REVIEW-HYPOTHESIS: Glycosylphosphatidylinositol-Anchored Proteins as Regulators of Cortical Cytoskeleton
G. V. Sharonov1,2*, M. N. Balatskaya1, and V. A. Tkachuk1
1Lomonosov Moscow State University, Faculty of Medicine, 119992 Moscow, Russia; fax: +7 (495) 932-9904; E-mail: sharonov@gmail.com2Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Structural Biology Department, 117997 Moscow, Russia
* To whom correspondence should be addressed.
Received November 4, 2015; Revision received April 3, 2016
Glycosylphosphatidylinositol-anchored proteins (GPI-AP) are important players in reception and signal transduction, cell adhesion, guidance, formation of immune synapses, and endocytosis. At that, a particular GPI-AP can have different activities depending on a ligand. It is known that GPI-AP oligomer creates a lipid raft in its base on plasma membrane, which serves as a signaling platform for binding and activation of src-family kinases. Yet, this does not explain different activities of GPI-APs. Meanwhile, it has been shown that short-lived actomyosin complexes are bound to GPI-APs through lipid rafts. Here, we hypothesize that cell cortical cytoskeleton is the main target of GPI-AP signaling. Our hypothesis is based on the fact that the GPI-AP-induced lipid raft bound to actin filaments and anionic lipids of this raft is known to interact with and activate various actin-nucleating factors, such as formins and N-WASP. It is also known that these and other actin-regulating proteins are activated by src-family kinases directly or through their effectors, such as cortactin and abl-kinases. Regulation of cytoskeleton by GPI-APs may have impact on morphogenesis, cell guidance, and endocytosis, as well as on signaling of other receptors. To evaluate our hypothesis, we have comprehensively considered physiological activities of two GPI-APs – urokinase receptor and T-cadherin.
KEY WORDS: glycosylphosphatidylinositol, cytoskeleton, actin, lipid rafts, uPAR, T-cadherinDOI: 10.1134/S0006297916060110
Abbreviations: DRM, detergent resistant membrane; EGFR, epidermal growth factor receptor; Gαi2, αi2 subunit of heterotrimeric G-protein; GEEC, GPI-APs enriched early endosomal compartments; GPCR, G-protein coupled receptor; GPI, glycosylphosphatidylinositol; GPI-AP, glycosylphosphatidylinositol-anchored proteins; LDL, low density lipoproteins; NgR, Nogo66 receptor; PI(x)P, phosphatidylinositol(x)phosphate; PLC, phospholipase C; PM, plasma membrane; PS, phosphatidylserine; RTK, receptor tyrosine kinase; SFK, src-family kinases; uPAR, urokinase receptor.
Modification with glycosylphosphatidylinositol (GPI) is an irreversible
glycolipid modification of the protein C-terminus (Fig. 1) that is initiated in endoplasmic reticulum by
substitution of C-terminal signal peptide – the signal of
GPI-modification – with the pro-form of GPI-anchor, and it
is followed by modification of the lipid part of the GPI-anchor in
Golgi [1]. In the UNIPROT database
(www.uniprot.org), there are 124 GPI-APs in the human genome, while
this number is reduced to 83 after combining homologous proteins.
According to the same database, the main function of all GPI-APs
appears on a surface of a cell, which suggests that the GPI-anchor is a
signal of localization on the outer surface of the plasma membrane
(PM). Based on GPI-AP activities, it is possible to distinguish their
major types: enzymes, navigation cues and navigation receptors,
adhesion molecules, growth factors receptors and coreceptors, and
receptors participating in formation of immune complexes. For
functioning of some GPI-APs, namely enzymes and navigation cues, a
GPI-AP serves mainly as a signal of PM localization. For the GPI-APs
that were shown to activate intracellular signaling, existence of some
transmembrane adapter that mediates its signal transduction is usually
assumed. Still, there are numerous indications that GPI-APs have their
own receptor function. The most prominent example is that
oligomerization of different GPI-APs with antibodies induces the same
type of cell response: activation of src-family kinases (SFK) and
phospholipase Cγ (PLCγ), phosphoinositide turnover, and
elevation of intracellular calcium [2-5]. Another example is potentiation of T-cell
activation by oligomerization of different GPI-APs with antibodies or
their crosslinking with other receptors and activation inhibition by
blocking of oligomerization [6]. However, it is
unclear how oligomerization with antibodies is related to the
activation of GPI-APs with physiological ligands.
Fig. 1. Structure of a mammalian GPI-AP. From the protein to lipid part, GPI-AP consists of ethanolamine, phosphate, three mannose moieties that can be modified with additional sugar chains (R), glucosamine, inositol, and phospholipid. The most abundant alkyl substituents are stearic acid residues C18:0 and, less often, in the sn-1 position are monounsaturated oleic acid (C18:1 as depicted) or saturated palmitic acid (C16:0) residues.
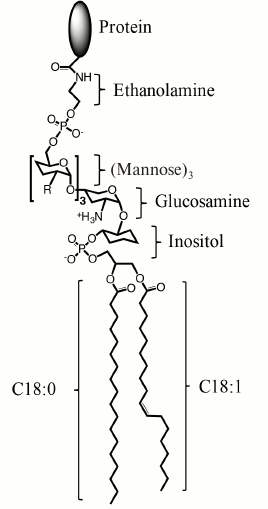
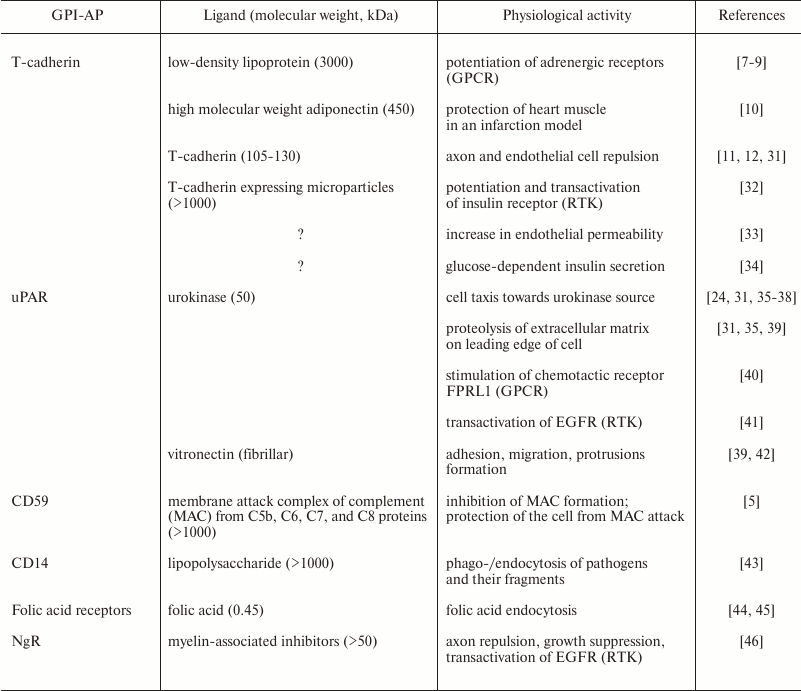
Upon activation by physiological ligands, GPI-APs have a broad spectrum of activities; moreover, one receptor can induce different and even opposite types of cell responses (Table 1). Thus, in studies led by V. A. Tkachuk, it has been shown that binding of low-density lipoproteins (LDLs) with T-cadherin on smooth muscle cells potentiates adrenergic receptors and vessel contraction, i.e. exacerbating stress outcomes [7-9]. At the same time, B. Ranscht’s group has shown that a high molecular weight form of adiponectin (a fat tissue hormone that has multiple beneficial physiological effects that is most prominent for the high molecular weight form) also acts through T-cadherin, but contrary to LDLs, it protects heart muscle from damage in a model of myocardial infarction [10]. Moreover, T-cadherin may act as a cell guidance receptor that prevents nerve and vessel growth on T-cadherin-expressing tissues [11, 12]. The T-cadherin example reflects the activity spectrum inherent for GPI-APs, namely signaling potentiation and effects on cell cytoskeleton in such processes as cell migration, cell–cell contacts, tissue boundary formation, and cell guidance. This list should be complemented by the ability of GPI-APs to promote endocytosis, as for CD14-dependent pathogen endocytosis and folic acid endocytosis by its receptor (Table 1).
Table 1. GPI-APs discussed in this paper.
