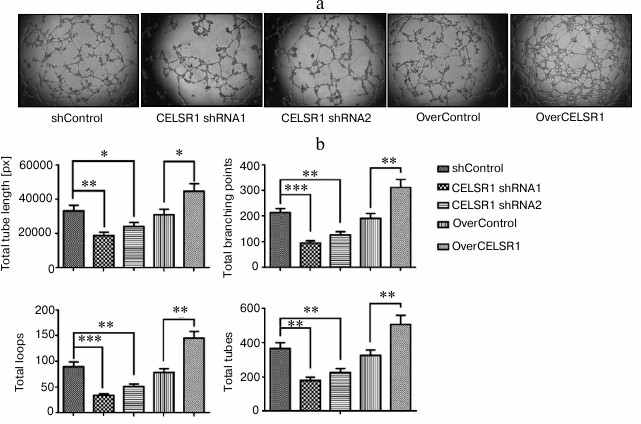
Fig. 6. CELSR1 critically regulates in vitro endothelial tube formation. The capability of HAEC with altered CELSR1 expression to form a tubular network was measured by a tubulogenesis assay on Matrigel. a) Representative images for endothelial tubes formed from indicated groups of cells. b) Quantification of total tube length in pixels (px), total number of branching points, total number of loops, and total number of tubes from three independent experiments were compared between different groups; * p < 0.05, ** p < 0.01, *** p < 0.001, compared to the corresponding control cells.
Dvl3 is a critical regulator for CELSR1-modulated endothelial cell behaviors. To explore the molecular mechanism underlying CELSR1-modulated endothelial behavior, we focused on Dvl3, a key regulator in Wnt/PCP signaling [1]. When we knocked down Dvl3 in OverCELSR1 cells, there was decreased cell migration and tubulogenesis. In contrast, overexpression of Dvl3 in CELSR1shRNA cells significantly boosted migration and tubulogenesis (Fig. 7), suggesting that Dvl3 is a critical mediator for CELSR1-modulated endothelial cell migration and tubulogenesis.
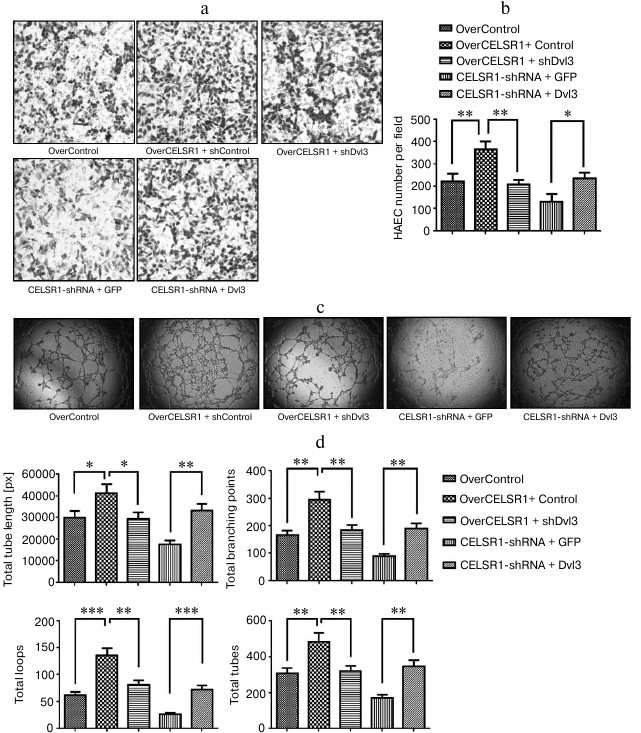
Fig. 7. Dvl3 mediates CELSR1-regulated endothelial migration and tube formation. a, b) Migration of indicated cells was measured by transwell migration assay, with represented images of the transwell insert from each group after the migration shown in (a) and the quantification of the number of migrated cells per field from three independent experiments shown in (b). Knockdown of Dvl3 abolished the migration of HAEC induced by CELSR1 overexpression, while ectopically expressing Dvl3 in CELSR1shRNA cells significantly boosted the migration. c, d) Tube formation of indicated cells was examined by tubulogenesis assay on Matrigel, with representative images of tube formation from each group shown in (c) and the quantification of total tube length in pixels (px), total number of branching points, total number of loops, and total number of tubes from three independent experiments shown in (d). shDvl3 reduced tubulogenesis in OverCELSR1 cells, while Dvl3 expression significantly increased tube formation in CELSR1-shRNA cells. * p < 0.05, ** p < 0.01, *** p < 0.001, compared to the corresponding control cells.
