REVIEW: Mechanisms of Changes in Immune Response during Bacterial Coinfections of the Respiratory Tract
E. N. Sviriaeva1,2, K. V. Korneev1,2, M. S. Drutskaya1,2, and D. V. Kuprash1,2*
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russia; E-mail: kuprash@gmail.com2Lomonosov Moscow State University, Faculty of Biology, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received July 17, 2016; Revision received August 19, 2016
Acute diseases of the respiratory tract are often caused by viral pathogens and accompanying secondary bacterial infections. It is known that the development of such bacterial complications is caused mainly by a decreased infiltration with immune system cells and by suppressed inflammation in the lungs. There are significant advances in understanding the mechanisms of secondary infections, although many details remain unclear. This review summarizes current knowledge of the molecular and cellular changes in the host organism that can influence the course of bacterial coinfections in the respiratory tract.
KEY WORDS: secondary bacterial coinfection, influenza, bacteria, pneumonia, macrophages, Toll-like receptorsDOI: 10.1134/S0006297916110110
Abbreviations: CXCL, chemokines, (C-X-C motif) ligand; ICAM-1, intercellular adhesion molecule 1; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; IL, interleukin; NA, neuraminidase; NF-κB, transcription factor κB; PPARγ, peroxisome proliferator-activated receptor gamma; RSV, respiratory syncytial virus; TGF-β, transforming growth factor beta; TLRs, Toll-like receptors; TNF, tumor necrosis factor.
Notwithstanding the high level of current medicine, severe and sometimes
lethal consequences of infection diseases remain an urgent problem. In
search for effective approaches against infections, great attention is
being given to creating experimental models in which a specific
pathogen under study would be isolated from the environment. However,
such approach has some limitations, in particular, it does not
reproduce the natural situation when various pathogens are coexisting
in the host organism and can aggravate pathological effects of each
other. Most frequently, such situation occurs in barrier tissues, such
as the respiratory tract, skin, and the digestive and urogenital
tracts, which normally are inhabited with symbiotic bacteria, fungi,
and other microorganisms. The intestinal microbiome is best studied; it
is known to influence many processes in the body, and its adequate
composition is beneficial for maintaining homeostasis of the immune
system [1]. A microbiome also exists in the lungs,
but its role was underestimated for a long time [2], although some representatives of the normal
microbiome, e.g. the bacteria Moraxella catarrhalis, are
opportunistic and capable of causing severe infections [3]. The majority of deaths during influenza pandemics
of 1918, 1957, and 1968 were caused by secondary bacterial infections
[4, 5]. Although infections of
the lower respiratory pathways are significantly behind cardiovascular
and oncologic diseases as causes of death, they are still among the ten
leading lethality causes even in developed countries, notwithstanding
impressive progress in therapy. Secondary bacterial infections on the
background of primary viral disease of the respiratory tract often
occur in current clinical practice, e.g. on the background of infection
with influenza virus or respiratory syncytial virus (RSV) [6-10]. In addition to
viral–bacterial coinfections, there are also cases of viral
diseases on the background of primary bacterial diseases [11].
In the present review, available data on viral–bacterial infections are considered, and special attention is given to diseases of the respiratory tract as the most widespread and the best studied. Infiltration of an infection site with cells of immune system, a decrease in the functional activity of these cells, an increase in immunosuppression due to production of glucocorticoids and immunosuppressive cytokines, and the role of Toll-like receptors (TLRs) in inflammatory processes during viral–bacterial coinfections are discussed.
INCIDENCE OF SECONDARY BACTERIAL INFECTIONS ON THE BACKGROUND OF
VIRAL DISEASES: CLINICAL CASES
Data on the incidence of secondary bacterial infections vary widely, from 4 to 66% [8, 12], and the range of the coinfection incidence observed in adults is much stronger (4-30%) than in children (22-33%) [12]. In addition to the patients’ age, there are other parameters obviously influencing the incidence of secondary coinfections: social status of the patients, geographic location, genetic predisposition, and the adopted standards of diagnosis and treatment. These parameters vary even within the same geographical region. Therefore, the wide range of the available data exemplified in the table is not surprising. The high adaptability of the widespread pathogens also plays an important role. Although during seasonal influenza epidemics of 2003-2008 in the USA, bacterial infections were observed in only 2% of patients younger than 18 years old [13], the number of recorded secondary bacterial infections with lethal outcome for this short period increased six-fold (from 6 to 34%). Meanwhile, the incidence of lethal infection with Staphylococcus aureus increased 15-fold (from 2 to 30%), and two thirds of these cases were represented by a highly pathogenic meticillin-resistant strain of S. aureus [14]. Most frequently, secondary bacterial infections were caused by microbes “classical” for viral–bacterial coinfections (Streptococcus pneumoniae, S. aureus, and Haemophilus influenzae), and by Moraxella catarrhalis, which is a frequent member of the normal microbiome of the upper respiratory pathways. Complications caused by secondary bacterial infections of upper respiratory pathways (sinusitis, bronchitis, genyantritis) were found in Russia in 32% of men hospitalized because of acute respiratory viral infection, and pneumonia developed in 25% of these patients [15].
