Mitochondrial Dysfunction in Neocortex and Hippocampus of Olfactory Bulbectomized Mice, a Model of Alzheimer’s Disease
A. V. Avetisyan1*, A. N. Samokhin2, I. Y. Alexandrova2, R. A. Zinovkin1, R. A. Simonyan1, and N. V. Bobkova2
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: +7 (495) 939-0338; E-mail: avetis@genebee.msu.ru2Institute of Cell Biophysics, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia
* To whom correspondence should be addressed.
Received November 17, 2015; Revision received March 16, 2016
Structural and functional impairments of mitochondria in brain tissues in the pathogenesis of Alzheimer’s disease (AD) cause energy deficiency, increased generation of reactive oxygen species (ROS), and premature neuronal death. However, the causal relations between accumulation of beta-amyloid (Aβ) peptide in mitochondria and mitochondrial dysfunction, as well as molecular mechanisms underlying deleterious effects of both these factors in sporadic AD, the most common form in humans, remain unknown. Here we used olfactory bulbectomized (OBX) mice of NMRI strain as a model for sporadic AD. Five weeks after surgery, the OBX mice developed major behavioral and biochemical features of AD neurodegeneration, including spatial memory loss, increased brain levels of Aβ, and energy deficiency. Mitochondria isolated from the neocortex and hippocampus of OBX mice displayed severe functional impairments, such as low NADH oxidation rate, reduced transmembrane potential, and decreased cytochrome c oxidase (complex IV) activity that correlated with high levels of soluble Aβ1-40. Mitochondria from OBX mice showed increased contents of lipid peroxidation products, indicative of the development of oxidative stress. We found that neurodegeneration caused by olfactory bulbectomy is accompanied by energy metabolism disturbances and oxidative stress in brain mitochondria similar to those occurring in transgenic animals – familial AD models and patients with sporadic AD. Therefore, OBX mice can serve as a valid AD model for investigating the mechanisms of AD neurodegeneration, drug testing, and development of therapeutic strategies for AD treatment.
KEY WORDS: Alzheimer’s disease, olfactory bulbectomized mice, mitochondrial dysfunction, oxidative stressDOI: 10.1134/S0006297916060080
Abbreviations: Aβ, beta-amyloid protein; AD, Alzheimer’s disease; APP, amyloid precursor protein; OBX, olfactory bulbectomy; ROS, reactive oxygen species; SO, sham-operated (mice); TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine.
Alzheimer’s disease (AD) is an incurable neurodegenerative
disorder that affects millions of elderly humans around the world. AD
is characterized by memory loss and dementia that accompany brain
atrophy, ultimately leading to the patient’s death. Major
morphological features of AD are extracellular accumulation of the
fibrillar beta-amyloid protein (Aβ) in cortical senile plaques and
intraneuronal formation of neurofibrillary tangles from phosphorylated
tau-protein [1, 2]. Unlike
hereditary AD, sporadic AD is the clinically predominant form with
mostly late onset. The reasons for Aβ accumulation in sporadic AD
and the mechanisms of Aβ neuronal toxicity remain unknown. One of
the difficulties in early diagnostics and timely treatment of sporadic
AD is a limited number of valid AD animal models with major features of
AD pathogenesis. The development of such models is hindered by the fact
that mice and rats do not form classic senile plaques in the process of
brain aging due to changes in the Aβ primary structure [3]. It was recently found that the neurotoxicity in AD
could be attributed mostly to soluble Aβ oligomers, while senile
plaques represent a product of the neuronal compensatory response and
can be used as AD markers, in addition to some other features [4, 5].
In this work, we used mice with surgically removed olfactory bulbs as a model of sporadic AD. It is known that olfactory bulbectomy (OBX) in rodents (mice, rats) results in the development of symptoms similar to those observed in AD neurodegeneration [6]. OBX animals display loss of spatial memory, increased brain levels of Aβ, tau-protein hyperphosphorylation, inhibition of long-term potentiation of hippocampal synaptic transmission, neuronal death in the cortex and hippocampus, and deficiency of the acetylcholinergic and serotonergic systems in the brain [7-9]. However, it remains unclear where OBX affects energy metabolism and bioenergetic characteristics of mitochondria. Mitochondrial dysfunctions caused by Aβ import during development of sporadic AD in animal models need further study.
Recently, a large body of evidence has been obtained that demonstrates the importance of mitochondria and Aβ-induced oxidative stress in AD pathogenesis in patients and transgenic animal models of hereditary AD [10, 11]. Mitochondria play a significant role in AD onset and development – they act simultaneously as targets and members in the neurodegenerative process in early AD stages, i.e. before Aβ aggregation into insoluble senile plaques [12, 13]. The mechanism of Aβ translocation from the cytoplasm into mitochondria has been widely discussed in recent publications. It is believed that this translocation occurs via the protein import TOM complex and results in inhibition of energy metabolism, impairments of oxidative phosphorylation, disturbances in activities of respiratory chain complexes, generation of reactive oxygen species (ROS), and eventually, induction of apoptosis in neurons [14]. These data have been obtained in various transgenic rodent models overproducing amyloid precursor protein (APP), presenilin, and tau-protein and in cell lines expressing mutant human APP [15, 16].