DISCUSSION
In this study, we have demonstrated for the first time that CELSR1, a seven-transmembrane cadherin protein, is a critical and positive regulator of proliferation, migration, and tubulogenesis in vascular endothelial cells in vitro.
The noncanonical Wnt/PCP pathway controls cell morphology, polarized cell movements, and tissue morphogenesis [18-20]. Recent studies by several groups point to the importance of the Wnt/PCP pathway in endothelial cell proliferation and angiogenesis [4-7]. As a core component of the noncanonical Wnt/PCP pathway, CELSR1 orchestrates the establishment of polarized cell–cell junctions across proximodistal cell boundaries by recruiting an asymmetric complex consisting of Strabismus/Van Gogh-like (Vangl) and Frizzled to the membrane [21, 22]. However, CELSR1 has not been studied in the context of vascular endothelial cells concerning its regulation of angiogenesis, a precisely engineered process coordinated by endothelial cell differentiation, proliferation, migration, and apoptosis. In this study, we provide the first evidence that CELSR1 is essential for in vitro vascular network formation.
As the initial step to study the correlation between CELSR1 and vascular endothelial cells, we first examined CELSR1 expression in vascular endothelial cells. We detected positive and comparable expression of CELSR1 in both arterial endothelial cells (HAEC) and venous endothelial cells (HUVEC). These data suggest a potential functional involvement of CELSR1 in both HAEC and HUVEC. In both cell lines, CELSR1 was detected as two isoforms, a full-length 400-kDa protein and a proteolytic 85-kDa product. A previous study showed that the 400- and 85-kDa isoforms are both present at the cell surface, yet the exact generating mechanism and functional significance for the smaller CELSR1 isoform remain unknown [1].
With the confirmation of positive CELSR1 expression in vascular endothelial cells, we altered the endogenous level of CELSR1 with either TALE technology (for upregulation) or shRNA (for downregulation). CELSR1 cDNA is approximately 9 kb in length, which is challenging for cloning and expression studies. In contrast, TALE is a newly developed tool for targeted genome engineering. TALE is easy to use and has been widely applied for a wide variety of genome engineering applications including transcriptional modulation and genome editing [14, 23]. Traditional ectopic expression involves subcloning a target cDNA into a plasmid and transfecting the plasmid into cells, which can result in plasmid insertion into random locations within the cellular genome and is often associated with overexpression at non-physiological high levels. The TALE technique is more specific and will only introduce a transcriptional activator into the endogenous promoter of a target gene and alter the target expression within the physiological range. Consistently, we found that upregulation of CELSR1 following TALE-CELSR1 was approximately fourfold higher than control cells. By upregulating and downregulating CELSR1 in HAEC, we noticed that both the 400- and 85-kDa isoforms were altered to similar extents. Therefore, the functional readouts from this study represent those from altering both isoforms in the same direction.
The formation of new vessels requires both endothelial cell proliferation and migration to enable sprout growth and the formation of a branched network. Here we demonstrated that reducing the expression of endogenous CELSR1 dramatically mitigated HAEC proliferation, migration, and endothelial tube formation, whereas increasing the endogenous CELSR1 expression enhanced the proliferative, migratory, and vascular network-forming capability of HAEC. These observations, although all obtained from in vitro analyses, strongly support CELSR1 as an essential regulator of angiogenesis in vivo.