Incidence of secondary bacterial infection on the background of viral
disease
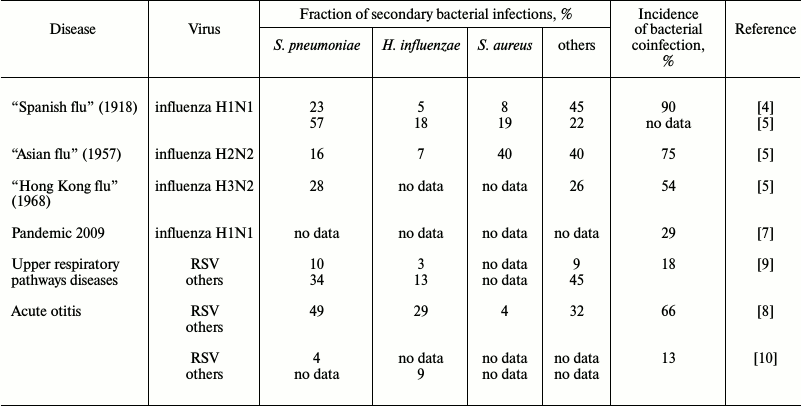
SECONDARY BACTERIAL INFECTIONS INCREASE RISK OF LETHAL OUTCOME IN
INFLUENZA PANDEMICS
The greatest attention was given to the viral–bacterial coinfection phenomenon during the influenza pandemic in 1918-1919 known as “Spanish flu”: ~50 million people died during 18 months because of the H1N1 subtype of influenza virus A [16]. Later it was found that ~90% of the deaths during this pandemic were associated with the presence in the pulmonary tissue of a “standard set” of pathogenic bacteria: Streptococcus pyogenes, S. pneumoniae, S. aureus, H. influenzae, and some others (table). Such high lethality because of secondary bacterial infections could be explained by the absence of antibiotics, but even after their appearance during the “Asian influenza” pandemic in 1957 (subtype H2N2 of influenza virus A) three quarters of the lethal cases were caused by bacterial complications [17]. The majority were caused by the same pathogens (table), although the ratio of pathogenic bacteria changed in comparison with that during the 1918 pandemic, and this could be associated with the greater resistance to antibiotics of bacteria of the Staphylococcus genus. In 1968 during the “Hong Kong influenza” pandemic on the background of infection with the H3N2 subtype of influenza virus A, the secondary infection most frequently was caused by S. pneumoniae. These bacteria were found in 27.8% of the cases, more often than all other bacteria together (table). In 2009 in the USA, there was a repeated pandemic of influenza virus A subtype H1N1, and notwithstanding the high level of the current medicine, secondary bacterial infections developed on influenza background in 29% of the cases (table).
ARE THERE STABLE “VIRUS—BACTERIUM” PAIRS AS
FREQUENT AGENTS OF COINFECTIONS?
There is a question whether a secondary infection with a particular bacterial species depends on the nature of the primary viral disease. There is no unambiguous answer to this question, but one can follow the tendency for development of particular coinfections in dependence on the patients’ age. In children, secondary bacterial infections most often develop on the background of infection with RSV, whereas in adults – on the background of influenza virus [12]. This can be associated with a partial retention in adults of immunity to RSV due to infection during their childhood. Influenza virus is known to be highly variable, which promotes seasonal epidemics [18], and can be the cause of a high incidence of coinfections in adults on the background of influenza virus. The existence of stable “virus–bacterium” pairs in secondary infections can be confirmed by the observation during the influenza pandemic of 2009: the secondary infection with S. aureus was more frequent in teenagers, whereas coinfection with S. pneumoniae was prevalent in adults [19].
IMMUNOLOGIC MECHANISMS RESPONSIBLE FOR DEVELOPMENT OF
VIRAL–BACTERIAL COINFECTIONS
The majority of viral infections of the respiratory tract are caused by so-called respiratory viruses: influenza virus, rhinoviruses, and RSV [20]. All respiratory viruses are RNA viruses characterized by a high rate of mutations and, as a consequence, by high variability. Just this feature of respiratory viruses, which is clearly pronounced and especially well studied in influenza virus, makes it impossible to form long-term immunity both natural and via vaccination. From the immune system standpoint, every cold, influenza, or rhinitis can have the same symptoms as previously, but be caused by a virus not encountered by the organism earlier.
There is an opinion formed historically that secondary bacterial infections are mainly caused by direct damage of the pulmonary tissue because of the viral infection [21], because many viruses, including influenza virus, are lytic. Secondary infections develop more rapidly than the immune response: mice are the most sensitive to secondary bacterial infections on the 7th day after infection with a virus, whereas one-to-two weeks are required for development of a stable adaptive immune response on the background of the virus-induced suppression of immunity [22]. Therefore, the innate immunity mechanisms play an important role in the development of coinfections, and it will be considered further.
Direct influence of viruses on state of respiratory tract tissues. Viruses influence development of secondary bacterial infection: neuraminidase (NA), a protein of the virus envelope, is responsible for hydrolysis of sialic acid residues on the surface of the host’s cells [23], and its activity releases hidden sites of bacterial adhesion on lung epithelial cells [24, 25] and also activates TGF-β [26]. During viral infection, increased expression of the platelet activation factor receptor (PAFR), which is also a receptor for adhesion of S. pneumoniae, is observed [27, 28]. Thus, viruses can directly promote the development of secondary bacterial infection (Fig. 1).
Fig. 1. Changes in immune response on successive infection with viruses and bacteria. PAFR, platelet activation factor receptor; T, T cell; B, B cell; MP, macrophage; AA-MP, alternatively activated macrophage; NP, neutrophil; Th2, T-helper type 2; Th17, T-helper type 17; Treg, regulatory T cell; γδT, gamma-delta T cell.
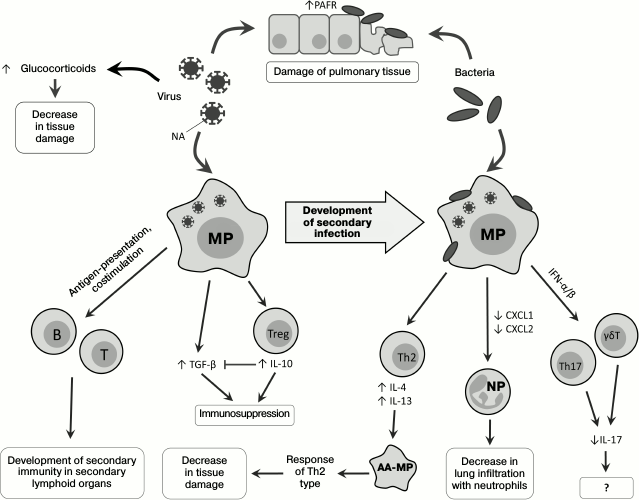
Infection of mice with influenza virus leads to damage of the pulmonary epithelium with subsequent development of a lethal infection by the gram-negative bacterium Legionella pneumophila [29]. During the secondary bacterial infection, an important role belongs to glucocorticoids, whose level significantly increases upon viral infection [30], possibly due to increase in production of proinflammatory cytokines in response to the viral infection [31, 32]. Glucocorticoids act systemically, suppressing immune response and decreasing tissue damage caused by the host’s immune cells, but not by the infecting agents during coinfections. In mice coinfected with influenza virus and Listeria monocytogenes, the levels of IFN-γ, IL-6, chemokines CXCL1, CXCL2 (Fig. 1), and adhesion molecules (ICAM-1) are decreased in comparison with mice infected only with the bacterium. After adrenalectomy, preventing the production of glucocorticoids, the coinfected mice did not differ in phenotype from the non-operated mice infected only by the bacterium: in these mice, the production of IL-6, chemokines, and adhesion molecules recovered, but the IFN-γ expression remained decreased [30].