The goal of this study was to investigate the relations between mitochondria dysfunctions and development of AD neurodegeneration in OBX animals as a valid model of sporadic AD. We compared Aβ accumulation, functional state of the respiratory chain, and the level of oxidative stress in mitochondria isolated from neocortex and hippocampus of OBX and sham-operated mice (SO). We found that functional impairments in the brain mitochondria of OBX mice resemble those observed earlier in the brain structures responsible for learning and memory in transgenic AD animal models (AβPP, AβPP/PS1, AβPP/PS1/Tau, 5xFAD, etc.) and AD patients [17].
The results of the study show that OBX mice can be used as a valid model of sporadic AD and demonstrate common mechanisms of energy metabolism disturbances in brain mitochondria in both familial and sporadic AD forms.
MATERIALS AND METHODS
Animals. Olfactory bulbectomy. All procedures were performed on three-month-old male mice (NMRI strain) weighing 20 ± 5 g. The mice were kept at 21-23°C and were given food and water ad libitum. Mice of the experimental group were anesthetized with barbital (40 mg/kg, i.p.); Novocain (0.3 ml of 0.5% solution, s.c.) was administered as a local analgesic. Following the exposure of the skull, the olfactory bulbs were removed bilaterally by aspiration through a hole in the skull with coordinates A-3, L-0, H-3 as described earlier [18]. Sham-operated (SO) mice underwent a similar surgical procedure except for the removal of the olfactory bulbs.
All procedures and experiments were performed in accordance with the European Union Directive 86/609/EEC on protection of animals used for experimental and other scientific purposes and approved by the Moscow State University Ethics Committee.
Spatial memory testing. Four weeks after OBX, the mice were tested for their ability to swim, the absence of initial preference for one of the Morris water maze sectors, and vision impairments (surgery side effect). Selected animals were trained to locate a hidden platform submerged under water in one of the pool sectors (target sector). Four training sessions were administered every day for five consecutive days. The mice were allowed a maximum of 60 s to find the platform. The latency to reach the platform (escape latency) was recorded as a parameter of learning ability. On day 6, a 60-s probe trial was performed, in which the platform was removed, and the number of entries and time spent in each sector were analyzed.
After completion of behavioral experiments, the mice were decapitated under deep Nembutal anesthesia (60 mg/kg, i.p.). The brain was removed and examined for the absence of abscesses and extent of olfactory bulb removal.
Isolation of mitochondria from mouse neocortex and hippocampus. Mitochondria were isolated from the brain tissues by a standard procedure using differential centrifugation in a Percoll gradient [19]. After decapitation, the brain was isolated; neocortex and hippocampus were separated on a chilled glass. The tissues were homogenized in 2 ml of ice-cold isolation medium containing 225 mM mannitol, 75 mM sucrose, 10 mM HEPES (pH 7.4), 1 mM EGTA, and 2 mg/ml BSA and centrifuged in 2-ml microtubes in a refrigerated centrifuge (Eppendorf, Germany). The last mitochondrial pellet was resuspended in 0.1 ml of the isolation medium without BSA and stored on ice. In each experiment, mitochondrial fractions were isolated from neocortex and hippocampus of the OBX and SO mice (two mice per group). Protein concentration was determined by the Lowry method using BSA as a standard.
Beta-amyloid protein 1-40 (Aβ1-40) level in mitochondrial fractions. Changes in the levels of Aβ1-40 were measured by enzyme-linked immunosorbent assay (ELISA) using an Amyloid beta 40 ELISA Kit (Invitrogen, USA) as recommended by the manufacturer. Optical density was measured at 450 nm with a BioRad IFA reader (USA). The Aβ1-40 content was determined in all mitochondrial fractions isolated from neurons and glia of the neocortices and hippocampi of OBX and SO animals. The data were normalized to the protein content and presented in ng/mg total protein.
NADH oxidase activity. The functional state of isolated mitochondria was estimated from the oxidation rate of reduced nicotinamide adenine dinucleotide (NADH), a natural substrate of mitochondrial complex I. Since the inner mitochondrial membrane is impermeable to NADH, measurements were performed in the presence of the pore-forming antibiotic alamethicin (25 µg/ml), which provided substrate access into the matrix to the NADH dehydrogenase subunit of complex I. The standard incubation medium contained 225 mM mannitol, 75 mM sucrose, 10 mM HEPES (pH 7.6), 1 mM EGTA, 4 mM magnesium chloride, 5 mM potassium phosphate, and 2 mg/ml BSA. The rate of NADH (0.2 mM) oxidation was measured at 25°C by a decrease in absorption at 340/420 nm using an Aminco DW-2000 (USA) dual-wavelength spectrophotometer and expressed in nmol/min per mg protein. The molar extinction coefficient for NADH is 6.22 mM–1·cm–1. Mitochondria were added at a concentration of 0.2 mg protein/ml.
Mitochondrial membrane potential. The membrane potential (ΔΨ) was estimated from the distribution of the potential-sensitive indicator safranin O [20] by measuring safranin O absorption ratio at 555/523 nm with the Aminco DW-2000 dual-wavelength spectrophotometer in medium containing 225 mM mannitol, 75 mM sucrose, 10 mM HEPES (pH 7.6), 1 mM EGTA, 1 mg/ml BSA, 2 µM rotenone, 2 µg/ml oligomycin, and 10 µM safranin O at 25°C. The inner mitochondrial membrane potential was generated by addition of 5 mM succinate (respiratory chain complex II substrate). Mitochondria were added at the concentration of 0.2 mg protein/ml.
To estimate the basal level of safranin absorption (deenergized mitochondria), the protonophoric uncoupler FCCP was added at the concentration of 2 µM. Relative ΔΨ is presented as the ratio between the maximal and basal safranin absorption normalized to the mitochondrial protein content.