Recent studies demonstrated a role of PCP in endothelial cell proliferation and angiogenesis [4-7]. Genetic approaches have previously shown that Dvl3 is involved in PCP signaling during gastrulation and that it has a functionally redundant role during development [4-7]. Interestingly, Dvl3 mutants exhibited craniorachischisis, which is often associated with PCP signaling disruption. Using in vitro assays, we report that cooperation of CELSR1 with Dvl3 is required for endothelial cell migration and tube formation in the Wnt/PCP signaling pathway. Our data further demonstrate that CELSR1 may be a major component of the Wnt/PCP pathway in angiogenesis.
In summary, the present study provides the first evidence of an active angiogenic role for CELSR1 in human vascular endothelial cells. We found that CELSR1 promotes the in vitro proliferation, migration, and tubulogenesis of HAEC, suggesting its functional significance in endothelial biology.
This study was supported by grants from the National Natural Science Foundation of China (81000567) and the Science and Technology Program of Xiamen (3502Z20154009), China.
REFERENCES
1.Formstone, C. J., Moxon, C., Murdoch, J., Little,
P., and Mason, I. (2010) Basal enrichment within neuroepithelia
suggests novel function(s) for CELSR1 protein, Mol. Cell.
Neurosci., 44, 210-222.
2.Crompton, L. A., Du Roure, C., and Rodriguez, T. A.
(2007) Early embryonic expression patterns of the mouse Flamingo and
Prickle orthologues, Dev. Dyn., 236, 3137-3143.
3.Yates, L. L., Schnatwinkel, C., Murdoch, J. N.,
Bogani, D., Formstone, C. J., Townsend, S., Greenfield, A., Niswander,
L. A., and Dean, C. H. (2010) The PCP genes Celsr1 and Vangl2 are
required for normal lung branching morphogenesis, Hum. Mol.
Genet., 19, 2251-2267.
4.Cirone, P., Lin, S., Griesbach, H. L., Zhang, Y.,
Slusarski, D. C., and Crews, C. M. (2008) A role for planar cell
polarity signaling in angiogenesis, Angiogenesis, 11,
347-360.
5.Descamps, B., Sewduth, R., Ferreira Tojais, N.,
Jaspard, B., Reynaud, A., Sohet, F., Lacolley, P., Allieres, C.,
Lamaziere, J. M., Moreau, C., Dufourcq, P., Couffinhal, T., and Duplaa,
C. (2012) Frizzled 4 regulates arterial network organization through
noncanonical Wnt/planar cell polarity signaling, Circ. Res.,
110, 47-58.
6.Ju, R., Cirone, P., Lin, S., Griesbach, H.,
Slusarski, D. C., and Crews, C. M. (2010) Activation of the planar cell
polarity formin DAAM1 leads to inhibition of endothelial cell
proliferation, migration, and angiogenesis, Proc. Natl. Acad. Sci.
USA, 107, 6906-6911.
7.Masckauchan, T. N., Agalliu, D., Vorontchikhina,
M., Ahn, A., Parmalee, N. L., Li, C. M., Khoo, A., Tycko, B., Brown, A.
M., and Kitajewski, J. (2006) Wnt5a signaling induces proliferation and
survival of endothelial cells in vitro and expression of MMP-1
and Tie-2, Mol. Biol. Cell, 17, 5163-5172.
8.Yamada, Y., Fuku, N., Tanaka, M., Aoyagi, Y.,
Sawabe, M., Metoki, N., Yoshida, H., Satoh, K., Kato, K., Watanabe, S.,
Nozawa, Y., Hasegawa, A., and Kojima, T. (2009) Identification of
CELSR1 as a susceptibility gene for ischemic stroke in Japanese
individuals by a genome-wide association study, Atherosclerosis,
207, 144-149.