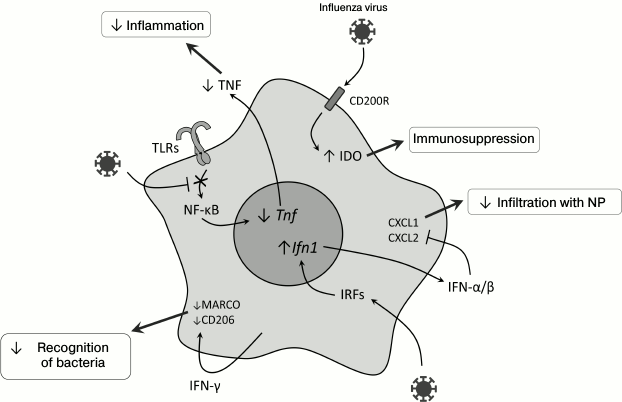
Macrophages and their role in development of secondary bacterial infections. Macrophages play an important role in the immune defense against bacteria penetrating into the lungs [33], and in the case of viral infection their ability for Fc-receptor-mediated phagocytosis is partially suppressed [34]. Pneumonia caused by influenza virus is associated with Th1-type differentiation of CD4+ cells and with production of IFN-γ required for effective struggle against intracellular pathogens [35]. However, in the case of infection with extracellular bacteria, e.g. S. pneumoniae, increased production of IFN-γ on the primary viral infection background suppresses Th2-differentiation and decreases the effectiveness of the antibacterial response [36]. Moreover, IFN-γ decreases expression of the innate immunity receptor MARCO, which recognizes pneumococcus, and of the mannose receptor, which triggers the complement system [36] (Fig. 2). Vaccination against influenza leads to increase in migration of the CD4+-T cells into the lungs, weakening IFN-γ production and normalizing functions of phagocytes [35].
However, it has been shown that infection with RSV results in the alternative, rather than classical, activation of macrophages, notwithstanding the production of IFN-γ [37, 38]. RSV stimulates the generation of IL-4 and IL-13, which are required alternative differentiation of macrophages. Alternatively activated macrophages, or M2-macrophages [39], do not produce reactive nitrogen species, and this critically lowers their bactericidal activity. On the other hand, a signal from IL-4Rα (the common subunit of IL-4 and IL-13 receptors) displaces the immune response towards Th2, which decreases pulmonary tissue damage [38] (Fig. 1). The bactericidal activity of alveolar macrophages decreased in mice as a result of viral infection could be additionally stimulated by exogenous activators of the innate immunity, e.g. by the granulocyte-macrophage colony-stimulating factor (GM-CSF) [40].
Are natural killers another potentially important participant of the inflammatory process? There are also data about the role of natural killers (NK cells) in the development of secondary bacterial infections, but it should be noted that mechanisms of NK cell participation in bacterial infections are not quite clear. Thus, mice with depleted NK cells were more resistant to influenza virus than wild type mice [41], which could be explained by a decrease in pulmonary tissue damage because of absence of virus-infected cell elimination. However, when mice infected with influenza virus were infected with S. aureus, a decrease in lung infiltration with NK cells aggravated the pathology. This was associated with a disturbance of NK cell activation by alveolar macrophages and by an insufficient production of tumor necrosis factor (TNF) by NK cells. The adoptive transfer of NK cells into the respiratory pathways of mice with influenza restored the production of TNF and promoted resistance to secondary bacterial infection [42].
Role of Toll-like receptors in the development of viral–bacterial coinfections. Innate immunity cells can recognize pathogen-associated molecular patterns (PAMPs), i.e. conservative pathogen components, such as lipopolysaccharide (LPS), single-stranded RNA, and double-stranded RNA [43]. This recognition occurs due to binding of PAMPs with innate immunity receptors, including the family of Toll-like receptors (TLRs) [44].
After infection with influenza virus, a long-term (up to 6 months) TLR-dependent desensitization was observed, i.e. loss of sensitivity of alveolar macrophages to bacterial ligands – LPS, flagellin, and lipoteichoic acids [45] – and this was not associated with suppressed expression of TLR on the cell surface but with a decrease in transcription factor NF-κB translocation into the nucleus (Fig. 2). Concurrently, poor migration into the lungs of the immune system cells (mainly of neutrophils) and decreased production of CXCL1, CXCL2, and TNF in response to the secondary bacterial infection on the background of viral infection were observed [45]. It was also shown that successive activation of mouse macrophages with TLR7 and TLR4 ligands decreased the expression of proinflammatory cytokine genes, which suggested a possible influence of juxtaposition of innate immunity receptor signals on desensitization of macrophages [46]. Inactivation of the TLR2-signaling pathway did not provide increased defense against secondary bacterial infection with S. pneumoniae [47], but S. pneumoniae could be recognized by the immune system through TLR4, which interacted with pneumolysin [48, 49] and explained the resistance of TLR2-deficient mice to infection with S. pneumoniae. Activation of the signaling TLR4 signaling pathway by monoclonal antibodies decreased pathologic effects caused by coinfection with the influenza virus and S. pneumoniae due to an increase in CXCL2 production and, consequently, attracting more macrophages into the lungs [50].
Fig. 2. Molecular events in myeloid cells during viral infection in the lungs, which can lead to weakening of the innate antibacterial response. CD200R, glycoprotein; Tnf, tumor necrosis factor gene; Ifn1, type I interferon genes; NP, neutrophils; MARCO, macrophage receptor with a collagen structure; CD206, mannose receptor; IRFs, interferon-regulatory factors.