Ligands of GPI-APs are given with their physiological activities
exerted through the GPI-AP
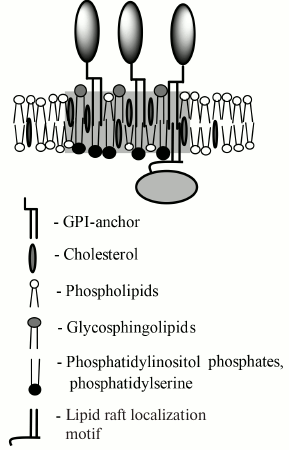
All GPI-APs have a marked affinity to nonionic detergent-resistant fractions of PM (DRM) that are commonly intermixed with liquid ordered domains of PM, also referred to as lipid rafts. For association with a lipid raft, a GPI-AP should contain two saturated or monounsaturated lipid moieties of 16 or more carbon atoms [13, 14]. In mammalian cells, GPI-APs usually contain two saturated substituent stearic acid residues (C18:0) or, less often, a monounsaturated oleic (C18:1) or saturated palmitic acid (C16:0) residue instead of one of the former in the sn-1 position of the glycerol base [1]. Lipid rafts differ from the rest of membrane by increased concentration of saturated charged lipids and cholesterol, increased by 1 nm (or ~15%) thickness, ordered hydrophobic phase, and slowed by a factor of 2 to 3 lateral diffusion of lipids [15-17]. The major saturated lipids of the outer PM leaflet are glycosphingolipids (gangliosides, sphingomyelin, ceramides), and their concentration in lipid rafts is approximately 1.5-fold higher than in the rest of the membrane (Fig. 2) [15]. For cholesterol, this factor is 2. From the inner side of the PM, the lipid raft is enriched in phosphatidylserine (PS) by a factor of 2-3, as well as in other anionic lipids – phosphatidylinositols that are specific to certain types of lipid rafts [16, 18, 19]. In biological membranes of living cells, lipid rafts have significantly shorter lifetime and smaller size than in model membranes [20-22], which indicates that in living cells they are actively regulated by constituent proteins and bound cytoskeleton. According to this view, the works of K. Suzuki’s and A. Kusumi’s groups have shown that GPI-AP oligomer induces formation of lipid raft in its base that binds the αi2 subunit of G-protein (Gαi2) and SFK Lyn and activates the latter. However, this does not explain the differences in activities of GPI-APs ligands and the absence of such activation in the rest of liquid ordered membrane that, for example, occupies 76% of the PM area in T-cells [23].
Fig. 2. Schematic drawing of a GPI-AP-containing lipid raft. The raft region is marked in gray. The raft is enriched with cholesterol, glycosphingolipids outside of the cell, and anionic lipids inside the cell, and it has increased thickness compared to the rest of the membrane. From the cytoplasmic side, the lipid raft interacts with proteins having a lipid raft-binding motif. Usually, this motif consists of lipid modifications with palmitoyl or myristoyl alkyl chains and/or polycationic amino acid sequence.
In this work, we suggest a working hypothesis that the activity of GPI-APs is dependent on their ability to organize cortical cytoskeleton that, in turn, is dependent on the oligomer properties of the GPI-AP. The first indications of the interaction of GPI-APs with cytoskeleton were obtained in the late 1990s [24-26]. A group of Indian scientists lead by S. Mayor [14, 27-30] studied this interaction most extensively. In their works, it was shown that GPI-AP oligomerization dynamics on PM is dependent on actin polymerization and myosin activity, and that the same dynamics is inherent to a transmembrane protein directly linked to actin. They also revealed that in the case of GPI-APs this link is mediated by lipid rafts and PS [14]. Based on the existence of the link between GPI-AP and cytoskeleton, we hypothesize that GPI-AP oligomerization should have an effect on the structure and dynamics of cortical cytoskeleton. As we suppose, a GPI-AP-induced lipid raft affects orientation, polymerization, and branching of actin filaments. This effect is mediated by actin-binding and actin-regulating proteins, most of which also bind to lipid rafts and are activated by SFK or through their direct effectors. Hence, we expect that the properties of a newly formed structure depend on the size, shape, and lifetime of a GPI-AP oligomeric complex. Induced cytoskeleton may serve as an active scaffold for assembly of signaling complexes and directly affect the cell shape. Further, we will consider data that underlie our hypothesis, propose a mechanism of GPI-AP effect on cytoskeleton, and use it to explain physiological activities of some GPI-APs.
ACTIVATION OF INTRACELLULAR SIGNALING BY A GPI-AP OLIGOMER
A valuable contribution to understanding GPI-AP signaling was made by the works of Japan scientists lead by K. Suzuki and A. Kusumi, where dynamics of a single GPI-AP was analyzed with nanometer spatial and millisecond temporal resolution on PM of living cells together with its relation to the movement and activation of intracellular effectors. It was shown that in the absence of stimuli, GPI-APs diffuse freely on PM and form transient short-lived (~0.15 s) dimers and (less often) trimers that are stabilized by intermolecular interaction of ectodomains and by lipid rafts [5]. We should note that dimerization dependence of lipid rafts does not exclude participation of cytoplasmic molecules and cytoskeleton in stabilization of this raft. GPI-AP dimers retain high mobility and do not induce any intracellular signaling.
Oligomerization of CD59 by a nanoparticle with specific antibodies activates SFK Lyn, leads to formation of inositol triphosphate by PLCγ and increase of intracellular calcium concentration [3, 4]. It has been revealed that oligomeric GPI-APs do not diffuse on PM freely, but appear immobilized (immobilization periods of ~0.5 s) for ~30% of the time. Alteration of a relative immobilization period by cholesterol depletion or repletion induces a proportional change of intracellular signaling intensity, indicating that it is induced by an immobilized oligomer. It appears that CD59 immobilization requires activity of Lyn and the αi2 subunit of G-protein (Gαi2), but does not require PLCγ. Overlay of a GPI-AP oligomer trace by traces of each of these molecules showed that immobilization is preceded by a short period (~0.13 s) of colocalization of Gαi2 with CD59 oligomer (both still moving), which is followed by Gαi2 dissociation and immobilization of the complex. The traces of Lyn and CD59 oligomer coincide frequently for periods of ~0.2 s, but these periods do not correlate with immobilization. In contrast, PLCγ appears to be colocalized with the CD59 cluster only during the periods of immobilization. It has been also ascertained that Lyn activation does not require cytoskeleton integrity, but PLCγ does. On the basis of these data, a model has been suggested where a lipid raft that is created/stabilized by CD59 oligomer promotes a colocalization within the raft of Gαi2 and Lyn, their interaction, and activation of Lyn that leads to immobilization. In turn, immobilized Lyn complex activates a further signaling cascade, particularly immobilization and activation of PLCγ.
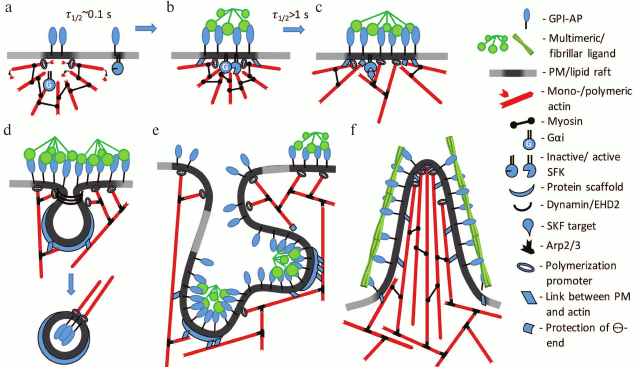
As follows from the works of Suzuki et al., lipid rafts mediate interaction of GPI-APs with intracellular signaling molecules, but this interaction is relatively weak and allows holding in place Lyn and Gαi2 only for 0.2 and 0.13 s, respectively. Lyn retention in a lipid raft is mediated by the N-terminal fragment that contains two modifications, by saturated fatty acids, palmitoleic and myristoleic, and three cationic lysines in the vicinity to this anchor, as well as by protein–protein interactions with some other components of the raft [3]. An analogous N-terminal motif with two lipid modifications but less prominent cationic part is present in Gαi2, this explaining its appearance in the lipid raft. It is remarkable that Gαi2 does not diffuse over the PM as Lyn does, but contacts it for short periods (<0.1 s), and points of contact are distributed on PM nonrandomly: they are placed near the GPI-AP cluster or even coincide with it. This indicates the directionality of Gαi2 motion in the cytoplasm towards the GPI-AP oligomer, and it is tempting to speculate that this directionality is mediated by cytoskeleton (Fig. 3, a and b). Besides the immobilization of GPI-AP oligomer and directed motion of Gαi2, it has also shown that PLCγ activation requires intact cytoskeleton [4].
Fig. 3. Hypothetical model of signaling activation and cytoskeleton rearrangement by GPI-APs. a) Metastable complex of actin filaments and myosin that is linked with GPI-AP through a lipid raft. The Gα-subunit of G-protein is also bound with actin filaments. Some GPI-APs and SFKs are localized in short-lived rafts that are not connected to cytoskeleton. b) Oligomerization of GPI-AP by multivalent ligand promotes coalescence of rafts and increase of their lifetime. Filament motion towards the center of the complex stimulates interaction of G-protein with SFK, which activates SFK. c) GPI-AP-stabilized lipid raft and activated SFK activate actin-regulating proteins such as cortactin, formins, WASP, and others (Table 2). This induces actin network growth and polymerization in the region of the GPI-AP oligomer. d) GPI-AP oligomers that are in the vicinity of caveolae induce caveolae budding or flattening due to polymerization or contraction of caveolae-associated cytoskeleton, respectively. e, f) Massive cytoskeleton rearrangement in the region of large and stable GPI-AP oligomeric complex that induces changes in PM shape. Depending on a ligand, particularly on its shape, cell invagination (GEEC) or protrusions (membrane ruffles or thin spikes) are formed.