Cytochrome c oxidase (complex IV) activity. Cytochrome c oxidase activity was determined from oxidation of the synthetic penetrating mediator N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD), which acts as an electron donor to cytochrome c. Cyanide-sensitive TMPD oxidation was measured from the increase in TMPD absorption at 612/630 nm at 25°C using the Aminco DW-2000 spectrophotometer. The reaction rate was calculated from the initial linear section of the reaction curve using the molar extinction coefficient of 11.6 mM–1·cm–1 and expressed in nmol/min mg protein. The reaction mixture contained 225 mM mannitol, 75 mM sucrose, 20 mM HEPES (pH 7.4), 1 mM EGTA, 4 mM magnesium chloride, and 5 mM potassium phosphate at 25°C. After 0.5 mM TMPD was added to the reaction mixture, the rate of TMPD autooxidation was recorded, and the reaction was then initiated by adding 0.1 mg/ml mitochondria.
Lipid peroxidation products in mitochondria. The level of lipid peroxidation in mitochondria was estimated by the accumulation of thiobarbituric acid reactive substances (TBARS) using absorption measurements at 535/572 nm with the Aminco DW-2000 spectrophotometer [21]. The suspension of mitochondria was mixed with 6 mM butylhydroxytoluene and 0.2 mM orthophosphoric acid, and 0.11 mM thiobarbituric acid (TBA) was added. The reaction mixture was incubated on a water bath at 90°C for 45 min; TBARS were then extracted with 0.5 ml of n-butanol. After centrifugation at 12,000g per 10 min, the optical density of the upper phase was measured, and the content of TBARS was expressed in nmol/mg protein, using 156 mM–1·cm–1 extinction coefficient.
Statistical analysis. The difference between OBX and SO groups (spatial memory, mitochondrial bioenergetic parameters) was evaluated using one-way analysis of variance (One-Way ANOVA) with subsequent Tukey’s post hoc test (Statistica 6). The results are presented as mean ± SEM. The difference is considered statistically significant at p < 0.05.
RESULTS
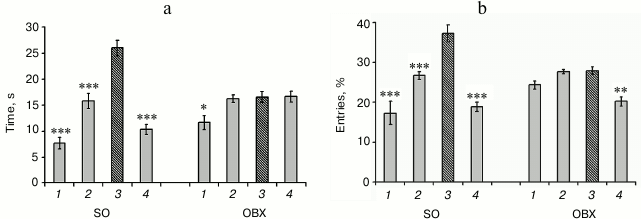
Spatial memory in OBX and SO mice. The loss of spatial memory is a major symptom of AD. We found that one month after surgery bilateral olfactory bulbectomy resulted in deterioration of spatial memory and learning ability (Fig. 1). Compared to the SO mice, OBX mice demonstrated significantly impaired ability for spatial learning and could not locate the sector that contained the escape platform during the training period. While the SO mice demonstrated clear preference for the target sector (3), both in the number of entries (Fig. 1b) and time spent in this sector (Fig. 1a), the OBX mice did not distinguish between the target and nontarget sectors.
Fig. 1. Behavioral characteristics of OBX and sham-operated (SO) mice. Spatial memory was assayed by the Morris water maze test: a) time spent in each sector; b) percentage of entries in each sector; 3) target sector (hatched column); 1, 2, 4) nontarget sectors (gray columns). The data are presented as mean ± SEM; * p < 0.05; ** p < 0.01; *** p < 0.001; n = 6.
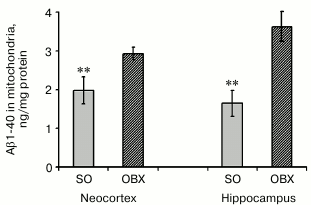
Aβ1-40 levels in mitochondrial fractions from neocortex and hippocampus. We demonstrated earlier that hippocampal and neocortical extracts from OBX mice display considerably higher levels of endogenous soluble Aβ1-40 five weeks after bulbectomy, i.e. when neurodegeneration of AD type is mostly pronounced [22]. Here we found that mitochondria isolated from these two brain regions of OBX mice contain significantly higher amounts of soluble Aβ1-40 than mitochondria from the SO animals (Fig. 2). According to previously published data, after translocation from the cytoplasm into mitochondria by the mitochondrial system of protein transport, soluble Aβ1-40 localizes to cristae and matrix and can cause mitochondrial dysfunction [15, 23].
Fig. 2. Endogenous Aβ1-40 in mitochondria from neocortex and hippocampus of SO (gray columns) and OBX (hatched columns) mice. The data are presented as mean ± SEM; ** p < 0.01; n = 6.
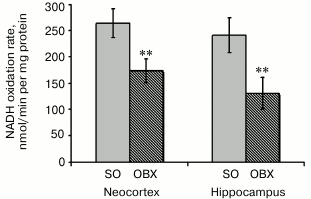
Activity of the mitochondrial respiratory chain. To confirm the correlation between Aβ1-40 accumulation in mitochondria of OBX mice and mitochondrial dysfunction, we compared the rates of NADH oxidation by mitochondria from the neocortex and hippocampus of OBX and SO mice. NADH is a natural substrate of complex I, and the rate of NADH oxidation reflects the activity of the entire respiratory chain that transports electron from complex I to cytochrome c oxidase with reduction of oxygen to water. NADH oxidation was monitored in the presence of the pore-forming antibiotic alamethicin that provided substrate entry into the mitochondrial matrix. Uncoupling of mitochondria by alamethicin allowed us to register the maximum rates of the respiratory chain redox reactions. Rotenone, an inhibitor of NADH:quinone oxidoreductase, prevented NADH oxidation in all cases (data not shown).