As discussed earlier, alternatively activated macrophages counteract the development of secondary bacterial infection on viral infection background virtually without damaging tissues, and this mechanism is controlled through IL-4Rα, TLR4, and IFN-β [38]. TLR4 via an unknown mechanism induces production of the nuclear receptor PPARγ, which suppresses innate immunity due to binding with another nuclear protein, NCoR, which stabilizes the NF-κB complex with gene promoters [51]. In PPARγ-deficient mice, the response is shifted to Th1-cells, and lung tissues are damaged more than in wild type mice [38].
Activation of the TLR3-signaling pathway with involvement of polyinosinic-polycytidylic acid (polyIC, an analog of double-stranded viral RNA) before infection of mice with gram-positive bacteria worsens their condition and increases the disease duration via a mechanism depending on type I interferons (type I IFN) [52]. Infection of human dendritic cells in vitro with influenza virus and then with S. pneumoniae also increases the TLR3-dependent production of IL-12 [53]. The exact mechanism of this effect is unclear; supposedly, production of type I IFN in response to influenza virus increases TLR3 expression in endosomes, whereas RNA of S. pneumoniae released from the bacteria on the late endosomes stage activates TLR3 and thus increases the production of IL-12 [53, 54]. TLR3 also participates in secondary rhinoviral infections on the background of infection with H. influenzae [55], as discussed further in the next section.
Increase in production of immunosuppressive mediators during coinfection. Immunosuppression develops during primary viral infection and decreases inflammation-caused tissue damage, and this immunosuppression plays an important role in the development of secondary bacterial infection. As discussed earlier, NA of the influenza virus is able to activate TGF-β, which acts as a tissue protection factor but concurrently reduces the efficiency of virus elimination [56]. Th1 cells can counteract this action of the virus due to production of IL-10, which plays a key role in their regulation. During the early stage of the viral infection, IL-10 inhibits the TGF-β-mediated immunosuppression and restores the function of T cells. Later, when the viral NA becomes inactive, IL-10 begins to act as an immunosuppressor [57].
Infection with influenza virus is accompanied by activation of the signaling pathway from CD200R, a glycoprotein, which is expressed on the surface of the majority of myeloid and lymphoid cells and acts as an immunosuppressor in allergic and autoimmune reactions [58]. This leads to activation of the immunomodulatory enzyme indoleamine 2,3-dioxygenase (IDO), and the inflammatory response is suppressed due to production of IL-10 [28, 59, 60]. Such inhibition of inflammation together with desensitization of TLR (see above) decreases the damage of lung tissues and promotes their rapid recovery. Activation of IDO during viral infection (Fig. 2) leads to production of the immunosuppressive IL-10 [28], suppression of neutrophil functioning and the immune response in the lungs [61], and stimulates the accumulation of immunosuppressive myeloid cells [62].
Type I interferons are important “players” during coinfection. Type I IFNs are among the most important mediators of viral infection [63, 64]. Their generation increases expression of genes responsible for the antiviral response [65, 66], but type I IFNs suppress expression of neutrophil chemoattractants CXCL1 and CXCL2 (Fig. 2). As a result, mice deficient in the type I interferon receptor (Ifnar–/–) infected with influenza virus are more resistant to secondary bacterial infection with S. pneumoniae [67]. The role of type I IFNs in this mechanism was confirmed by data on the similar effect observed after the injection of polyIC, which is a ligand to TLR3 [52].
Type I IFNs also suppress the activation of Th17 cells, which are necessary for elimination of bacteria in the lungs: Th17 cells produce IL-17, IL-22, and IL-23, and IL-23 overexpression significantly improves the state of coinfected mice [68]. Moreover, type I IFNs suppress the ability of γδT cells to express IL-17 [69], which plays an important role during infection with influenza virus [70] (Fig. 1).
Secondary bacterial infections and adaptive immunity. Secondary bacterial infection can immediately influence the adaptive antiviral response of the immune system. During secondary bacterial infection with S. pneumoniae on the background of influenza virus, a strong stable B-cell response was observed in the mediastinal lymph nodes and spleen, and the blood serum level of IgG increased [71]. However, during the infection of mice with S. pneumoniae on the background of infection with influenza virus, the number of CD4+-T and B cells decreased and the titers of antiviral IgG, IgM, and IgA antibodies also decreased compared to mice infected only with the virus [72]. In the case of development of secondary infection with influenza virus after infection with S. pneumoniae, the primary B-cell response could be detected only in the local lymph nodes and spleen, but it was not retained for a long time and failed to protect against the virus [71].
Development of secondary viral infections on the background of primary bacterial infections and mechanisms involved in this process. It should be noted that only few cases of secondary viral diseases on the background of bacterial infections are described in the literature. Nevertheless, it was shown that infection with H. influenzae increased the expression of ICAM-1 and TLR3 interacting with the rhinovirus that promoted its penetration into cells [55]. Moreover, in vitro infection with human metapneumovirus (HMPV) occurred more often after infection caused by S. pneumoniae, but not by H. influenzae, M. catarrhalis, or S. aureus [73]. Infection with S. pneumoniae also increased the replication level of RSV and production of syncytial cells in vitro and in vivo [11], but on successive infection in vitro of epithelial cell lines with S. pneumoniae and the influenza virus, influenza virus replication did not increase [74].
Clinical data indicate that secondary bacterial infections are a great threat for human health and life. According to the data presented in this review, pneumonia often leads to lethal outcome. The upper respiratory pathways are connected with the middle ear; therefore, acute respiratory diseases can be complicated by otitis, which is often accompanied by viral–bacterial coinfections and results in hearing loss in children [75]. However, despite efforts of many research groups throughout the world, details of the mechanisms underlying secondary bacterial infections are still unclear.
An important role of the innate immunity during the development of viral–bacterial coinfections in respiratory diseases has been shown in many works. In particular, a decrease in the entrance of immune cells into the lungs is extremely important for the development of coinfections. This decrease can be caused by a weakened production of chemokines and proinflammatory cytokines in the lung tissues, mainly those mediated by type I IFN and IFN-γ. Concurrently, production of anti-inflammatory cytokines is increased.