It should be noted that the requirement of cytoskeleton or other protein scaffold emerges from consideration of physicochemical properties of lipid rafts. So, the retention of various lipids including PS in lipid rafts lasts only for 10-20 ms [47, 48], that is, one order of magnitude lower than the retention time of intracellular effectors. This shows that lipid order and slowed diffusion in the lipid raft are not the major factors of protein retention within it. Therefore, this means that the lipid raft can catalyze just an initial phase of complex formation, after which it does not require the lipid raft (but still may require PM anchorage). This is illustrated by data on PLCγ activation that, in contrast to Lyn and Gαi2, does not contain lipid modifications and hence should have weaker interaction with lipid rafts. Nevertheless, its retention in CD59-associated lipid raft is longer-lived and amounts to 0.3 s, which is longer than that for Lyn and Gαi2. Such prolonged colocalization with lipid raft is necessary for induction of Ca2+ response and requires intact cytoskeleton.
GPI-APs INTERACTION WITH CYTOSKELETON
Effects of GPI-APs on cytoskeleton with requirements of G-proteins and SKF were shown in the 1990s [24, 25]. At that time, it was assumed that there was some transmembrane adaptor that mediates this link, like integrins for urokinase receptor (uPAR), for example [49, 50]. However, later it was shown that the effect of uPAR on cell morphology and motility could be transduced independently of integrins and other possible coreceptors [42]. Existence of a link between GPI-AP and cytoskeleton mediated by a lipid raft was supposed by Indian scientists lead by S. Mayor, who analyzed oligomerization dynamics of GPI-AP on PM. In was found that folic acid receptor forms oligomers of from two to four molecules that do not follow thermodynamic rules and are energetically unfavorable [27, 28]. It has been shown that: (i) oligomer dissociation rate is independent of the temperature (above 28°C); (ii) and oligomer/monomer ratio is independent of GPI-AP expression level; and (iii) frequencies of homo- and heterooligomer formation are equal. Similar results were obtained for other GPI-APs: CD55 and model fluorescent GPI-APs. It turned out that GPI-AP dynamics and oligomerization are critically dependent on actin polymerization and myosin activity, and that is clear evidence for their association with actin cytoskeleton. Later, it was shown that this association is mediated by lipid rafts and PS [14] with the involvement of the actin-nucleating factor formin [30].
While searching for possible cytoskeleton structures with dynamics corresponding to GPI-AP oligomerization, Mayor and coworkers suggested their link to short dynamic filaments [29]. Existence of such filaments has been shown by several groups using cryoelectron tomography, which allows analysis of thick specimens (whole cells or slices up to 1 µm) with spatial resolution in all dimensions (down to 5 nm) and retention of native structure [51, 52]. Despite a limited number of these works related to technical difficulties of the method, all of them clearly show high density of actin filament in the cortical region of eukaryotic cells with the most filaments being 200- to 500-nm long [53-56]. Decrease of the filament length hinders its identification by cryoelectron tomography, which may lead to overestimation of the filament mean length [57]. In the works of Mayor’s group, the mean filament length was evaluated by analysis of diffusion of fluorescent actin-binding domain of ezrin and was estimated to be ~250 nm [29].
Computational modeling by Mayor’s group showed that in the system of short actin filaments (~200 nm) and myosin, metastable and dynamic aster-like structures are formed, wherein filaments continuously move towards the center of the aster [29] (Fig. 3, a and b). It the model, they took into account basic actin dynamics: continuous polymerization/depolymerization, association/dissociation with PM, crosslinking, and movement by myosin motors. It appeared that the dynamics and the temperature dependency of GPI-AP oligomerization correspond well to the dynamics of formation of asters with lifetime of ~0.1 s and decay determined by “active noise”, which accumulates due to stochasticity of myosin binding and direction of related forces.
Mayor et al. considered GPI-APs as passive molecules that are bound to a cytoskeleton, but not affecting filament dynamics. Nevertheless, it is obvious that as far as the link between GPI-APs and cytoskeleton exists, the impact on GPI-AP will be transduced to the cytoskeleton. Indeed, Suzuki and Sheetz measured mechanical resistance during movement of GPI-AP oligomer over PM with an optical tweezer [58]. While doing so, they observed static barriers that were reproduced during repetitive passages, as well as dynamic barriers that were always different during each repetitive passage. The static barriers correspond to stable cytoskeleton filaments, while dynamic ones are likely to be represented by metastable structures of dynamic cortical cytoskeleton.
MECHANISM OF GPI-AP EFFECT ON CORTICAL ACTIN CYTOSKELETON: A
HYPOTHESIS
It has been shown that many GPI-APs (ephrins-A, semaphorins, contactins, Thy-1, netrins-G, repulsive guidance molecule, T-cadherin, uPAR, and others [31]) have important functions in morphogenesis, especially in neural tissue development. Morphogenesis is closely related to cytoskeleton remodeling, and as mentioned earlier, GPI-AP oligomerization induces SFK- and G-protein-mediated accumulation of cortical actin [24, 25]. Based on the data of K. Suzuki et al. and S. Mayor’s team, we supposed that there are two major components of the effect of GPI-APs on cortical actin cytoskeleton (Fig. 3). First, a GPI-AP-induced lipid raft is enriched in anionic lipids, which interact with a number of actin-nucleating and actin-binding proteins (Table 2), leading to their local concentration together with actin filaments in the region of GPI-AP oligomerization, their activation, and growth of an actin network at this place. We should note that cytoskeleton reorganization is possible even without polymerization, but by means of local concentration and/or alignment of dynamic filaments, which can result in directional flow, contraction, or severing of filaments (due to action of myosins) [59, 60]. We assume that this mechanism can be realized in some cases of cortical contraction, which is common for axon growth cone repulsion and for tissue border formation during development [61, 62]. Cortical contraction also may affect properties of other signaling systems, in particular through the impact on caveolae (see below).
Second, oligomerization of GPI-APs activates SFKs, and their major targets are key regulators of actin cytoskeleton (Table 2): cortactin, catenins, abl-kinases, focal adhesion kinase, vinculin, talin, and others [63, 64]. Activation of these proteins results in conversion of dynamic actin filaments to a stable branched and/or bundled actin network. This is mediated by Arp2/3 nucleating complex, bundling and polymerization promoter formins, and adapters between actin and PM (Fig. 3 and Table 2). Such actin network rearrangement results in change of the cell shape, motility, and endocytosis, as well as in regulation of signaling [65, 66].
Table 2. Some possible mediators of GPI-APs
and cytoskeleton
GPI-APs IN REGULATION OF CAVEOLAE
A common feature of almost all GPI-APs is their abundance in caveolin-containing DRM fractions of PM and endosomal compartments [73, 74]. Caveolae are Ω-shaped invaginations of PM, 60-80 nm in diameter, scaffolded by oligomeric complex of ~140-150 caveolins and ~12 cavins [75]. On the basis on caveolae lipid composition and their requirement for cholesterol, they are commonly considered as a subset of lipid rafts [76]. Many receptors and intracellular effectors interact with caveolae and caveolin-1, but more often, this leads to inhibition of their activity, as in the case of SFKs, MAP-kinases, endothelial NO-synthase, and some GPCRs (G-protein coupled receptors) and RTKs (receptor tyrosine kinases) [77]. Accordingly, caveolae can be considered as a scaffold that serves for assembly of receptor/signaling complex but holds this complex inactive until an appropriate stimulus is received.
It is now assumed that the ability of caveolae to respond rapidly to mechanical stimulus and PM damage underlies most of their biological activities [78-80]. Thus, PM tension induces caveolae coalescence and flattening, while compression induces their formation and budding [78, 79]. Cell attack by pore-forming toxins or PM mechanical damage induces intense caveolae budding that is believed to internalize a damaged part of the PM [78, 80]. It is quite natural that linkage of caveolae to mechanical membrane properties is mediated by actin cytoskeleton [79]. Several ways of their interaction have been revealed: through dynamin family proteins, that are responsible for membrane fission during budding, through myosin Myo1c, and through mechanosensitive actin-bound protein filamin-A [80]. It was also shown that actin polymerization supports uniform distribution of caveolae on PM, and their budding requires activation of formin mDia1 by Abl-kinase [81]. In contrast, hyperactivation of mDia1 induces caveolae flattening accompanied by formation of stress fibrils and cell stretching.
Fluorescence microscopy analysis has confirmed association of GPI-APs with caveolae [82, 83]. Ultrastructural analysis by electron microscopy has revealed that very few GPI-APs can be found inside caveolae cavity, while their majority is located on PM in the vicinity of caveolae attachment to PM [82, 84] (Fig. 3d). Moreover, these regions, as well as other regions of GPI-APs clustering without caveolae, have increased electronic density, indicating possibly cytoskeleton condensation there [84]. After oligomerization with antibodies, GPI-APs are translocated to caveolae, but it does not change the number of caveolae [82]. Inhibition of GPI-AP biosynthesis increases caveolae number [85], indicating that biogenesis of caveolae does not depend on GPI-APs, yet pointing out the activation of some compensatory mechanisms in order to compensate (partial) dysfunction of caveolae in the absence of GPI-APs.
Since SFKs are among the major activators of Abl-kinases [64], we suppose that GPI-APs regulate dynamics of caveolae through activation of SFKs, Abl-kinases, and actin polymerization in the region of caveolae attachment of PM (Fig. 3d). It is also possible to assume that effects of GPI-APs on caveolae are mediated by cortical cytoskeleton contraction induced by alignment of dynamic filaments. As mentioned above, caveolae play an important role in regulating activity of various receptor-signaling complexes, thus an impact on caveolae is certainly reflected on signaling of these complexes.