The measured rates of NADH oxidation indicate significantly lower respiratory chain electron-transport activity in the neocortex and hippocampus of OBX mice as compared to the SO mice (Fig. 3). The limiting stage of respiratory chain activity in the OBX mice is most probably catalyzed by complex IV, which is known to be inactivated in all experimental AD animal models and in AD patients [24, 25].
Based on considerable difference between NADH oxidation rates in the mitochondrial fractions from the OBX and SO mice, we suggest a cytotoxic effect of mitochondrially accumulated Aβ1-40.
Fig. 3. Rates of NADH oxidase reaction as determined spectrophotometrically in mitochondrial fractions from brains of SO (gray columns) and OBX (hatched columns) mice. Mitochondria were added to medium containing 0.2 mM NADH. The data are presented as mean ± SEM; ** p < 0.01; n = 12.
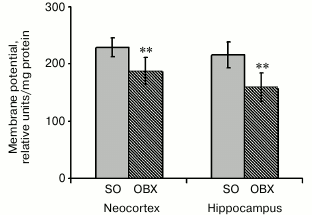
Membrane potential (ΔΨ) in neocortical and hippocampal mitochondria. Since safranin is known to inhibit complex I, we used the complex II substrate succinate to generate membrane potential on the inner mitochondrial membrane [20]. The incubation medium contained rotenone (a complex I inhibitor) to prevent reverse electron flow to complex I and oligomycin (an ATP synthase inhibitor) to inhibit ΔΨ decrease resulting from ATP synthesis. Energization of mitochondria by succinate resulted in rapid ΔΨ generation, which caused safranin transport into mitochondria by the membrane potential gradient and could be monitored by a decrease in safranin absorbance. Figure 4 shows that the mitochondrial membrane potential in OBX animals was decreased compared to the SO mice, probably due to lower respiratory chain electron-transport activity or partial uncoupling of the inner mitochondrial membrane.
Fig. 4. Relative values of generated membrane potential (ΔΨ) upon mitochondria energization with succinate in neocortex and hippocampus of SO (gray columns) and OBX (hatched columns) mice; ** p < 0.01; n = 12.
Activity of cytochrome c oxidase (respiratory chain complex IV). Cytochrome c oxidase is the terminal complex of the aerobic respiratory chain. It consists of 13 subunits, catalyzes electron transfer from cytochrome c to oxygen with the formation of a water molecule, and generates transmembrane proton gradient required for ATP synthesis. Complex IV plays a key role in the regulation of the entire respiratory chain activity and energy production [26]. Insufficient cytochrome c oxidase activity is as indicative AD feature as accumulation of Aβ and phosphorylated tau-protein. This enzyme was found to be inactivated in brain and platelet mitochondria in AD transgenic models and AD patients [27, 28].
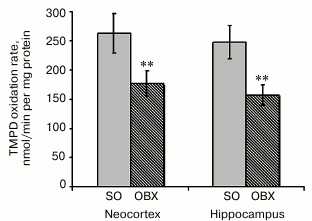
To study functional deficit of cytochrome c oxidase in OBX mice, we measured the activity of complex IV separately from the respiratory chain by the synthetic electron donor TMPD (Fig. 5). Since phospholipid membrane is permeable for TMPD, it rapidly penetrates the membrane and reduces cytochrome c, a natural electron donor for cytochrome c oxidase. Cytochrome c oxidase activity is proportional to the rate of TMPD oxidation, which can be measured spectrophotometrically. Figure 5 shows that neurodegeneration in the OBX mice was accompanied by a considerable decrease in cytochrome c oxidase activity in the neocortical and hippocampal mitochondria, which is similar to the data obtained with other AD animal models [13, 29]. It remains to be elucidated if the observed cytochrome c oxidase activity deficit is related to the decrease in the content of enzymatic complexes in the inner mitochondrial membrane or results from downregulation of their activity.
Fig. 5. Rates of enzymatic oxidation of the synthetic electron donor TMPD by respiratory chain complex IV from neocortical and hippocampal mitochondria of SO (gray columns) and OBX (hatched columns) mice; ** p < 0.01; n = 18.
Oxidative stress and lipid peroxidation in mitochondria. AD pathogenesis is accompanied by active ROS generation, with mitochondria as one of the main ROS sources [30]. An increased content of lipid peroxidation products confirms oxidative stress enhancement in blood samples from AD patients [31]. The toxicity of mitochondrially accumulated Aβ1-40 (Fig. 2) might be mediated by stimulation of ROS production and subsequent damage to electron-transport complexes.
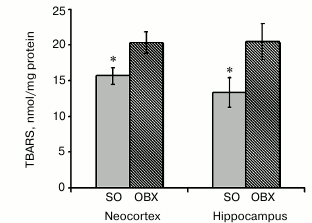
The levels of oxidative stress were estimated from accumulation of lipid peroxidation products capable of reacting with TBA. We measured the contents of TBA reactive substances (TBARS), as indicators of free-radical lipid oxidation, in all mitochondrial fractions and found that mitochondrial fractions from the hippocampus and neocortex of OBX mice contained significantly higher levels of TBARS than similar fractions from SO animals (Fig. 6). These results unambiguously show that neurodegeneration in OBX mice causes the development of oxidative stress in mitochondria.