Adaptive immune reactions have not been studied comprehensively during coinfections, although in the pneumonia model in mice it has been shown that numbers of T and B cells in the lungs can be significantly decreased in coinfections compared to mice infected only with bacteria [72]. It is interesting if there are qualitative changes in the repertoires of T- and B-lymphocyte receptors in viral–bacterial coinfections in comparison with solitary infections. Deep sequencing methods can be of help for answering these questions [76]. It is also interesting if the allelic set of the HLA-locus genes of patients can influence the possibility of secondary infection development on the background of viral diseases [19].
One of the major problems in the identification of viral–bacterial coinfections is the absence of a reliable method for determination of infection in a patient. Recently, a research group from Stanford University proposed an approach for discriminating viral and bacterial diseases using a simple test on expression of a set of 11 genes [77]. Moreover, this test allows researchers to differentiate infection with the influenza virus from infections with other viruses [78]. This approach is promising not only for accurate statistical analysis of the expansion of viral–bacterial coinfections, but also for choosing the necessary treatment. However, changes in the expression of these genes have not been studied yet in viral–bacterial infections and are interesting in the context of the theme under discussion. It can be expected that in the development of personalized medicine the above-mentioned advances in molecular-genetic analysis of adaptive immunity components will result in rapid progress in understanding the individual reactions of patients to coinfections.
Acknowledgements
We are grateful to Prof. S. A. Nedospasov for his valuable remarks.
This work was supported by the Russian Federation President for the Russian Federation Leading Scientific Schools (NS-10014.2016.4) and by the Russian Foundation for Basic Research (project No. 16-34-01087 mol_a).
REFERENCES
1.Nellore, A., and Fishman, J. A. (2016) The
microbiome, systemic immune function, and allotransplantation, Clin.
Microbiol. Rev., 29, 191-199.
2.Beck, J. M., Young, V. B., and Huffnagle, G. B.
(2012) The microbiome of the lung, Transl. Res., 160,
258-266.
3.Murphy, T. F., and Parameswaran, G. I. (2009)
Moraxella catarrhalis, a human respiratory tract pathogen,
Clin. Infect. Dis., 49, 124-131.
4.Morens, D. M., Taubenberger, J. K., and Fauci, A.
S. (2008) Predominant role of bacterial pneumonia as a cause of death
in pandemic influenza: implications for pandemic influenza
preparedness, J. Infect. Dis., 198, 962-970.
5.Wang, X. Y., Kilgore, P. E., Lim, K. A., Wang, S.
M., Lee, J., Deng, W., Mo, M. Q., Nyambat, B., Ma, J. C., Favorov, M.
O., and Clemens, J. D. (2011) Influenza and bacterial pathogen
coinfections in the 20th century, Interdiscip. Perspect. Infect.
Dis., 2011, 146376.
6.Thorburn, K., Harigopal, S., Reddy, V., Taylor, N.,
and Van Saene, H. K. (2006) High incidence of pulmonary bacterial
coinfection in children with severe respiratory syncytial virus (RSV)
bronchiolitis, Thorax, 61, 611-615.
7.Centers for Disease Control and Prevention (2009)
Bacterial coinfections in lung tissue specimens from fatal cases of
2009 pandemic influenza A (H1N1) United States, May-August 2009,
MMWR Morb. Mortal. Wkly Rep., 58, 1071-1074.
8.Ruohola, A., Meurman, O., Nikkari, S., Skottman,
T., Salmi, A., Waris, M., Osterback, R., Eerola, E., Allander, T.,
Niesters, H., Heikkinen, T., and Ruuskanen, O. (2006) Microbiology of
acute otitis media in children with tympanostomy tubes: prevalences of
bacteria and viruses, Clin. Infect. Dis., 43,
1417-1422.
9.Lehtinen, P., Jartti, T., Virkki, R., Vuorinen, T.,
Leinonen, M., Peltola, V., Ruohola, A., and Ruuskanen, O. (2006)
Bacterial coinfections in children with viral wheezing, Eur. J.
Clin. Microbiol. Infect. Dis., 25, 463-469.
10.Pettigrew, M. M., Gent, J. F., Pyles, R. B.,
Miller, A. L., Nokso-Koivisto, J., and Chonmaitree, T. (2011)
Viral–bacterial interactions and risk of acute otitis media
complicating upper respiratory tract infection, J. Clin.
Microbiol., 49, 3750-3755.
11.Nguyen, D. T., Louwen, R., Elberse, K., Van
Amerongen, G., Yuksel, S., Luijendijk, A., Osterhaus, A. D., Duprex, W.
P., and De Swart, R. L. (2015) Streptococcus pneumoniae enhances
human respiratory syncytial virus infection in vitro and in
vivo, PLoS One, 10, e0127098.
12.Pavia, A. T. (2013) What is the role of
respiratory viruses in community-acquired pneumonia? What is the best
therapy for influenza and other viral causes of community-acquired
pneumonia? Infect. Dis. Clin. North Am., 27, 157-175.
13.Dawood, F. S., Fiore, A., Kamimoto, L., Nowell,
M., Reingold, A., Gershman, K., Meek, J., Hadler, J., Arnold, K. E.,
Ryan, P., Lynfield, R., Morin, C., Baumbach, J., Zansky, S., Bennett,
N. M., Thomas, A., Schaffner, W., Kirschke, D., Finelli, L., and
Emerging Infections Program Network (2010) Influenza-associated
pneumonia in children hospitalized with laboratory-confirmed influenza,
2003-2008, Pediatr. Infect. Dis. J., 29, 585-590.
14.Finelli, L., Fiore, A., Dhara, R., Brammer, L.,
Shay, D. K., Kamimoto, L., Fry, A., Hageman, J., Gorwitz, R., Bresee,
J., and Uyeki, T. (2008) Influenza-associated pediatric mortality in
the United States: increase of Staphylococcus aureus
coinfection, Pediatrics, 122, 805-811.