GPI-APs ENDOCYTOSIS
Despite the close relation of GPI-APs with caveolae, their endocytosis via caveolae is still debated [78, 86]. At the same time, GPI-APs have a marked ability to internalize extracellular fluid phase and large oligomeric complexes by clathrin-dependent and clathrin- and dynamin-independent mechanisms [73]. Clathrin-dependent internalization of GPI-APs occurs after their interaction with classical endocytosis regulators such as LDL-receptor like protein (LRP1) and lipopolysaccharide binding protein (LBP) [86]. In the absence of such lateral associations, GPI-APs are internalized by specific clathrin- and dynamin-independent mechanisms with formation of so-called GPI-AP-enriched early endosomal compartments (GEECs). Unlike other types of endocytosis, GEECs do not have specialized coat proteins and therefore have large variation in shape and size and frequently stay attached to PM as tubular structures [87, 88]. In the absence of specialized structural proteins, the main driving force of GEECs formation is polymerization and contraction of cortical cytoskeleton [89] (Fig. 3e). Compensation of membrane area during stretching and regulation of activity of surface proteins are thought to be the main functions of GEECs [87].
GPI-AP oligomerization induces formation of GEECs. This process has been studied at a single molecule level [90]. GEECs are originated in the region of dense actin patches with size less than 1 µm, indicating the crucial role of actin cytoskeleton not only in maturation, but also in initiation of GEECs. GEEC formation is strictly dependent on cholesterol, pointing out the importance of lipid rafts in their biogenesis. Then, the small GTPase Cdc42 that belongs to the Rho GTPase family binds in the place of GEEC initiation. Cdc42, in turn, activates WASP, which induces actin polymerization and branching. Cdc42 associates with PM in its active GTP-bound form, and the duration of this association has a crucial effect on further cytoskeleton structure. It has been found that Cdc42 activity is stopped by ADP-ribosylation factor ARF1, while inactivation of ARF1, as well as the use of constitutively active Cdc42, induces the formation of cortical spike-like structures instead of invaginations [91] (Fig. 3f). Switching from invagination to spikes occurs when Cdc42 residence time in the complex is increased from 2 to 4 s.
ANALOGY OF GPI-AP-INDUCED CYTOSKELETON WITH IMMUNE SYNAPSE
T-cell activation is accompanied by formation of immune synapse – a region of tight contact between the T-cell and antigen-presenting cell wherein many receptors and signaling molecules are concentrated [92, 93]. The cytoskeleton structure in an immune synapse is similar to the aster-like structure underlying GPI-AP oligomer, proposed by Mayor’s group, but it is 10 times larger. It consists of radially allocated filaments where polymerization of barbed ends pointing outward causes retrograde filament flow towards the center of the synapse. This similarity suggests an analogy between immune synapse formation and GPI-AP-induced response. As in the case of GPI-AP oligomer, T-cell signaling is initiated by SFK (Lck) [93], and this activation is critically dependent on actin cytoskeleton and myosin-II activity [92, 93]. There are several models of actin cytoskeleton interplay with T-cell signaling that are also plausible for GPI-AP signaling. According to these models, the cytoskeleton serves for: (i) concentrating of T-cell receptor and intracellular signaling molecule in the synapse; (ii) stabilization of lipid raft in the synapse region, which promotes activation of Lck and exclusion of phosphatases; (iii) T-cell receptor activation by mechanical tension [93]. Although structural data on mechanosensitivity of T-cell receptors are still lacking, mechanical action of cytoskeleton is considered as one of the crucial steps in T-cell activation. Mechanical tension in the complex of T-cell receptor with an antigen (in the complex with the main histocompatibility complex) provides high specificity and sensitivity of T-cell activation down to activation by a single antigen molecule [94].
Mechanisms of cytoskeleton rearrangement and tension generation in the region of the immune synapse are still elusive. Integrins, particularly LFA-1, play an important role in adhesion initiation, but integrins need mechanical tension for activation [95]. Upon formation of an immune synapse, actin filaments polymerize outwards, causing retrograde actin filament flow towards the center of the synapse [96]. It is thought that this retrograde flow is responsible for the observed motion of integrins and T-cell receptor towards the center of the synapse, but their movement is several-fold slower than the retrograde flow. To explain this discrepancy, existence of a frictional but not rigid link between these molecules and cytoskeletons has been suggested, although molecular details of this link are not known [93]. As we suppose, GPI-APs are able to align filaments and induce their polymerization. In addition, it can be expected that their lipid-mediated link to cytoskeleton has frictional property, so GPI-APs may have an important function in formation of immune synapse cytoskeleton. Frequent appearance of GPI-APs in immune synapses [97] favors this assumption. Oligomerization of various GPI-APs by antibodies potentiates T-cell activation substantially and can even activate it without additional stimuli in some cases [6].
DESCRIPTION OF PHYSIOLOGICAL ACTIVITIES OF uPAR AND T-CADHERIN
ACCORDING TO THE SUGGESTED MODEL
uPAR. One of the GPI-APs that regulate lymphocyte adhesion and migration is uPAR [35]. uPAR expression is rather limited in all tissues except leukocytes, but it increases significantly during tissue remodeling, particularly in wound healing, vessels and nerve growth, as well as in cancer [36]. uPAR ligands are the urokinase and extracellular matrix components vitronectin and fibulin V [31, 37, 38]. A close relation between uPAR and integrins has been established, but their direct interaction is still under debate [98]. uPAR was among the first GPI-APs that were shown to affect cytoskeleton [25, 99]. In those experiments, uPAR activation by urokinase or its kinase-deficient analog induced directed cell migration towards increasing urokinase concentration. This process was accompanied by redistribution of uPAR, SFKs, and integrins to the leading edge and condensation of cytoskeleton in this region, indicating directed transport of these molecules. As in the case of immune synapse (see above), it is assumed that the integrins are responsible for uPAR-induced chemotaxis through interacting with uPAR [100]. Further, the mechanism of cytoskeleton rearrangement triggering at an integrin adhesion site resembles GPI-AP signaling: activation of SFK by Gα-subunit of G-protein and further activation of SFK substrates [101]. Thus, uPAR may serve for triggering cytoskeletal rearrangement at adhesion contact. This assumption is supported by data on inhibition of uPAR-induced chemotaxis by pertussis toxin that inhibits G-proteins [25] and consequently the initial stage of GPI-AP signaling as well.
Morphological analysis of cell structures that are formed with the aid of uPAR and integrin complex provides more evidence that in this complex uRAR is responsible for cytoskeleton activation, while integrin – for connection to extracellular matrix. In adhesion contacts, filament ends are attached to extracellular matrix through integrins. uPAR activation can induce formation of filaments that have no points of attachment to extracellular matrix. In the work of Degrise et al., in the first 5 min after uPAR stimulation, marked accumulation of actin in cell periphery was observed, and after 30 min it was transformed into ring-like filaments [25]. At the cell–matrix contact, the effect of uPAR depends on matrix composition. On vitronectin, which is a ligand for uPAR, cells are spread as wide as possible irrespectively of integrins, and uPAR is fully colocalized with vitronectin [102]. This indicates that uPAR may act as an adhesion molecule. On fibronectin and collagen that bind to integrins but not to uPAR, cells are significantly less spread than on vitronectin, and they form filopodia. Analysis of distribution of uPAR and integrins in filopodia shows that near its base uPAR and integrins are colocalized and filopodia stay straight, but at remote sites integrin disappears and filopodia form disperse bundle and terminate. These data support our hypothesis that cytoskeleton rearrangement is initiated not by integrins, but by uPAR. The fundamental difference of such activation stimulus is that it is present over the whole length of filopodia, not only on its end, as in the case of integrins, and at that the underlying filaments retain the ability to slide along the membrane and extracellular matrix.
Besides its role in chemotaxis, uPAR is able to induce release of intracellular vesicle contents, in particular neutrophil degranulation, and to transactivate epidermal growth factor receptor (EGFR) [61, 103]. Neutrophil degranulation has been shown to be the result of PLCγ-mediated increase of intracellular calcium concentration [103]. That is in agreement with the common perception of exocytosis as Ca2+-dependent vesicle fusion with PM mediated by SNARE-family proteins [104]. However, it now becomes more evident that cytoskeleton activity is needed for exocytosis, particularly for a vesicle to approach the PM [105]. In turn, calcium activates myosin light chain kinase, which activates myosin and induces its motion [106]. In this regard, it is reasonable to ask whether calcium is necessary for GPI-AP-induced cytoskeletal rearrangement and what is the source of this calcium? PLCγ activation that occurs due to GPI-AP oligomerization is known to stimulate phosphatidylinositol production and calcium release from endoplasmic reticulum. This calcium probably serves as positive feedback for enhancement of cortical contraction, yet calcium signaling may participate in earlier GPI-AP signaling stages before activation of PLCγ. So, transactivation of EGFR has been shown for several GPI-APs besides uPAR (Table 1), particularly for receptor of myelin inhibitor Nogo-66 (NgR) [46]. In the case of NgR, transactivation did not depend on PLCs, SFKs, and G-proteins, but it was blocked almost completely by chelation of cytoplasmic calcium [46]. Calcium simulates calmodulin binding to EGFR, which stabilizes its active conformation, but this occurs after stimulation with EGF. What the activating stimulus is during transactivation of EGFR by GPI-APs is still unknown. One can speculate that cortical tension induced by filament alignment with GPI-AP oligomer activates EGFR through caveolae flattening or by direct mechanical tension. The hypothesis of receptor activation by mechanical tension was proposed earlier, particularly for RTKs [107].