Fig. 6. Lipid peroxidation in mitochondrial fractions from SO (gray columns) and OBX (hatched columns) mice as assayed by the TBARS; * p < 0.05; n = 6.
DISCUSSION
Dysfunction of the olfactory system is one of the early symptoms of Alzheimer’s and Parkinson’s diseases. In both cases, deterioration of olfaction is observed before manifestation of typical clinical symptoms and, therefore, can be used as an early disease marker [32]. The removal of olfactory bulbs in adult rodents (olfactory bulbectomy, OBX) induces a cascade of neurodegenerative processes similar to those observed in AD, including loss of spatial memory, death of neurons in brain structures responsible for learning and memory, and increased APP synthesis with subsequent formation and accumulation of Aβ [33, 34].
To model the sporadic form of AD in adult NMRI mice, we surgically removed the olfactory bulbs from adult animals, as this procedure is known to cause neurodegeneration of AD type in brains 4-5 weeks after surgery. The goal of this study was to elucidate whether OBX-induced neurodegeneration is accompanied by brain mitochondrial dysfunctions similar to those observed in transgenic AD animal models.
It has been shown in transgenic animals that energy metabolism deficit and mitochondria dysfunctions appear at the early stages of AD and result in decrease in the efficiency of oxidative phosphorylation, ATP depletion, and development of oxidative stress [13, 25, 29]. Despite numerous studies, it remains unknown if ROS generation by damaged mitochondria leads to the activation of Aβ synthesis and accumulation, or accumulation of Aβ soluble oligomers in mitochondria inhibits key mitochondrial enzymes and causes oxidative stress and neuronal death. According to the “mitochondrial cascade” hypothesis, mitochondrial dysfunction observed in sporadic AD precedes and initiates Aβ synthesis in brain cells and its accumulation in the cytoplasm and organelles, including mitochondria [35].
Dysfunctional mitochondria are typical for transgenic animal models of familial AD and neuronal cell cultures overexpressing APP. Mitochondrial dysfunctions can also be induced by treatment of isolated mitochondria with exogenous Aβ in vitro [16, 23, 29, 36]. These experimental models are characterized by high Aβ levels that are typical for familial AD and considered a cause for rapid development of deleterious processes in cells and mitochondria. However, sporadic AD attracts the most interest now, since it is not related to mutations observed in familial cases of AD. In addition to increased Aβ levels, sporadic AD is characterized by synaptic dysfunctions, massive death of neurons in the neocortex and hippocampus, and cell cycle deterioration [2]. Patients with sporadic AD display impaired mitochondrial energy metabolism, in particular, decreased activity of cytochrome c oxidase resulting from low enzyme expression and/or insufficient number of subunits of the complex [24, 28]. Therefore, studying the state of energy metabolism in brain mitochondria of OBX animals as the most relevant model of sporadic AD is a problem of utmost importance.
Here we studied functional impairments of mitochondria from brain tissues of OBX mice. These animals exhibit neurodegenerative processes similar to those observed in sporadic AD [37]. OBX mice showed impairment of spatial memory and learning when tested in the Morris water maze (Fig. 1). Based on these results, we studied OBX mice for the presence of mitochondrial dysfunctions, since cerebral mitochondria have not been investigated earlier for this AD model. Mitochondria were isolated from neocortices and hippocampi of OBX mice five weeks after surgery, i.e. when animals displayed pronounced features of AD. In addition to memory loss and Aβ accumulation in brain tissues, we found that mitochondria of OBX mice accumulated neurotoxic soluble amyloid-beta 1-40 (Aβ1-40) (Fig. 2), which has been described earlier for transgenic AD models [11, 25].
Measuring metabolic activity of mitochondria (NADH oxidation) revealed low activity of the electron transport chain in OBX mice as compared to the sham-operated control animals (Fig. 3). Earlier published data showed that energy metabolism deficit in brain tissues, lymphocytes, and platelets of AD patients is caused mostly by the low activity of the mitochondrial respiratory chain complex IV [24, 27, 38]. We demonstrated that the activity of this terminal complex of the respiratory chain in mitochondria from the neocortex and hippocampus of OBX mice is considerably lower than in the sham-operated control animals (Fig. 5). The decrease in complex IV activity was proportional to the inhibition of NADH oxidase activity, which indicates the key role of cytochrome c oxidase in inhibition of respiration.
It has been shown earlier that deficit of cytochrome c oxidase activity might result from either low expression or decreased content of protein complexes, but also from direct enzyme inhibition by Aβ dimer that forms in the presence of Cu2+ [39, 40]. Further studies of mitochondria from the neocortex and hippocampus of OBX mice will elucidate the mechanism of complex IV inhibition during neurodegeneration.
Low activity of the respiratory chain, particularly its terminal complex, results in generation of insufficient value of the transmembrane proton gradient and therefore decreased membrane potential (ΔΨ) in OBX mitochondria (Fig. 4). Other reasons for low ΔΨ might be increased permeability of the inner mitochondrial membrane for protons (uncoupling) due to lipid peroxidation and/or the effect of membrane-bound Aβ oligomers. Thus, it has been shown that oligomers of human Aβ1-40 increase ion permeability of bilayer lipid membranes (BLM) [41].
In this study, we have demonstrated for the first time that development of AD-type neurodegeneration in OBX mice is accompanied by mitochondrial dysfunction in the hippocampus and neocortex. In both hippocampal and neocortical mitochondrial fractions from OBX animals, the respiratory chain was inhibited, and at least one of its complexes (cytochrome c oxidase) exhibited decreased activity (Fig. 5) typical for brain tissues of AD patients.