15.Khaitov, R. M., and Ataullakhanov, R. I. (2012)
Immunotherapy: A Guidebook [in Russian], GEOTAR-Media,
Moscow.
16.Taubenberger, J. K., and Morens, D. M. (2006)
1918 influenza: the mother of all pandemics, Emerg. Infect.
Dis., 12, 15-22.
17.Louria, D. B., Blumenfeld, H. L., Ellis, J. T.,
Kilbourne, E. D., and Rogers, D. E. (1959) Studies on influenza in the
pandemic of 1957-1958. II. Pulmonary complications of influenza, J.
Clin. Invest., 38, 213-265.
18.Lofgren, E., Fefferman, N. H., Naumov, Y. N.,
Gorski, J., and Naumova, E. N. (2007) Influenza seasonality: underlying
causes and modeling theories, J. Virol., 81,
5429-5436.
19.Louie, L. G., Hartogensis, W. E., Jackman, R. P.,
Schultz, K. A., Zijenah, L. S., Yiu, C. H., Nguyen, V. D., Sohsman, M.
Y., Katzenstein, D. K., and Mason, P. R. (2004) Mycobacterium
tuberculosis/HIV-1 coinfection and disease: role of human leukocyte
antigen variation, J. Infect. Dis., 189, 1084-1090.
20.Hament, J. M., Kimpen, J. L., Fleer, A., and
Wolfs, T. F. (1999) Respiratory viral infection predisposing for
bacterial disease: a concise review, FEMS Immunol. Med.
Microbiol., 26, 189-195.
21.MacCallum, W. G. (1921) Pathological anatomy of
pneumonia associated with influenza, Johns Hopkins Hosp. Rep.,
20, 149-249.
22.McCullers, J. A., and Rehg, J. E. (2002) Lethal
synergism between influenza virus and Streptococcus pneumoniae:
characterization of a mouse model and the role of platelet-activating
factor receptor, J. Infect. Dis., 186, 341-350.
23.Air, G. M., and Laver, W. G. (1989) The
neuraminidase of influenza virus, Proteins, 6,
341-356.
24.McCullers, J. A., and Bartmess, K. C. (2003) Role
of neuraminidase in lethal synergism between influenza virus and
Streptococcus pneumoniae, J. Infect. Dis., 187,
1000-1009.
25.Siegel, S. J., Roche, A. M., and Weiser, J. N.
(2014) Influenza promotes pneumococcal growth during coinfection by
providing host sialylated substrates as a nutrient source, Cell Host
Microbe, 16, 55-67.
26.Carlson, C. M., Turpin, E. A., Moser, L. A.,
O’Brien, K. B., Cline, T. D., Jones, J. C., Tumpey, T. M., Katz,
J. M., Kelley, L. A., Gauldie, J., and Schultz-Cherry, S. (2010)
Transforming growth factor-β: activation by neuraminidase and role
in highly pathogenic H5N1 influenza pathogenesis, PLoS Pathog.,
6, e1001136.
27.Ishizuka, S., Yamaya, M., Suzuki, T., Takahashi,
H., Ida, S., Sasaki, T., Inoue, D., Sekizawa, K., Nishimura, H., and
Sasaki, H. (2003) Effects of rhinovirus infection on the adherence of
Streptococcus pneumoniae to cultured human airway epithelial
cells, J. Infect. Dis., 188, 1928-1939.
28.Van der Sluijs, K. F., Nijhuis, M., Levels, J.
H., Florquin, S., Mellor, A. L., Jansen, H. M., Van der Poll, T., and
Lutter, R. (2006) Influenza-induced expression of indoleamine
2,3-dioxygenase enhances interleukin-10 production and bacterial
outgrowth during secondary pneumococcal pneumonia, J. Infect.
Dis., 193, 214-222.
29.Jamieson, A. M., Pasman, L., Yu, S., Gamradt, P.,
Homer, R. J., Decker, T., and Medzhitov, R. (2013) Role of tissue
protection in lethal respiratory viral–bacterial coinfection,
Science, 340, 1230-1234.
30.Jamieson, A. M., Yu, S., Annicelli, C. H., and
Medzhitov, R. (2010) Influenza virus-induced glucocorticoids compromise
innate host defense against a secondary bacterial infection, Cell
Host Microbe, 7, 103-114.
31.Bethin, K. E., Vogt, S. K., and Muglia, L. J.
(2000) Interleukin-6 is an essential, corticotropin-releasing
hormone-independent stimulator of the adrenal axis during immune system
activation, Proc. Natl. Acad. Sci. USA, 97,
9317-9322.
32.Dunn, A. J., and Vickers, S. L. (1994)
Neurochemical and neuroendocrine responses to Newcastle disease virus
administration in mice, Brain Res., 645, 103-112.
33.Green, G. M., and Kass, E. H. (1964) The role of
the alveolar macrophage in the clearance of bacteria from the lung,
J. Exp. Med., 119, 167-176.
34.Nickerson, C. L., and Jakab, G. J. (1990)
Pulmonary antibacterial defenses during mild and severe influenza virus
infection, Infect. Immun., 58, 2809-2814.
35.Sun, K., Ye, J., Perez, D. R., and Metzger, D. W.
(2011) Seasonal FluMist vaccination induces cross-reactive T-cell
immunity against H1N1 (2009) influenza and secondary bacterial
infections, J. Immunol., 186, 987-993.
36.Sun, K., and Metzger, D. W. (2008) Inhibition of
pulmonary antibacterial defense by interferon-γ during recovery
from influenza infection, Nat. Med., 14, 558-564.
37.Mosser, D. M. (2003) The many faces of macrophage
activation, J. Leukoc. Biol., 73, 209-212.
38.Shirey, K. A., Pletneva, L. M., Puche, A. C.,
Keegan, A. D., Prince, G. A., Blanco, J. C., and Vogel, S. N. (2010)
Control of RSV-induced lung injury by alternatively activated
macrophages is IL-4Rα-, TLR4-, and IFN-β-dependent,
Mucosal Immunol., 3, 291-300.