T-cadherin. T-cadherin belongs to the cadherin family of cell–cell adhesion molecules, but unlike other members of this family, it is devoid of the cytoplasmic part of a GPI-AP, and that is why it has obtained the prefix “T” (from “truncated”) [108]. T-cadherin is expressed mainly in cardiovascular and nerve tissues [31, 109]. T-cadherin was discovered as a guidance cue that restricts motor neurons from T-cadherin expressing tissue regions [11]. Ligands for most of cadherins, including T-cadherin, are cadherins themselves, and their interaction occurs in trans-conformation, i.e. while interacting cadherins are located on neighboring cells [109]. Accordingly, it was shown that axon repulsion from T-cadherin expressing tissues is mediated by T-cadherin as a receptor on the axon [110]. As noted above, cell guidance and repulsion is inherent for many GPI-APs. Repulsion is driven by contraction of cell cortex as a consequence of cell guidance receptor activation [31, 61, 62]. As far as in both contacting cells T-cadherin has high lateral diffusion, their interaction results in T-cadherin accumulation in the contact region. According to our hypothesis, this promotes alignment of cortical filaments, which induces contraction and consequent repulsion.
In studies of V. A. Tkachuk’s group, it was shown that T-cadherin mediates effects of LDLs on activity of adrenergic receptors [7, 111, 112]. The dissociation constant of LDL with T-cadherin was estimated as 45 µg/ml [9]. It is known that the dissociation constant is the ratio of rate constants of reverse (unbinding) and forward (binding) reactions, while the forward reaction constant is determined by diffusion and usually lies in the range of 106-108 M–1·s–1. This allows one to estimate the unbinding rate constant, which comprises ~0.5 s–1, i.e. the T-cadherin/LDL complex lifetime is ~2 s. This factor probably determines the outcome of T-cadherin oligomerization with LDL since it limits the size and the lifetime of underling cytoskeleton structure to be induced according to our hypothesis. This time is longer than the immobilization time required to activate PLCγ by GPI-AP oligomer (~0.5 s) [4]. Indeed, it was shown that LDL induces calcium response either alone or in the presence of other agonists [8, 113], and this requires G-protein activity [9]. It is also possible that potentiation of adrenergic receptors by LDL/T-cadherin oligomer does not require cytoskeleton. In this case, the lipid raft in the base of this oligomer may favor G-protein and GPCR complex formation, as it does for G-protein and SFK complex (Fig. 4).
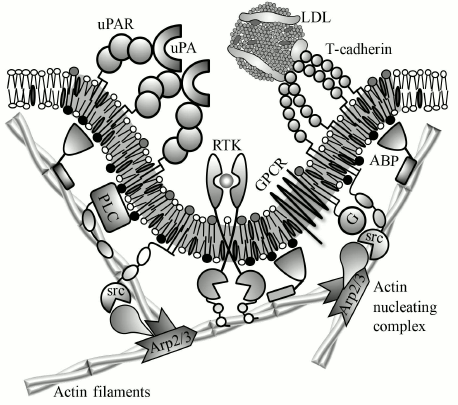
Fig. 4. Schematic representation of suggested mechanist of uPAR and T-cadherin effect on membrane properties and activity of transmembrane receptors. Urokinase (uPA) and LDLs induce oligomerization of uPAR and T-cadherin, respectively. Oligomeric receptor induces lipid raft formation (gray membrane region) that binds proteins from the cytoplasmic leaflet based on a raft-localization motif (see annotations from Fig. 2). These proteins include G-proteins (G), src-family kinases (src), and actin-binding proteins (ABP). T-cadherin-induced lipid raft may potentiate GPCRs by facilitating its interaction with G-proteins. ABP links short dynamic actin filaments with the lipid raft, which creates a scaffold for signaling complex. This scaffold is required for activation of phospholipase C (PLC) and probably other receptors, particularly receptor tyrosine kinases (RTKs). Other possible functions of GPI-AP-induced cytoskeleton are concentrating of receptors and signaling molecules, their activation by mechanical tension, change of membrane shape, and assembly/disassembly of caveolae.
In 2004, one more ligand of T-cadherin was discovered – a high molecular weight form of adiponectin [114]. T-cadherin and adiponectin knockout mice have similar increase of infarct size during ischemia–reperfusion injury [10]. Adiponectin is bound to T-cadherin on PM of cardiomyocytes, while adiponectin knockout results in disappearance of T-cadherin. It should be noted that T-cadherin is the most abundant GPI-AP on cardiac sarcolemma [115]. According to our hypothesis, substantial stability and large size of adiponectin/T-cadherin oligomer should result in formation of condensed cytoskeletal structures underneath this oligomer. Indeed, stimulation of cardiomyocytes with adiponectin activates Rho GTPases, deactivates cofilin, a protein that severs actin filaments, and induces actin polymerization and formation of membrane ruffles [116, 117]. Membrane ruffles are used as a membrane container; they are flattened during tension and compensate increase in cell surface density, preventing membrane rupture [87]. Actin polymerization and formation of membrane ruffles by T-cadherin/adiponectin complex may protect cardiomyocyte from mechanical damage.
The data now available allow one to say confidently that GPI-APs are competent receptors that fulfil important physiological functions of signal reception and transduction. Since GPI-APs are devoid of intracellular domain, they transmit signals by affecting properties of the membrane. Spatial reorganization of GPI-APs on a cell surface can affect lipid bilayer properties, which in turn trigger intracellular signaling. With the aid of long and saturated lipid moieties, GPI-APs are able to create and stabilize a liquid ordered phase – a lipid raft that serves as a platform for binding of many effectors including G-proteins and SFK, whose interaction results in activation of SFK. By analogy, it can be supposed that a GPI-AP-induced lipid raft promotes association of G-protein with GPCR, thus potentiating GPCR activity as in the case of adrenergic receptors potentiation by T-cadherin (Fig. 4). At the same time, many types of GPI-AP activities, such as regulation of adhesion, cell polarity, migration, and endocytosis, point on formation of more stable molecular complexes that can be stabilized by a lipid raft.
A lipid raft is a highly dynamic structure with high lateral diffusion of lipids – the mean retention time of saturated lipids is 10-20 ms. However, the GPI-AP-induced lipid raft is able to retain signaling proteins for more than 100 ms. This indicates that the lipid raft catalyzes just the initial stage of complex formation. Prolonged stabilization may be achieved with the aid of cytoskeleton, as in the case of the GPI-AP and PLCγ complex. We suppose that assembly of this cytoskeleton scaffold is initiated by GPI-AP oligomerization that consolidates cytoskeleton machinery within the lipid raft. Short actin filaments (through actin-binding proteins) and actin-regulating proteins such as cortactin, formins, and WASP interact with lipid rafts and are activated by SFK directly or through its effectors (Fig. 4). This structure is dependent on GPI-AP oligomer stability due to the necessity of membrane binding for activity of these proteins. It can also be expected that the structure of induced cytoskeleton depends on the size and the shape of GPI-AP oligomer, for example, through an effect of membrane curvature that defines the choice between cell protrusions or invaginations. It is well known that cytoskeleton plays an important role in activity of many receptors by regulating their traffic, membrane distribution, and interactions between each other and with scaffold proteins, by exerting mechanical tension, as well as by affecting caveolae and membrane lipid composition.
This work was financially supported by the Russian Science Foundation (project No. 14-24-00086).
REFERENCES
1.Kinoshita, T. (2014) Biosynthesis and deficiencies
of glycosylphosphatidylinositol, Proc. Jpn. Acad. Ser. B,
90, 130-143.
2.Stefanova, I., Horejsi, V., Ansotegui, I. J.,
Knapp, W., and Stockinger, H. (1991) GPI-anchored cell-surface
molecules complexed to protein tyrosine kinases, Science,
254, 1016-1019.
3.Suzuki, K. G. N., Fujiwara, T. K., Sanematsu, F.,
Iino, R., Edidin, M., and Kusumi, A. (2007) GPI-anchored receptor
clusters transiently recruit Lyn and G alpha for temporary cluster
immobilization and Lyn activation: single-molecule tracking study 1,
J. Cell Biol., 177, 717-730.
4.Suzuki, K. G. N., Fujiwara, T. K., Edidin, M., and
Kusumi, A. (2007) Dynamic recruitment of phospholipase C gamma at
transiently immobilized GPI-anchored receptor clusters induces
IP3-Ca2+ signaling: single-molecule tracking study 2, J.
Cell Biol., 177, 731-742.
5.Suzuki, K. G. N., Kasai, R. S., Hirosawa, K. M.,
Nemoto, Y. L., Ishibashi, M., Miwa, Y., Fujiwara, T. K., and Kusumi, A.
(2012) Transient GPI-anchored protein homodimers are units for raft
organization and function, Nat. Chem. Biol., 8,
774-783.