The observed dysfunctions of the electron transport chain in the OBX mitochondria were accompanied by development of oxidative stress, as demonstrated by increased lipid peroxidation – ROS-induced oxidative degradation of polyunsaturated fatty acids and formation of TBA-reactive compounds (Fig. 6). Lipid peroxidation disturbs lipid homeostasis of mitochondrial and cell membranes and changes membrane structure and properties, which impairs membrane function and promotes cell apoptosis [42]. The observed dysfunction of the electron transport chain might result from the damaging effect of mitochondrially accumulated neurotoxic Aβ1-40 or from oxidative stress, which activates lipid peroxidation and amyloidogenesis [43, 44].
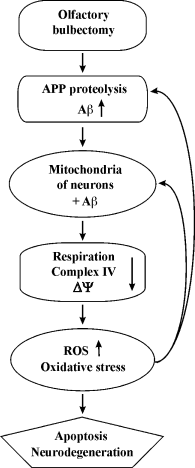
Revealing the correlation between the functional impairments, Aβ1-40 accumulation, and oxidative stress development in mitochondria from the neocortex and hippocampus of OBX mice will bring us closer to understanding causative relations in AD pathogenesis. Based on our data, we suggest the following mechanism of development of neurodegeneration in OBX mice: 1) olfactory bulbectomy induces APP proteolysis and accumulation of soluble Aβ oligomers with their subsequent translocation into mitochondria; 2) Aβ accumulation in the matrix and the coupling membrane damages the respiratory chain and the mitochondrial membrane; 3) mitochondrial dysfunctions cause generation of ROS and oxidative stress; 4) oxidative stress exacerbates ROS generation and mitochondrial dysfunctions. This forms a “vicious circle” of the Aβ cascade amplification, oxidative stress, and mitochondrial dysfunction, which eventually ends in apoptosis and neurodegeneration (Fig. 7).
Fig. 7. Schematic diagram of putative neurobiological processes underlying the development of AD in OBX – cause–effect relations between mitochondrial energy metabolism disturbances and amyloidogenesis.
In this work, we showed, based on observed behavioral, mitochondrial, and biochemical parameters of OBX mice, that olfactory bulbectomy model is a valid model of sporadic AD. OBX mice deserve serious attention of researchers and can be used for development of drugs for treating and slowing progress of AD, as well as for AD prevention in humans.
This work was supported by grants from the Russian Foundation for Fundamental Research 13-04-00982 and the Russian Science Foundation 14-50-00029 (functional characteristics of mitochondria).
REFERENCES
1.Hardy, J., and Selkoe, D. J. (2002) The amyloid
hypothesis of Alzheimer’s disease: progress and problems on the
road to therapeutics, Science, 297, 353-356.
2.Atwood, C. S., and Bowen, R. L. (2015) A unified
hypothesis of early- and late-onset Alzheimer’s disease
pathogenesis, J. Alzheimer’s Dis., 47, 33-47.
3.Bobkova, N. V., Nesterova, I. V., Medvinskaya, N.
I., Aleksandrova, I. Y., Samokhin, A. N., Gershovich, Y. G.,
Gershovich, P. M., and Yashin, V. A. (2005) Possible role of olfactory
system in Alzheimer’s disease genesis, in Alzheimer’s
and Parkinson’s Disease – AD/PD (Hanin, L., and
Fisher, A., eds.) Monduzzi Medimond, pp. 91-95.
4.Walsh, D. M., and Selkoe, D. J. (2004) Deciphering
the molecular basis of memory failure in Alzheimer’s disease,
Neuron, 44, 181-193.
5.Kayed, R., Head, E., Thompson, J. L., McIntire, T.
M., Milton, S. C., Cotman, C. W., and Glabe, C. G. (2003) Common
structure of soluble amyloid oligomers implies common mechanism of
pathogenesis, Science, 300, 486-489.
6.Bobkova, N. V., Nesterova, I. V., Dana, R., Dana,
E., Nesterov, V. I., Aleksandrova, I. Y., Medvinskaya, N. I., and
Samokhin, A. N. (2004) Morphofunctional changes in neurons in the
temporal cortex of the brain in relation to spatial memory in
bulbectomized mice after treatment with mineral ascorbates,
Neurosci. Behav. Physiol., 34, 671-676.
7.Hu, J., Wang, X., Liu, D., Wang, Q., and Zhu, L. Q.
(2012) Olfactory deficits induce neurofilament hyperphosphorylation,
Neurosci. Lett., 506, 180-183.
8.Hozumi, S., Nakagawasai, O., Tan-No, K., Niijima,
F., Yamadera, F., Murata, A., Arai, Y., Yasuhara, H., and Tadano, T.
(2003) Characteristics of changes in cholinergic function and
impairment of learning and memory-related behavior induced by olfactory
bulbectomy, Behav. Brain Res., 138, 9-15.
9.Moriguchi, S., Han, F., Nakagawasai, O., Tadano,
T., and Fukunaga, K. M. (2006) Decreased calcium/calmodulin-dependent
protein kinase II and protein kinase C activities mediate impairment of
hippocampal long-term potentiation in the olfactory bulbectomized mice,
J. Neurochem., 97, 22-29.
10.Wang, X., Wang, W., Li, L., Perry, G., Lee, H.