39.Schwartz, Y. Sh., and Svistelnik, A. V. (2012)
Functional phenotypes of macrophages and the M1-M2 polarization
concept. Part I. Proinflammatory phenotype, Biochemistry
(Moscow), 77, 246-260.
40.Subramaniam, R., Barnes, P. F., Fletcher, K.,
Boggaram, V., Hillberry, Z., Neuenschwander, P., and Shams, H. (2014)
Protecting against post-influenza bacterial pneumonia by increasing
phagocyte recruitment and ROS production, J. Infect. Dis.,
209, 1827-1836.
41.Abdul-Careem, M. F., Mian, M. F., Yue, G.,
Gillgrass, A., Chenoweth, M. J., Barra, N. G., Chew, M. V., Chan, T.,
Al-Garawi, A. A., Jordana, M., and Ashkar, A. A. (2012) Critical role
of natural killer cells in lung immunopathology during influenza
infection in mice, J. Infect. Dis., 206, 167-177.
42.Small, C. L., Shaler, C. R., McCormick, S.,
Jeyanathan, M., Damjanovic, D., Brown, E. G., Arck, P., Jordana, M.,
Kaushic, C., Ashkar, A. A., and Xing, Z. (2010) Influenza infection
leads to increased susceptibility to subsequent bacterial
superinfection by impairing NK cell responses in the lung, J.
Immunol., 184, 2048-2056.
43.Akira, S., Uematsu, S., and Takeuchi, O. (2006)
Pathogen recognition and innate immunity, Cell, 124,
783-801.
44.Akira, S. (2006) TLR signaling, Curr. Top.
Microbiol. Immunol., 311, 1-16.
45.Didierlaurent, A., Goulding, J., Patel, S.,
Snelgrove, R., Low, L., Bebien, M., Lawrence, T., Van Rijt, L. S.,
Lambrecht, B. N., Sirard, J. C., and Hussell, T. (2008) Sustained
desensitization to bacterial Toll-like receptor ligands after
resolution of respiratory influenza infection, J. Exp. Med.,
205, 323-329.
46.Sviriaeva, E. N.., Korneev, K. V., Drutskaya, M.
S., Nedospasov, S. A., and Kuprash, D. V. (2016) Modelling of
bacterial, viral co-infection at the molecular level using receptor
agonists innate immunity, Dokl. Akad. Nauk, 471, in
press.
47.Dessing, M. C., Van der Sluijs, K. F., Florquin,
S., Akira, S., and Van der Poll, T. (2007) Toll-like receptor 2 does
not contribute to host response during postinfluenza pneumococcal
pneumonia, Am. J. Respir. Cell. Mol. Biol., 36,
609-614.
48.Malley, R., Henneke, P., Morse, S. C.,
Cieslewicz, M. J., Lipsitch, M., Thompson, C. M., Kurt-Jones, E.,
Paton, J. C., Wessels, M. R., and Golenbock, D. T. (2003) Recognition
of pneumolysin by Toll-like receptor 4 confers resistance to
pneumococcal infection, Proc. Natl. Acad. Sci. USA, 100,
1966-1971.
49.Dessing, M. C., Florquin, S., Paton, J. C., and
Van der Poll, T. (2008) Toll-like receptor 2 contributes to
antibacterial defence against pneumolysin-deficient pneumococci,
Cell Microbiol., 10, 237-246.
50.Tanaka, A., Nakamura, S., Seki, M., Fukudome, K.,
Iwanaga, N., Imamura, Y., Miyazaki, T., Izumikawa, K., Kakeya, H.,
Yanagihara, K., and Kohno, S. (2013) Toll-like receptor 4 agonistic
antibody promotes innate immunity against severe pneumonia induced by
coinfection with influenza virus and Streptococcus pneumoniae,
Clin. Vaccine Immunol., 20, 977-985.
51.Wahli, W., and Michalik, L. (2012) PPARs at the
crossroads of lipid signaling and inflammation, Trends Endocrinol.
Metab., 23, 351-363.
52.Nakamura, S., Davis, K. M., and Weiser, J. N.
(2011) Synergistic stimulation of type I interferons during influenza
virus coinfection promotes Streptococcus pneumoniae colonization
in mice, J. Clin. Invest., 121, 3657-3665.
53.Spelmink, L., Sender, V., Hentrich, K., Kuri, T.,
Plant, L., and Henriques-Normark, B. (2016) Toll-Like receptor
3/TRIF-dependent IL-12p70 secretion mediated by Streptococcus
pneumoniae RNA and its priming by influenza A virus coinfection in
human dendritic cells, MBio, 7, e00168-16.
54.Kuri, T., Sorensen, A. S., Thomas, S., Karlsson
Hedestam, G. B., Normark, S., Henriques-Normark, B., McInerney, G. M.,
and Plant, L. (2013) Influenza A virus-mediated priming enhances
cytokine secretion by human dendritic cells infected with
Streptococcus pneumoniae, Cell Microbiol., 15,
1385-1400.
55.Sajjan, U. S., Jia, Y., Newcomb, D. C., Bentley,
J. K., Lukacs, N. W., LiPuma, J. J., and Hershenson, M. B. (2006) H.
influenzae potentiates airway epithelial cell responses to
rhinovirus by increasing ICAM-1 and TLR3 expression, FASEB J.,
20, 2121-2123.
56.Williams, A. E., Humphreys, I. R., Cornere, M.,
Edwards, L., Rae, A., and Hussell, T. (2005) TGF-β prevents
eosinophilic lung disease but impairs pathogen clearance, Microbes
Infect., 7, 365-374.
57.Dutta, A., Huang, C. T., Chen, T. C., Lin, C. Y.,
Chiu, C. H., Lin, Y. C., Chang, C. S., and He, Y. C. (2015) IL-10
inhibits neuraminidase-activated TGF-β and facilitates Th1
phenotype during early phase of infection, Nat. Commun.,
6, 6374.
58.Gorczynski, R. M. (2005) CD200 and its receptors
as targets for immunoregulation, Curr. Opin. Investig. Drugs,
6, 483-488.