6.Loertscher, R., and Lavery, P. (2002) The role of
glycosyl phosphatidyl inositol (GPI)-anchored cell surface proteins in
T-cell activation, Transpl. Immunol., 9, 93-96.
7.Tkachuk, V. A., Bochkov, V. N., Philippova, M. P.,
Stambolsky, D. V., Kuzmenko, E. S., Sidorova, M. V., Molokoedov, A. S.,
Spirov, V. G., and Resink, T. J. (1998) Identification of an atypical
lipoprotein-binding protein from human aortic smooth muscle as
T-cadherin, FEBS Lett., 421, 208-212.
8.Bochkov, V. N., Tkachuk, V. A., Hahn, A. W.,
Bernhardt, J., Buhler, F. R., and Resink, T. J. (1993) Concerted
effects of lipoproteins and angiotensin II on signal transduction
processes in vascular smooth muscle cells, Arterioscler.
Thromb., 13, 1261-1269.
9.Bochkov, V. N., Tkachuk, V. A., Kuzmenko, Y. S.,
Borisova, Y. L., Buhler, F. R., and Resink, T. J. (1994)
Characteristics of low and high-density lipoprotein binding and
lipoprotein-induced signaling in quiescent human vascular smooth muscle
cells, Mol. Pharmacol., 45, 262-270.
10.Denzel, M. S., Scimia, M.-C., Zumstein, P. M.,
Walsh, K., Ruiz-Lozano, P., and Ranscht, B. (2010) T-cadherin is
critical for adiponectin-mediated cardioprotection in mice, J. Clin.
Invest., 120, 4342-4352.
11.Fredette, B. J., and Ranscht, B. (1994)
T-cadherin expression delineates specific regions of the developing
motor axon-hindlimb projection pathway, J. Neurosci., 14,
7331-7346.
12.Rubina, K., Kalinina, N., Potekhina, A.,
Efimenko, A., Semina, E., Poliakov, A., Wilkinson, D. G., Parfyonova,
Y., and Tkachuk, V. (2007) T-cadherin suppresses angiogenesis in
vivo by inhibiting migration of endothelial cells,
Angiogenesis, 10, 183-195.
13.Benting, J., Rietveld, A., Ansorge, I., and
Simons, K. (1999) Acyl and alkyl chain length of GPI-anchors is
critical for raft association in vitro, FEBS Lett.,
462, 47-50.
14.Raghupathy, R., Anilkumar, A. A., Polley, A.,
Singh, P. P., Yadav, M., Johnson, C., Suryawanshi, S., Saikam, V.,
Sawant, S. D., Panda, A., Guo, Z., Vishwakarma, R. A., Rao, M., and
Mayor, S. (2015) Transbilayer lipid interactions mediate nanoclustering
of lipid-anchored proteins, Cell, 161, 581-594.
15.Pike, L. J. (2009) The challenge of lipid rafts,
J. Lipid Res., 50, S323-328.
16.Binder, W. H., Barragan, V., and Menger, F. M.
(2003). Domains and rafts in lipid membranes, Angew. Chem. Int.
Ed., 42, 5802-5827.
17.Hancock, J. F. (2006) Lipid rafts: contentious
only from simplistic standpoints, Nat. Rev. Mol. Cell Biol.,
7, 456-462.
18.Viola, A., and Gupta, N. (2007) Tether and trap:
regulation of membrane-raft dynamics by actin-binding proteins, Nat.
Rev. Immunol., 7, 889-896.
19.Zhou, Y., and Hancock, J. F. (2015) Ras
nanoclusters: versatile lipid-based signaling platforms, Biochim.
Biophys. Acta, 1853, 841-849.
20.Simons, K., and Sampaio, J. L. (2011) Membrane
organization and lipid rafts, Cold Spring Harb. Perspect. Biol.,
3, a004697.
21.Saha, S., Lee, I.-H., Polley, A., Groves, J. T.,
Rao, M., and Mayor, S. (2015) Diffusion of GPI-anchored proteins is
influenced by the activity of dynamic cortical actin, Mol. Biol.
Cell, 25, 4033-4045.
22.Lee, I.-H., Saha, S., Polley, A., Huang, H.,
Mayor, S., Rao, M., and Groves, J. T. (2015) Live cell plasma membranes
do not exhibit a miscibility phase transition over a wide range of
temperatures, J. Phys. Chem. B, 119, 4450-4459.
23.Owen, D. M., Williamson, D. J., Magenau, A., and
Gaus, K. (2012) Sub-resolution lipid domains exist in the plasma
membrane and regulate protein diffusion and distribution, Nat.
Commun., 3, 1256.
24.Harder, T., and Simons, K. (1999) Clusters of
glycolipid and glycosylphosphatidylinositol-anchored proteins in
lymphoid cells: accumulation of actin regulated by local tyrosine
phosphorylation, Eur. J. Immunol., 29, 556-562.
25.Degryse, B., Resnati, M., Rabbani, S. A., Villa,
A., Fazioli, F., and Blasi, F. (1999) Src-dependence and
pertussis-toxin sensitivity of urokinase receptor-dependent chemotaxis
and cytoskeleton reorganization in rat smooth muscle cells,
Blood, 94, 649-662.
26.Varma, R., and Mayor, S. (1998) GPI-anchored
proteins are organized in submicron domains at the cell surface,
Nature, 394, 798-801.
27.Sharma, P., Varma, R., Sarasij, R. C., Ira,
Gousset, K., Krishnamoorthy, G., Rao, M., and Mayor, S. (2004)
Nanoscale organization of multiple GPI-anchored proteins in living cell
membranes, Cell, 116, 577-589.
28.Goswami, D., Gowrishankar, K., Bilgrami, S.,
Ghosh, S., Raghupathy, R., Chadda, R., Vishwakarma, R., Rao, M., and
Mayor, S. (2008) Nanoclusters of GPI-anchored proteins are formed by
cortical actin-driven activity, Cell, 135, 1085-1097.
29.Gowrishankar, K., Ghosh, S., Saha, S. C. R.,
Mayor, S., and Rao, M. (2012) Active remodeling of cortical actin
regulates spatiotemporal organization of cell surface molecules,
Cell, 149, 1353-1367.
30.Saha, S., Lee, I.-H., Polley, A., Groves, J. T.,
Rao, M., and Mayor, S. (2015) Diffusion of GPI-anchored proteins is
influenced by the activity of dynamic cortical actin, Mol. Biol.
Cell, 26, 4033-4045.
31.Rubina, K. A., and Tkachuk, V. A. (2015) Guidance
receptors in the nervous and cardiovascular systems, Biochemistry
(Moscow), 80, 1235-1253.
32.Philippova, M., Joshi, M. B., Pfaff, D.,
Kyriakakis, E., Maslova, K., Erne, P., and Resink, T. J. (2012)
T-cadherin attenuates insulin-dependent signaling, eNOS activation, and
angiogenesis in vascular endothelial cells, Cardiovasc. Res.,
93, 498-507.
33.Semina, E. V., Rubina, K. A., Sysoeva, V. Y.,
Rutkevich, P. N., Kashirina, N. M., and Tkachuk, V. A. (2013) Novel
mechanism regulating endothelial permeability via T-cadherin-dependent
VE-cadherin phosphorylation and clathrin-mediated endocytosis, Mol.
Cell. Biochem., 387, 39-53.
34.Tyrberg, B., Miles, P., Azizian, K. T., Denzel,
M. S., Nieves, M. L., Monosov, E. Z., Levine, F., and Ranscht, B.
(2011) T-cadherin (Cdh13) in association with pancreatic β-cell
granules contributes to second phase insulin secretion, Islets,
3, 327-337.
35.Blasi, F., and Tjwa, M. (2002) uPAR: a versatile
signaling orchestrator, Nat. Rev. Mol. Cell Biol., 3,
932-943.
36.Smith, H. W., and Marshall, C. J. (2010)
Regulation of cell signaling by uPAR, Nat. Rev. Mol. Cell Biol.,
11, 23-36.
37.Stepanova, V. V., and Tkachuk, V. A. (2002)
Urokinase as a multidomain protein and polyfunctional cell regulator,
Biochemistry (Moscow), 67, 109-118.
38.Kapustin, A., Stepanova, V., Aniol, N., Cines, D.
B., Poliakov, A., Yarovoi, S., Lebedeva, T., Wait, R., Ryzhakov, G.,
Parfyonova, Y., Gursky, Y., Yanagisawa, H., Minashkin, M.,
Beabealashvilli, R., Vorotnikov, A., Bobik, A., and Tkachuk, V. (2012)
Fibulin-5 binds urokinase-type plasminogen activator and mediates
urokinase-stimulated β1-integrin-dependent cell migration,
Biochem. J., 443, 491-503.
39.Blasi, F., and Sidenius, N. (2010) The urokinase
receptor: focused cell surface proteolysis, cell adhesion and
signaling, FEBS Lett., 584, 1923-1930.
40.Resnati, M., Pallavicini, I., Wang, J. M.,
Oppenheim, J., Serhan, C. N., Romano, M., and Blasi, F. (2002) The
fibrinolytic receptor for urokinase activates the G protein-coupled
chemotactic receptor FPRL1/LXA4R, Proc. Natl. Acad. Sci. USA,
99, 1359-1364.