G., and Zhu, X. (2014) Oxidative stress and mitochondrial dysfunction
in Alzheimer’s disease, Biochim. Biophys. Acta,
1842, 1240-1247.
11.Manczak, M., Anekonda, T. S., Henson, E., Park,
B. S., Quinn, J., and Reddy, P. H. (2006) Mitochondria are a direct
site of A beta accumulation in Alzheimer’s disease neurons:
implications for free radical generation and oxidative damage in
disease progression, Hum. Mol. Genet., 15, 1437-1449.
12.Yao, J., Irwin, R. W., Zhao, L., Nilsen, J.,
Hamilton, R. T., and Brinton, R. D. (2009) Mitochondrial bioenergetic
deficit precedes Alzheimer’s pathology in female mouse model of
Alzheimer’s disease, Proc. Natl. Acad. Sci. USA,
106, 14670-14675.
13.Schmitt, K., Grimm, A., Kazmierczak, A.,
Strosznajder, J. B., Gotz, J., and Eckert, A. (2012) Insights into
mitochondrial dysfunction: aging, amyloid-β, and tau –
a deleterious trio, Antioxid. Redox Signal., 16,
1456-1466.
14.Hansson Petersen, C. A., Alikhani, N., Behbahani,
H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., Leinonen, V., Ito, A.,
Winblad, B., Glaser, E., and Ankarcrona, M. (2008) The amyloid
beta-peptide is imported into mitochondria via the TOM import machinery
and localized to mitochondrial cristae, Proc. Natl. Acad. Sci.
USA, 105, 13145-13150.
15.Eckert, A., Schmitt, K., and Gotz, J. (2011)
Mitochondrial dysfunction – the beginning of the end in
Alzheimer’s disease? Separate and synergistic modes of tau and
amyloid-β toxicity, Alzheimer’s Res. Ther., 3,
15.
16.Rhein, V., Baysang, G., Rao, S., Meier, F.,
Bonert, A., Muller-Spahn, F., and Eckert, A. (2009) Amyloid-beta leads
to impaired cellular respiration, energy production and mitochondrial
electron chain complex activities in human neuroblastoma cells,
Cell. Mol. Neurobiol., 29, 1063-1071.
17.Hroudova, J., Singh, N., and Fisar, Z. (2014)
Mitochondrial dysfunctions in neurodegenerative diseases: relevance to
Alzheimer’s disease, Biomed. Res. Int., 175062.
18.Kamynina, A. V., Volpina, O. M., Medvinskaya, N.
I., Aleksandrova, I. Ju., Volkova, T. D., and Koroev, D. O. (2010)
Vaccination with peptide 173-193 of acetylcholine receptor
α7-subunit prevents memory loss in olfactory bulbectomized mice,
J. Alzheimer’s Dis., 21, 249-261.
19.Chinopoulos, C., Zhang, S. F., Thomas, B., Ten,
V., and Starkov, A. A. (2011) Isolation and functional assessment of
mitochondria from small amounts of mouse brain tissue, Methods Mol.
Biol., 793, 311-324.
20.Krumschnabel, G., Eigentler, A., Fasching, M.,
and Gnaiger, E. (2014) Use of safranin for the assessment of
mitochondrial membrane potential by high-resolution respirometry and
fluorometry, Methods Enzymol., 542, 163-181.
21.Jentzsch, A. M., Bachmann, H., Furst, P., and
Biesalski, H. K. (1996) Improved analysis of malondialdehyde in human
body fluids, Free Radic. Biol. Med., 20, 251-256.
22.Aleksandrova, I. Yu., Kuvichkin, V. V.,
Kashparov, I. V., Medvinskaya, N. I., Nesterova, I. V., Lunin, S. V.,
Samokhin, A. N., and Bobkova, N. V. (2004) Increased levels of
beta-amylod protein in the brain of bulbectomized mice, Biochemistry
(Moscow), 69, 176-180.
23.Cha, M. Y., Han, S. H., Son, S. M., Hong, H. S.,
Choi, Y. J., Byun, J., and Mook-Jung, I. (2012) Mitochondria-specific
accumulation of amyloid β induces mitochondrial dysfunction
leading to apoptotic cell death, PLoS One, 7, e34929.
24.Parker, W. D., Jr., Parks, J., Filley, C. M., and
Kleinschmidt-DeMasters, B. K. (1994) Electron transport chain defects
in Alzheimer’s disease brain, Neurology, 44,
1090-1096.
25.Du, H., Guo, L., Yan, S., Sosunov, A. A.,
McKhann, G. M., and Yan, S. S. (2010) Early deficits in synaptic
mitochondria in an Alzheimer’s disease mouse model, Proc.
Natl. Acad. Sci. USA, 107, 18670-18675.
26.Wilson, D. F., and Vinogradov, S. A. (2014)
Mitochondrial cytochrome c oxidase: mechanism of action and role
in regulating oxidative phosphorylation, J. Appl. Physiol.,
117, 1431-1439.
27.Cardoso, S. M., Proenca, M. T., Santos, S.,
Santana, I., and Oliveira, C. R. (2004) Cytochrome c oxidase is
decreased in Alzheimer’s disease platelets, Neurobiol.
Aging, 25, 105-110.
28.Maurer, I., Zierz, S., and Moller, H. J. (2000) A
selective defect of cytochrome c oxidase is present in brain of
Alzheimer’s disease patients, Neurobiol. Aging, 21,
455-462.