59.Yoshida, R., Imanishi, J., Oku, T., Kishida, T.,
and Hayaishi, O. (1981) Induction of pulmonary indoleamine
2,3-dioxygenase by interferon, Proc. Natl. Acad. Sci. USA,
78, 129-132.
60.Hussell, T., and Cavanagh, M. M. (2009) The
innate immune rheostat: influence on lung inflammatory disease and
secondary bacterial pneumonia, Biochem. Soc. Trans., 37,
811-813.
61.De Vries, J. E. (1995) Immunosuppressive and
anti-inflammatory properties of interleukin 10, Ann. Med.,
27, 537-541.
62.Ponomarev, A. B. (2016) Myeloid suppressory
cells: general characteristics, Immunologiya, 37,
47-50.
63.Diebold, S. S., Montoya, M., Unger, H.,
Alexopoulou, L., Roy, P., Haswell, L. E., Al-Shamkhani, A., Flavell,
R., Borrow, P., and Reise Sousa, C. (2003) Viral infection switches
non-plasmacytoid dendritic cells into high interferon producers,
Nature, 424, 324-328.
64.Gonzalez-Navajas, J. M., Lee, J., David, M., and
Raz, E. (2012) Immunomodulatory functions of type I interferons,
Nat. Rev. Immunol., 12, 125-135.
65.Schneider, W. M., Chevillotte, M. D., and Rice,
C. M. (2014) Interferon-stimulated genes: a complex web of host
defenses, Annu. Rev. Immunol., 32, 513-545.
66.Floyd-Smith, G., Slattery, E., and Lengyel, P.
(1981) Interferon action: RNA cleavage pattern of a
(2′-5′)oligoadenylate-dependent endonuclease,
Science, 212, 1030-1032.
67.Shahangian, A., Chow, E. K., Tian, X., Kang, J.
R., Ghaffari, A., Liu, S. Y., Belperio, J. A., Cheng, G., and Deng, J.
C. (2009) Type I IFNs mediate development of postinfluenza bacterial
pneumonia in mice, J. Clin. Invest., 119, 1910-1920.
68.Kudva, A., Scheller, E. V., Robinson, K. M.,
Crowe, C. R., Choi, S. M., Slight, S. R., Khader, S. A., Dubin, P. J.,
Enelow, R. I., Kolls, J. K., and Alcorn, J. F. (2011) Influenza A
inhibits Th17-mediated host defense against bacterial pneumonia in
mice, J. Immunol., 186, 1666-1674.
69.Henry, T., Kirimanjeswara, G. S., Ruby, T.,
Jones, J. W., Peng, K., Perret, M., Ho, L., Sauer, J. D., Iwakura, Y.,
Metzger, D. W., and Monack, D. M. (2010) Type I IFN signaling
constrains IL-17A/F secretion by γδ T-cells during
bacterial infections, J. Immunol., 184, 3755-3767.
70.Li, W., Moltedo, B., and Moran, T. M. (2012) Type
I interferon induction during influenza virus infection increases
susceptibility to secondary Streptococcus pneumoniae infection
by negative regulation of γδ T cells, J. Virol.,
86, 12304-12312.
71.Wolf, A. I., Strauman, M. C., Mozdzanowska, K.,
Whittle, J. R., Williams, K. L., Sharpe, A. H., Weiser, J. N., Caton,
A. J., Hensley, S. E., and Erikson, J. (2014) Coinfection with
Streptococcus pneumoniae modulates the B cell response to
influenza virus, J. Virol., 88, 11995-12005.
72.Wu, Y., Tu, W., Lam, K. T., Chow, K. H., Ho, P.
L., Guan, Y., Peiris, J. S., and Lau, Y. L. (2015) Lethal coinfection
of influenza virus and Streptococcus pneumoniae lowers antibody
response to influenza virus in lung and reduces numbers of germinal
center B cells, T follicular helper cells, and plasma cells in
mediastinal lymph node, J. Virol., 89, 2013-2023.
73.Verkaik, N. J., Nguyen, D. T., de Vogel, C. P.,
Moll, H. A., Verbrugh, H. A., Jaddoe, V. W., Hofman, A., Van Wamel, W.
J., Van den Hoogen, B. G., Buijs-Offerman, R. M., Ludlow, M., De Witte,
L., Osterhaus, A. D., Van Belkum, A., and De Swart, R. L. (2011)
Streptococcus pneumoniae exposure is associated with human
metapneumovirus seroconversion and increased susceptibility to in
vitro HMPV infection, Clin. Microbiol. Infect., 17,
1840-1844.
74.Ouyang, K., Woodiga, S. A., Dwivedi, V.,
Buckwalter, C. M., Singh, A. K., Binjawadagi, B., Hiremath, J.,
Manickam, C., Schleappi, R., Khatri, M., Wu, J., King, S. J., and
Renukaradhya, G. J. (2014) Pretreatment of epithelial cells with live
Streptococcus pneumoniae has no detectable effect on influenza A
virus replication in vitro, PLoS One, 9,
e90066.
75.Paparella, M. M., Morizono, T., Le, C. T.,
Mancini, F., Sipila, P., Choo, Y. B., Liden, G., and Kim, C. S. (1984)
Sensorineural hearing loss in otitis media, Ann. Otol. Rhinol.
Laryngol., 93, 623-629.
76.Nazarov, V. I., Pogorelyy, M. V., Komech, E. A.,
Zvyagin, I. V., Bolotin, D. A., Shugay, M., Chudakov, D. M., Lebedev,
Y. B., and Mamedov, I. Z. (2015) tcR: an R package for T-cell receptor
repertoire advanced data analysis, BMC Bioinformatics,
16, 175.
77.Sweeney, T. E., Wong, H. R., and Khatri, P.
(2016) Robust classification of bacterial and viral infections via
integrated host gene expression diagnostics, Sci. Transl. Med.,
8, 346ra391.
78.Andres-Terre, M., McGuire, H. M., Pouliot, Y.,
Bongen, E., Sweeney, T. E., Tato, C. M., and Khatri, P. (2015)
Integrated, multi-cohort analysis identifies conserved transcriptional
signatures across multiple respiratory viruses, Immunity,
43, 1199-1211.