9.Gouveia, L. O., Sobral, J., Vicente, A. M., Ferro,
J. M., and Oliveira, S. A. (2011) Replication of the CELSR1 association
with ischemic stroke in a Portuguese case-control cohort,
Atherosclerosis, 217, 260-262.
10.Zhan, Y. H., Lin, Y., Tong, S. J., Ma, Q. L., Lu,
C. X., Fang, L., Wei, W., Cai, B., and Wang, N. (2015) The CELSR1
polymorphisms rs6007897 and rs4044210 are associated with ischaemic
stroke in Chinese Han population, Ann. Hum. Biol., 42,
26-30.
11.Curtin, J. A., Quint, E., Tsipouri, V., Arkell,
R. M., Cattanach, B., Copp, A. J., Henderson, D. J., Spurr, N.,
Stanier, P., Fisher, E. M., Nolan, P. M., Steel, K. P., Brown, S. D.,
Gray, I. C., and Murdoch, J. N. (2003) Mutation of Celsr1 disrupts
planar polarity of inner ear hair cells and causes severe neural tube
defects in the mouse, Curr. Biol., 13, 1129-1133.
12.Devenport, D., Oristian, D., Heller, E., and
Fuchs, E. (2011) Mitotic internalization of planar cell polarity
proteins preserves tissue polarity, Nat. Cell Biol.,
13, 893-902.
13.Tatin, F., Taddei, A., Weston, A., Fuchs, E.,
Devenport, D., Tissir, F., and Makinen, T. (2013) Planar cell polarity
protein CELSR1 regulates endothelial adherens junctions and directed
cell rearrangements during valve morphogenesis, Dev. Cell,
26, 31-44.
14.Maeder, M. L., Linder, S. J., Reyon, D.,
Angstman, J. F., Fu, Y., Sander, J. D., and Joung, J. K. (2013) Robust,
synergistic regulation of human gene expression using TALE activators,
Nat. Methods, 10, 243-245.
15.Livak, K. J., and Schmittgen, T. D. (2001)
Analysis of relative gene expression data using real-time quantitative
PCR and the 2(–ΔΔC(T)), Methods, 25,
402-408.
16.Van Meerloo, J., Kaspers, G. J., and Cloos, J.
(2011) Cell sensitivity assays: the MTT assay, Methods Mol.
Biol., 731, 237-245.
17.Justus, C. R., Leffler, N., Ruiz-Echevarria, M.,
and Yang, L. V. (2014) In vitro cell migration and invasion
assays, J. Vis. Exp., 88, doi: 10.3791/51046.
18.Goodrich, L. V., and Strutt, D. (2011) Principles
of planar polarity in animal development, Development,
138, 1877-1892.
19.Gray, R. S., Roszko, I., and Solnica-Krezel, L.
(2011) Planar cell polarity: coordinating morphogenetic cell behaviors
with embryonic polarity, Dev. Cell, 21, 120-133.
20.Seifert, J. R., and Mlodzik, M. (2007)
Frizzled/PCP signalling: a conserved mechanism regulating cell polarity
and directed motility, Nat. Rev. Genet., 8, 126-138.
21.Chen, P. L., and Clandinin, T. R. (2008) The
cadherin Flamingo mediates level-dependent interactions that guide
photoreceptor target choice in Drosophila, Neuron,
58, 26-33.
22.Usui, T., Shima, Y., Shimada, Y., Hirano, S.,
Burgess, R. W., Schwarz, T. L., Takeichi, M., and Uemura, T. (1999)
Flamingo, a seven-pass transmembrane cadherin, regulates planar cell
polarity under the control of Frizzled, Cell, 98,
585-595.
23.Joung, J. K., and Sander, J. D. (2013) TALENs: a
widely applicable technology for targeted genome editing, Nat. Rev.
Mol. Cell Biol., 14, 49-55.