41.Liu, D., Aguirre Ghiso, J., Estrada, Y., and
Ossowski, L. (2002) EGFR is a transducer of the urokinase receptor
initiated signal that is required for in vivo growth of a human
carcinoma, Cancer Cell, 1, 445-457.
42.Madsen, C. D., Ferraris, G. M. S., Andolfo, A.,
Cunningham, O., and Sidenius, N. (2007) uPAR-induced cell adhesion and
migration: vitronectin provides the key, J. Cell Biol.,
177, 927-939.
43.Zanoni, I., Ostuni, R., Marek, L. R., Barresi,
S., Barbalat, R., Barton, G. M., Granucci, F., and Kagan, J. C. (2011)
CD14 controls the LPS-induced endocytosis of Toll-like receptor 4,
Cell, 147, 868-880.
44.Sabharanjak, S., and Mayor, S. (2004) Folate
receptor endocytosis and trafficking, Adv. Drug Deliv. Rev.,
56, 1099-1109.
45.Bhagatji, P., Leventis, R., Comeau, J., Refaei,
M., and Silvius, J. R. (2009) Steric and not structure-specific factors
dictate the endocytic mechanism of
glycosylphosphatidylinositol-anchored proteins, J. Cell Biol.,
186, 615-628.
46Koprivica, V., Cho, K.-S., Park, J. B., Yiu, G., Atwal, J., Gore, B.,
Kim, J. A., Lin, E., Tessier-Lavigne, M., Chen, D. F., and He, Z.
(2005) EGFR activation mediates inhibition of axon regeneration by
myelin and chondroitin sulfate proteoglycans, Science,
310, 106-110.
47.Eggeling, C., Ringemann, C., Medda, R.,
Schwarzmann, G., Sandhoff, K., Polyakova, S., Belov, V. N., Hein, B.,
Von Middendorff, C., Schonle, A., and Hell, S. W. (2009) Direct
observation of the nanoscale dynamics of membrane lipids in a living
cell, Nature, 457, 1159-1162.
48.Kay, J. G., Koivusalo, M., Ma, X., Wohland, T.,
and Grinstein, S. (2012) Phosphatidylserine dynamics in cellular
membranes, Mol. Biol. Cell, 23, 2198-2212.
49.Wei, Y., Lukashev, M., Simon, D. I., Bodary, S.
C., Rosenberg, S., Doyle, M. V., and Chapman, H. A. (1996) Regulation
of integrin function by the urokinase receptor, Science,
273, 1551-1555.
50.Busso, N., Masur, S. K., Lazega, D., Waxman, S.,
and Ossowski, L. (1994) Induction of cell migration by pro-urokinase
binding to its receptor: possible mechanism for signal transduction in
human epithelial cells, J. Cell Biol., 126, 259-270.
51.Ben-Harush, K., Maimon, T., Patla, I., Villa, E.,
and Medalia, O. (2010) Visualizing cellular processes at the molecular
level by cryo-electron tomography, J. Cell Sci., 123,
7-12.
52.Lucic, V., Rigort, A., and Baumeister, W. (2013)
Cryo-electron tomography: the challenge of doing structural biology
in situ, J. Cell Biol., 202, 407-419.
53.Urban, E., Jacob, S., Nemethova, M., Resch, G.
P., and Small, J. V. (2010) Electron tomography reveals unbranched
networks of actin filaments in lamellipodia, Nat. Cell Biol.,
12, 429-435.
54.Medalia, O., Weber, I., Frangakis, A. S., and
Nicastro, D. (2002) Macromolecular architecture in eukaryotic cells
visualized by cryoelectron tomography, Science, 298,
1209-1213.
55.Medalia, O., Beck, M., Ecke, M., Weber, I., and
Neujahr, R. (2007) Organization of actin networks in intact filopodia,
Curr. Biol., 17, 79-84.
56.Maurer, U. E., Sodeik, B., and Grunewald, K.
(2008) Native 3D intermediates of membrane fusion in herpes simplex
virus 1 entry, Proc. Natl. Acad. Sci. USA, 105,
10559-10564.
57.Kudryashev, M., Lepper, S., Baumeister, W.,
Cyrklaff, M., and Frischknecht, F. (2010) Geometric constrains for
detecting short actin filaments by cryogenic electron tomography,
PMC Biophys., 3, 6.
58.Suzuki, K., and Sheetz, M. P. (2001) Binding of
cross-linked glycosylphosphatidylinositol-anchored proteins to discrete
actin-associated sites and cholesterol-dependent domains, Biophys.
J., 81, 2181-2189.
59.Sanchez, T., Chen, D. T. N., DeCamp, S. J.,
Heymann, M., and Dogic, Z. (2012) Spontaneous motion in hierarchically
assembled active matter, Nature, 491, 431-434.
60.Reymann, A.-C., Boujemaa-Paterski, R., Martiel,
J.-L., Guerin, C., Cao, W., Chin, H. F., La Cruz, De, E. M., Thery, M.,
and Blanchoin, L. (2012) Actin network architecture can determine
myosin motor activity, Science, 336, 1310-1314.
61.Dent, E. W., Gupton, S. L., and Gertler, F. B.
(2011) The growth cone cytoskeleton in axon outgrowth and guidance,
Cold Spring Harb. Perspect. Biol., 3, a001800.
62.Fagotto, F., Rohani, N., Touret, A.-S., and Li,
R. (2014) A molecular base for cell sorting at embryonic boundaries:
contact inhibition of cadherin adhesion by ephrin/Eph-dependent
contractility, Dev. Cell, 27, 1-16.
63.Reynolds, A. B., Kanner, S. B., Bouton, A. H.,
Schaller, M. D., Weed, S. A., Flynn, D. C., and Parsons, J. T. (2014)
SRChing for the substrates of Src, Oncogene, 33,
4537-4547.
64.Greuber, E. K., Smith-Pearson, P., Wang, J., and
Pendergast, A. M. (2013) Role of ABL family kinases in cancer: from
leukemia to solid tumors, Nat. Rev. Cancer, 13,
559-571.
65.Girao, H., Geli, M.-I., and Idrissi, F.-Z. (2008)
Actin in the endocytic pathway: from yeast to mammals, FEBS
Lett., 582, 2112-2119.
66.Herve, J.-C. (2014) Reciprocal influences between
cell cytoskeleton and membrane channels, receptors and transporters,
Biochim. Biophys. Acta, 1838, 511-513.
67.Gorelik, R., Yang, C., Kameswaran, V., Dominguez,
R., and Svitkina, T. (2011) Mechanisms of plasma membrane targeting of
formin mDia2 through its amino terminal domains, Mol. Biol.
Cell, 22, 189-201.
68.Blanchoin, L., Boujemaa-Paterski, R., Sykes, C.,
and Plastino, J. (2014) Actin dynamics, architecture, and mechanics in
cell motility, Physiol. Rev., 94, 235-263.
69.Takenawa, T., and Suetsugu, S. (2007) The
WASP-WAVE protein network: connecting the membrane to the cytoskeleton,
Nat. Rev. Mol. Cell Biol., 8, 37-48.
70.McConnell, R. E., and Tyska, M. J. (2010)
Leveraging the membrane – cytoskeleton interface with myosin-1,
Trends Cell Biol., 20, 418-426.
71.Clucas, J., and Valderrama, F. (2015) ERM
proteins in cancer progression, J. Cell Sci., 128,
1253-1253.
72.Saarikangas, J., Zhao, H., and Lappalainen, P.
(2010) Regulation of the actin cytoskeleton–plasma membrane
interplay by phosphoinositides, Physiol. Rev., 90,
259-289.
73.Mayor, S., and Riezman, H. (2004) Sorting
GPI-anchored proteins, Nat. Rev. Mol. Cell Biol., 5,
110-120.
74.Sprenger, R. R., Fontijn, R. D., Van Marle, J.,
Pannekoek, H., and Horrevoets, A. J. G. (2006) Spatial segregation of
transport and signaling functions between human endothelial caveolae
and lipid raft proteomes, Biochem. J., 400, 401-410.
75.Locker, J. K., and Schmid, S. L. (2013)
Integrated electron microscopy: super-duper resolution, PLoS
Biol., 11, e1001639
76.Tsui-Pierchala, B. A., Encinas, M., Milbrandt,
J., and Johnson, E. M. (2002) Lipid rafts in neuronal signaling and
function, Trends Neurosci., 25, 412-417.
77.Patel, H. H., Murray, F., and Insel, P. A. (2008)
Caveolae as organizers of pharmacologically relevant signal
transduction molecules, Annu. Rev. Pharmacol. Toxicol.,
48, 359-391.
78.Cheng, J. P. X., and Nichols, B. J. (2015)
Caveolae: one function or many? Trends Cell. Biol., 26,
1-13.
79.Echarri, A., and Del Pozo, M. A. (2015) Caveolae
– mechanosensitive membrane invaginations linked to actin
filaments, J. Cell Sci., 128, 2747-2758.
80.Andrews, N. W., Almeida, P. E., and Corrotte, M.
(2014) Damage control: cellular mechanisms of plasma membrane repair,
Trends Cell Biol., 24, 1-9.