29.Rhein, V., Song, X., Wiesner, A., Ittner, L. M.,
Baysang, G., Meier, F., Ozmen, L., Bluethmann, H., Drose, S., Brandt,
U., Savaskan, E., Czech, C., Gotz, J., and Eckert, A. (2009)
Amyloid-beta and tau synergistically impair the oxidative
phosphorylation system in triple transgenic Alzheimer’s disease
mice, Proc. Natl. Acad. Sci. USA, 106, 20057-20062.
30.Ma, T., Hoeffer, C. A., Wong, H., Massaad, C. A.,
Zhou, P., Iadecola, C., Murphy, M. P., Pautler, R. G., and Klann, E.
(2011) Amyloid β-induced impairments in hippocampal synaptic
plasticity are rescued by decreasing mitochondrial superoxide, J.
Neurosci., 31, 5589-5595.
31.Serra, J. A., Dominguez, R. O., Marschoff, E. R.,
Guareschi, E. M., Famulari, A. L., and Boveris, A. (2009) Systemic
oxidative stress associated with the neurological diseases of aging,
Neurochem. Res., 34, 2122-2132.
32.Devanand, D. P., Lee, S., Manly, J., Andrews, H.,
Schupf, N., Doty, R. L., Stern, Y., Zahodne, L. B., Louis, E. D., and
Mayeux, R. (2015) Olfactory deficits predict cognitive decline and
Alzheimer dementia in an urban community, Neurology, 84,
182-189.
33.Bobkova, N., Guzhova, I., Margulis, B.,
Nesterova, I., Medvinskaya, N., Samokhin, A., Alexandrova, I., Garbuz,
D., Nudler, E., and Evgen’ev, M. (2013) Dynamics of endogenous
Hsp70 synthesis in the brain of olfactory bulbectomized mice, Cell
Stress Chaperones, 18, 109-118.
34.Nesterova, I. V., Bobkova, N. V., Medvinskaya, N.
I., Samokhin, A. N., and Aleksandrova, I. Y. (2008) Morphofunctional
state of neurons in the temporal cortex and hippocampus in relation to
the level of spatial memory in rats after ablation of the olfactory
bulbs, Neurosci. Behav. Physiol., 38, 349-353.
35.Swerdlow, R. H., Burns, J. M., and Khan, S. M.
(2010) The Alzheimer’s disease mitochondrial cascade hypothesis,
J. Alzheimer’s Dis., 20, Suppl. 2, S265-279.
36.Aleardi, A. M., Benard, G., Augereau, O., Malgat,
M., Talbot, J. C., Mazat, J. P., Letellier, T., Dachary-Prigent, J.,
Solaini, G. C., and Rossignol, R. (2005) Gradual alteration of
mitochondrial structure and function by beta-amyloids: importance of
membrane viscosity changes, energy deprivation, reactive oxygen species
production, and cytochrome c release, J. Bioenerg.
Biomembr., 37, 207-225.
37.Bobkova, N. V. (2010) Bulbectomized animals as
models for sporadic Alzheimer’s desease, in Neurodegenerative
Disorders: Fundamental Aspects and Applications (Ugryumov, M. V.,
ed.) [in Russian], Nauka, Moscow, pp. 341-349.
38.Mutisya, E. M., Bowling, A. C., and Beal, M. F.
(1994) Cortical cytochrome oxidase activity is reduced in
Alzheimer’s disease, J. Neurochem., 63,
2179-2184.
39.Crouch, P. J., Blake, R., Duce, J. A.,
Ciccotosto, G. D., Li, Q. X., Barnham, K. J., Curtain, C. C., Cherny,
R. A., Cappai, R., Dyrks, T., Masters, C. L., and Trounce, I. A. (2005)
Copper-dependent inhibition of human cytochrome c oxidase by a
dimeric conformer of amyloid-beta1-42, J. Neurosci., 19,
672-679.
40.Pedros, I., Petrov, D., Allgaier, M., Sureda, F.,
Barroso, E., Beas-Zarate, C., Auladell, C., Pallas, M.,
Vazquez-Carrera, M., Casadesus, G., Folch, J., and Camins, A. (2014)
Early alterations in energy metabolism in the hippocampus of
APPswe/PS1dE9 mouse model of Alzheimer’s disease, Biochim.
Biophys. Acta, 1842, 1556-1566.
41.Schauerte, J. A., Wong, P. T., Wisser, K. C.,
Ding, H., Steel, D. G., and Gafni, A. (2010) Simultaneous
single-molecule fluorescence and conductivity studies reveal distinct
classes of Abeta species on lipid bilayers, Biochemistry,
13, 3031-3039.
42.Greilberger, J., Koidl, C., Greilberger, M.,
Lamprecht, M., Schroecksnadel, K., Leblhuber, F., Fuchs, D., and Oettl,
K. (2008) Malondialdehyde, carbonyl proteins and albumin-disulphide as
useful oxidative markers in mild cognitive impairment and
Alzheimer’s disease, Free Radic. Res., 42,
633-638.
43.Hernandez-Zimbron, L. F., and Rivas-Arancibia, S.
(2015) Oxidative stress caused by ozone exposure induces β-amyloid
1-42 overproduction and mitochondrial accumulation by activating the
amyloidogenic pathway, Neuroscience, 304, 340-348.
44.Jiao, Y., Zhang, Y., Wei, Y., Liu, Z., An, W.,
and Guo, M. (2012) Direct observation of internalization and ROS
generation of amyloid β-peptide in neuronal cells at subcellular
resolution, ChemBioChem, 13, 2335-2338.