81.Echarri, A., Muriel, O., Pavon, D. M., Azegrouz,
H., Escolar, F., Terron, M. C., Sanchez-Cabo, F., Martinez, F.,
Montoya, M. C., Llorca, O., and Del Pozo, M. A. (2012) Caveolar domain
organization and trafficking is regulated by Abl kinases and mDia1,
J. Cell Sci., 125, 3097-3113.
82.Mayor, S., Rothberg, K. G., and Maxfield, F. R.
(1994) Sequestration of GPI-anchored proteins in caveolae triggered by
cross-linking, Science, 264, 1948-1951.
83.Stahl, A., and Mueller, B. M. (1995) The
urokinase-type plasminogen activator receptor, a GPI-linked protein, is
localized in caveolae, J. Cell Biol., 129, 335-344.
84.Ying, Y. S., and Anderson, R. (1992) Each caveola
contains multiple glycosylphosphatidylinositol-anchored membrane
proteins, Cold Spring Harb. Symp. Quant. Biol., 57,
593-604.
85.Abrami, L., Fivaz, M., Kobayashi, T., Kinoshita,
T., Parton, R. G., and Van der Goot, F. G. (2001) Cross-talk between
caveolae and glycosylphosphatidylinositol-rich domains, J. Biol.
Chem., 276, 30729-30736.
86.Doherty, G. J., and McMahon, H. T. (2009)
Mechanisms of endocytosis, Annu. Rev. Biochem., 78,
857-902.
87.Gauthier, N. C., Masters, T. A., and Sheetz, M.
P. (2012) Mechanical feedback between membrane tension and dynamics,
Trends Cell Biol., 22, 527-535.
88.Kirkham, M., Fujita, A., Chadda, R., Nixon, S.
J., Kurzchalia, T. V., Sharma, D. K., Pagano, R. E., Hancock, J. F.,
Mayor, S., and Parton, R. G. (2005) Ultrastructural identification of
uncoated caveolin-independent early endocytic vehicles, J. Cell
Biol., 168, 465-476.
89.Johannes, L., Parton, R. G., Bassereau, P., and
Mayor, S. (2015) Building endocytic pits without clathrin, Nat. Rev.
Mol. Cell Biol., 16, 1-11.
90.Chadda, R., Howes, M. T., Plowman, S. J.,
Hancock, J. F., Parton, R. G., and Mayor, S. (2007)
Cholesterol-sensitive Cdc42 activation regulates actin polymerization
for endocytosis via the GEEC pathway, Traffic, 8,
702-717.
91.Kumari, S., and Mayor, S. (2008) ARF1 is directly
involved in dynamin-independent endocytosis, Nat. Cell Biol.,
10, 30-41.
92.Dustin, M. L., and Groves, J. T. (2012) Receptor
signaling clusters in the immune synapse, Annu. Rev. Biophys.,
41, 543-556.
93.Yu, Y., Smoligovets, A. A., and Groves, J. T.
(2013) Modulation of T-cell signaling by the actin cytoskeleton, J.
Cell Sci., 126, 1049-1058.
94.Liu, B., Chen, W., Evavold, B. D., and Zhu, C.
(2014) Accumulation of dynamic catch bonds between TCR and agonist
peptide-MHC triggers T-cell signaling, Cell, 157,
357-368.
95.Bakker, G. J., Eich, C., Torreno-Pina, J. A.,
Diez-Ahedo, R., Perez-Samper, G., Van Zanten, T. S., Figdor, C. G.,
Cambi, A., and Garcia-Parajo, M. F. (2012) Lateral mobility of
individual integrin nanoclusters orchestrates the onset for leukocyte
adhesion, Proc. Natl. Acad. Sci. USA, 109, 4869-4874.
96.Kaizuka, Y., Douglass, A. D., Varma, R., Dustin,
M. L., and Vale, R. D. (2007) Mechanisms for segregating T-cell
receptor and adhesion molecules during immunological synapse formation
in Jurkat T-cells, Proc. Natl. Acad. Sci. USA, 104,
20296-20301.
97.Ilangumaran, S., He, H. T., and Hoessli, D. C.
(2000) Microdomains in lymphocyte signaling: beyond GPI-anchored
proteins, Immunol. Today, 21, 2-7.
98.Blasi, F., and Sidenius, N. (2009) Efferocytosis:
another function of uPAR, Blood, 114, 752-753.
99.Blasi, F. (1988) A surface receptor for urokinase
plasminogen activator: a link between the cytoskeleton and the
extracellular matrix, Protoplasma, 145, 95-98.
100.Ossowski, L., and Aguirre-Ghiso, J. A. (2000)
Urokinase receptor and integrin partnership: coordination of signaling
for cell adhesion, migration and growth, Curr. Opin. Cell Biol.,
12, 613-620.
101.Shen, B., Delaney, M. K., and Du, X. (2012)
Inside-out, outside-in, and inside-outside-in: G protein signaling in
integrin-mediated cell adhesion, spreading, and retraction, Curr.
Opin. Cell Biol., 24, 600-606.
102.Zhu, S., Gladson, C. L., White, K. E., Ding,
Q., Stewart, J., Jin, T. H., Chapman, H. A., and Olman, M. A. (2009)
Urokinase receptor mediates lung fibroblast attachment and migration
toward provisional matrix proteins through interaction with multiple
integrins, Am. J. Physiol. Lung Cell. Mol. Physiol., 297,
L97-L108.
103.Sitrin, R. G., Pan, P. M., Harper, H. A., Todd,
R. F., Harsh, D. M., and Blackwood, R. A. (2000) Clustering of
urokinase receptors (uPAR; CD87) induces proinflammatory signaling in
human polymorphonuclear neutrophils, J. Immunol., 165,
3341-3349.
104.Chen, Y. A., and Scheller, R. H. (2001)
SNARE-mediated membrane fusion, Nat. Rev. Mol. Cell Biol.,
2, 98-106.
105.Porat-Shliom, N., Milberg, O., Masedunskas, A.,
and Weigert, R. (2012) Multiple roles for the actin cytoskeleton during
regulated exocytosis, Cell. Mol. Life Sci., 70,
2099-2121.
106.Somlyo, A. P., and Somlyo, A. V. (2003)
Ca2+ sensitivity of smooth muscle and nonmuscle myosin II:
modulated by G proteins, kinases, and myosin phosphatase, Physiol.
Rev., 83, 1325-1358.
107.Salaita, K., and Groves, J. T. (2010) Roles of
the cytoskeleton in regulating EphA2 signals, Commun. Integr.
Biol., 3, 454-457.
108.Ivanov, D. B., Philippova, M. P., and Tkachuk,
V. A. (2001) Structure and functions of classical cadherins,
Biochemistry (Moscow), 66, 1174-1186.
109.Rubina, K. A., Kalinina, N. I., Parfenova, E.
V., and Tkachuk, V. A. (2007) T-cadherin as a receptor involved in the
regulation of angiogenesis and the remodeling of blood vessels,
Biol. Membr. (Moscow), 24, 62-69.
110.Fredette, B. J., Miller, J., and Ranscht, B.
(1996) Inhibition of motor axon growth by T-cadherin substrata,
Development, 122, 3163-3171.
111.Kuzmenko, Y. S., Stambolsky, D., Kern, F.,
Bochkov, V. N., Tkachuk, V. A., and Resink, T. J. (1998)
Characteristics of smooth muscle cell lipoprotein binding proteins
(p105/p130) as T-cadherin and regulation by positive and negative
growth regulators, Biochem. Biophys. Res. Commun., 246,
489-494.
112.Bochkov, V. N., Voino-Yasenetskaya, T. A., and
Tkachuk, V. A. (1991) Epinephrine potentiates activation of human
platelets by low-density lipoproteins, Biochim. Biophys. Acta,
1097, 123-127.
113.Rubina, K., Talovskaya, E., Cherenkov, V.,
Ivanov, D., Stambolsky, D., Storozhevykh, T., Pinelis, V., Shevelev,
A., Parfyonova, Y., Resink, T., Erne, P., and Tkachuk, V. (2005) LDL
induces intracellular signaling and cell migration via atypical
LDL-binding protein T-cadherin, Mol. Cell. Biochem., 273,
33-41.
114.Hug, C., Wang, J., Ahmad, N. S., and Bogan, J.
S. (2004) T-cadherin is a receptor for hexameric and
high-molecular-weight forms of Acrp30/adiponectin, Proc. Natl. Acad.
Sci. USA, 101, 10308-10313.
115.Doyle, D. D., Goings, G. E., Upshaw-Earley, J.,
and Page, E. (1998) T-cadherin is a major glycophosphoinositol-anchored
protein associated with noncaveolar detergent-insoluble domains of the
cardiac sarcolemma, J. Biol. Chem., 273, 6937-6943.
116.Vu, V., Bui, P., Eguchi, M., Xu, A., and
Sweeney, G. (2013) Globular adiponectin induces LKB1/AMPK-dependent
glucose uptake via actin cytoskeleton remodeling, J. Mol.
Endocrinol., 51, 155-165.
117.Palanivel, R., Ganguly, R., Turdi, S., Xu, A.,
and Sweeney, G. (2014) Adiponectin stimulates Rho-mediated actin
cytoskeleton remodeling and glucose uptake via APPL1 in primary
cardiomyocytes, Metabolism, 63, 1363-1373.